

Abstract
Gene multiplications or point mutations in alpha (α)-synuclein are associated with familial and sporadic Parkinson’s disease (PD). An increase in copper (Cu) levels has been reported in the cerebrospinal fluid and blood of PD patients, while occupational exposure to Cu has been suggested to augment the risk to develop PD. We aimed to elucidate the mechanisms by which α-synuclein and Cu regulate dopaminergic cell death. Short-term overexpression of WT or A53T α-synuclein had no toxic effect in human dopaminergic cells and primary midbrain cultures, but it exerted a synergistic effect on Cu-induced cell death. Cell death induced by Cu was potentiated by overexpression of the Cu transporter protein 1 (Ctr1) and depletion of intracellular glutathione (GSH) indicating that the toxic effects of Cu are linked to alterations in its intracellular homeostasis. Using the redox sensor roGFP, we demonstrated that Cu-induced oxidative stress was primarily localized in the cytosol and not in the mitochondria. However, α-synuclein overexpression had no effect on Cu-induced oxidative stress. WT or A53T α-synuclein overexpression exacerbated Cu toxicity in dopaminergic cells and yeast in the absence of α-synuclein aggregation. Cu increased autophagic flux and protein ubiquitination. Impairment of autophagy by overexpression of a dominant negative Atg5 form or inhibition of the ubiquitin/proteasome system (UPS) with MG132 enhanced Cu-induced cell death. However, only inhibition of the UPS stimulated the synergistic toxic effects of Cu and α-synuclein overexpression. Our results demonstrate that α-synuclein stimulates Cu toxicity in dopaminergic cells independent from its aggregation via modulation of protein degradation pathways.
Keywords: α-Synuclein, A53T, Autophagy, Proteasome, Ubiquitination, Oxidative stress, AMPK, Copper and Parkinson’s disease
Introduction
The progressive neurodegeneration in Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons in the substantia nigra, and the presence of the fibrillar cytoplasmic aggregates of α-synuclein. Multiplication and mutations in α-synuclein (SNCA/PARK1) (Chartier-Harlin et al., 2004; Polymeropoulos et al., 1997) and its abnormal aggregation are implicated in the etiology of familial or sporadic PD, as well as in other α-synucleinopathies (Galvin et al., 2001; Stefanis, 2012). A53T mutation is the most frequent mutation with a penetrance of 85%, while duplications have only reduced penetrance suggesting that additional factors might contribute to disease progression. Gene dosage (number of SNCA copies) has been associated with an earlier onset, more severe phenotype, and faster disease progression (Klein and Westenberger, 2012; Rochet et al., 2012). Conformational changes in α-synuclein including oligomerization and fibrillogenesis are also associated with PD. α-synuclein aggregation is modulated by various mechanisms including phosphorylation, C-terminal truncation, interactions with metals or redox reactions (Breydo et al., 2012; Stefanis, 2012). However, the pathogenicity of these conformational changes is unclear as some studies suggest that mature fibrils might sequester toxic protofibrillar intermediates, and that aggregation of α-synuclein is required for its detoxification (Klucken et al., 2012). A variety of transgenic animal models overexpressing either WT or mutant α-synuclein have been established. Nevertheless, dopaminergic cell loss is poorly observed in most of these transgenic mouse models (Horowitz and Greenamyre, 2010). These observations suggest that other factors, including aging or environmental exposures might contribute to PD associated with alterations in α-synuclein (Cannon and Greenamyre, 2013; Horowitz and Greenamyre, 2010; Stefanis, 2012).
Only 10% of people with PD have a family history of this disorder (Klein and Westenberger, 2012). Most of PD occurs in a sporadic (idiopathic) form, without a defined genetic basis. Thus, it is considered that PD arises from the convergence of genetic susceptibility, environmental factors, and aging (Horowitz and Greenamyre, 2010; Vance et al., 2010). Epidemiological evidence suggests an involvement of heavy metals in the etiology of PD (Santner and Uversky, 2010). Dysregulation of intracellular metal homeostasis has been suggested as a contributor to PD progression (Ayton et al., 2012). An increase in copper (Cu) levels has been reported in the cerebrospinal fluid and blood of PD patients (Hozumi et al., 2011; Pall et al., 1987), while occupational exposure to Cu augments the risk to develop PD (Gorell et al., 1997, 1999; Rybicki et al., 1993; Santner and Uversky, 2010). In contrast, other studies have reported a decrease or no change in Cu levels in the serum of PD patients or in the substantia nigra of post-mortem PD brains (Dexter et al., 1989; Jimenez-Jimenez et al., 1992; Mariani et al., 2013).
Cu is an essential metal ion in the central nervous system. As a transition metal, Cu mediates oxidation-reduction reactions and participates in the generation of reactive oxygen species (ROS) and oxidative damage. Thus, a number of Cu-binding proteins and transporters have evolved to facilitate Cu-dependent enzymatic processes, maintain Cu homeostasis, and minimize its detrimental effects (Kim et al., 2008). An unbalanced intracellular Cu homeostasis induces profound defects in neurological function. Human genetic disorders involving dysfunction of Cu-transporting ATPases, such as Menke’s and Wilson’s disease, have demonstrated the importance of Cu homeostasis in brain function. For example, the Cu overload in Wilson’s disease leads to oxidative stress and progressive neurodegeneration that develops in parkinsonism (Gaier et al., 2013).
Previous studies have aimed to understand the mechanism by which Cu regulates α-synuclein aggregation and toxicity. In this study, we demonstrated that non-toxic α-synuclein overexpression can increase dopaminergic cell death induced by Cu, independent from oxidative stress or α-synuclein aggregation. The synergistic toxicity induced by Cu and α-synuclein overexpression was found to be linked to the regulation of protein degradation pathways, particularly the ubiquitin proteasome system (UPS). These results demonstrate that α-synuclein can stimulate Cu toxicity independent from its aggregation.
Materials and methods
Cell culture and reagents
The dopaminergic properties of the neuroblastoma cell line SK-N-SH and their cell culture have been detailed before (Garcia-Garcia et al., 2013). Wild type (WT), double knockout AMPK (DKO-AMPK−/−) and Ulk1 knockout (Ulk1−/−) mouse embryonic fibroblasts (MEFs) were kindly provided by Dr. Mondira Kundu (St. Jude Children’s Research Hospital) and Dr. Benoit Viollet (Institut Cochin INSERM) (J. Kim et al., 2011). MEFs were cultured in Dulbecco’s Modified Eagle: Nutrient Mixture F-12 (DMEM/F-12) media supplemented with 10% [v/v] fetal bovine serum (FBS), penicillin (200 U/ml), streptomycin (200 μg/ml), 2 mM L-glutamine, and 200 μM β-mercaptoethanol. Cell culture reagents were obtained from Thermo Scientific/Hyclone or Life Technologies/GIBCO. L-buthionine-sulfoximide (B2515) and Chloroquine (C6628) were obtained from Sigma/Aldrich. CuCl2, was from Thermo/Fisher Scientific. z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone, 219007) and Ac-DMQD-CHO (235421) were from EMD Millipore. MG132 (carbobenzoxy-L-leucyl-L-leucyl-L-leucinal, Z-LLL-CHO) was obtained from Cayman Chemical. When inhibitors were used, control samples contained equal volume or concentration of vehicle (DMSO), which never exceeded 1% (v/v). All other reagents were obtained from Sigma/Aldrich, Thermo/Fisher Scientific or Acros Organics.
Intracellular Cu analysis by inductively coupled plasma mass spectrometry (ICP-MS)
Cells with and without CuCl2 supplementation were harvested and washed twice in TBS + EDTA (10 mM) to remove Cu that had not been incorporated by cells. Cell pellets were dissolved in 20% [v/v] HNO3 overnight at room temperature. Cu levels were measured in an ICP-MS 7500cs (Agilent) coupled to an autosampler (Elemental Scientific). The instrument was operated in collision/reaction mode (He 3.5 ml/min) for the elimination of polyatomic species. The system used 2% [v/v] HNO3 as rinse and carrier fluid. 50 ppb gallium (Ga) was added to each sample as an internal standard. The concentrations of Cu were calculated from an external calibration curve yielding linear fits with correlation coefficients equal or better than 0.998. The total acquisition time was 65 s per sample and each isotope was read 3 times with dwell times of 0.1 s in triplicate. The results were normalized to protein concentration.
Recombinant adenoviral vectors
Replication-deficient recombinant adenoviruses (Ad5CMV) encoding WT and mutant A53T α-synuclein have been described previously (Liu et al., 2008a). Ad-dnAtg5, Ad-ATP7A-myc and Ad-GFP-LC3 were generously provided by Dr. Gökhan S. Hotamisligil (Harvard School of Public Health) (Yang et al., 2010), Dr. Giuseppe Inesi (California Pacific Medical Center Research Institute) (Liu et al., 2010), and Dr. Aviva M. Tolkovski (University of Cambridge) (Bampton et al., 2005), respectively. Adenovirus construct to overexpress human (h) CTR1 was generated using the Ad-Easy system (He et al., 1998) and hCTR1 cDNA (Lee et al., 2002). Adenovirus containing only the CMV promoter (Ad-Empty) was used as negative control. Adenoviruses were amplified and titered in HEK293T cells as described previously (Barde et al., 2010; Rodriguez-Rocha et al., 2012). Cells were infected with adenoviral particles at indicated multiplicity of infection (MOI), washed and treated under the specified experimental conditions at 24 h post-infection.
Cell death determination
Loss of cell viability was determined by measuring propidium iodide (PI, 1 μg/ml) (Life Technologies/Molecular Probes) uptake as a marker for plasma membrane integrity loss. PI fluorescence was detected using 488 nm excitation (ex), 695/40 nm emission (em) or 561 nm ex, 615/25 nm em filters. Apoptosis was determined by assessing the externalization of phosphatidylserine using Annexin V. When indicated, cells were washed in PBS and stained with Annexin V-Alexa Fluor 488 (1 μl/2.4 × 105 cells, Life Technologies/Molecular Probes) in binding buffer (150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM Hepes [pH 7.4]) for 20 min at RT. Alexa 488 fluorescence was recorded using 488 nm ex, 530/30 nm em filters. When indicated cell shrinkage (apoptotic volume decrease) was determined by flow cytometry as changes in the forward scatter light pattern of the cells (Franco et al., 2008). The forward-angle light scatter relates to cell diameter, i.e. cell shrinkage is reflected as a decrease in the amount of forward scattered light. Flow cytometry was performed in a BDFACSort (Cytek-DxP-10 upgrade). 10,000 cells were acquired per sample and data were analyzed using FlowJo 7.6.5 software. Alternatively, cell death was assessed using Calcein retention assay (Life Technologies/Molecular Probes), which measures both the number of cells attached and the integrity of the plasma membrane. Cells were incubated with 1 μg/ml Calcein-AM for 30 min prior to analysis. Then, cells were washed and analyzed at 485 nm ex and 520 nm em in a FLUOstar OPTIMA plate reader (BMG Labtech).
Oxidative stress
Oxidative stress was determined using dihydroethidine (DHE, 5 μM, Life Technologies/Molecular Probes), which after its oxidization to ethidium intercalates within the DNA and exhibits bright red fluorescence (Franco et al., 2007; Rodriguez-Rocha et al., 2013). Alterations in the redox state of both cytosol and mitochondrial compartments were monitored using the reduction-oxidation sensitive green fluorescent protein (roGFP) sensor as done before (Rodriguez-Rocha et al., 2013). When indicated, changes in GSH content were determined by flow cytometry using monochlorobimane (mBCl, 10 μM, Life Technologies/Molecular Probes). Apoptosis is a stochastic phenomenon (Messam and Pittman, 1998), and we have demonstrated that GSH depletion occurs as an asynchronous event during cell death progression (Franco and Cidlowski, 2012; Franco et al., 2007). As such, GSH depletion determined by flow cytometry is observed as the appearance of a population of cells with a decrease in intracellular GSH (i.e. low GSH levels or GSH depleted cells) and the concomitant decrease in the number of cells with high (basal) GSH levels. A detailed description of these methods can be found in (Franco et al., 2007; Rodriguez-Rocha et al., 2013).
Primary midbrain cultures experiments
Primary midbrain cultures were prepared via dissection of E17 embryos obtained from pregnant Sprague–Dawley rats (Harlan) as described previously (Liu et al., 2008b). All animal procedures were approved by the Purdue Animal Care and Use Committee. The mesencephalic region containing the substantia nigra and ventral tegmental area was isolated and the cells were dissociated with trypsin (26 μg/ml in 0.9% [w/v] NaCl). Cells were plated at a density of 5 × 105 cells in dishes pretreated with poly-l-lysine, 5 μg/ml. Culture media consisted of DMEM, 10% (v/v) FBS, 10% (v/v) horse serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). Five days after plating, cells were treated with cytosine arabinofuranoside (20 μM, 48 h, Sigma/Aldrich) to inhibit the growth of glial cells.
After treatment, cells were fixed in 4% (w/v) paraformaldehyde in PBS (pH 7.4) for 30 min and subsequently permeabilized for 1 h (PBS, pH 7.4, 0.3% [v/v] Triton X-100, 1% [w/v] bovine serum albumin (BSA), and 10% [v/v] FBS). Then, fixed cells were incubated overnight at 4 °C with primary antibodies against MAP2 (1:100, EnCor Biotechnology) and TH (1:500, Millipore), washed with PBS, and stained with anti-chicken Alexa Fluor 594 and anti-rabbit Alexa Fluor 488 (1:1000, Life Technologies/Molecular Probes) for 1 h at RT. ProLong Gold antifade reagent (Life Technologies/Molecular Probes) with the nuclear stain 4′,6-diamidino-2-phenylindole (DAPI) was applied before adding a coverslip (Liu et al., 2008a). Relative dopaminergic cell viability was determined by counting MAP2- and TH-immunoreactive neurons in a blinded manner using a Nikon TE2000-U inverted fluorescence microscope (Nikon Instruments) with a 20× objective. The cultures were scored for the percentage of MAP2+ neurons that were also TH+ (Liu et al., 2008a; Strathearn et al., 2014). Replicate experiments were carried out using embryonic cultures prepared from different pregnant rats. Approximately 500–1000 MAP2+ neurons were counted per experiment for each condition.
Yeast strains, plasmids, culturing conditions and growth assays
Saccharomyces cerevisiae W303-1A WT strain (MATa can1-100 ade2-1 his3-1,15 leu2-3,112 trp1-1 ura3-1) was used in this work. The galactose inducible GAL1 promoter-driven GFP and the human α-synuclein-GFP expressing strains were generated by transformation of WT cells with linearized pFA6a-TRP1-GAL1-GFP and YIp351-SYN-GFP integrative plasmids (Longtine et al., 1998; Ocampo et al., 2013; Wach et al., 1994) kindly provided by Drs. Audrey Atkin (University of Nebraska-Lincoln) and Antoni Barrientos (University of Miami) (Ocampo et al., 2013), respectively. Yeast cells were cultured in rich YP medium or supplemented with synthetic complete (SC) medium. The media contained 2% (w/v) raffinose or 2% (v/v) glycerol-2% (w/v) lactate-0.1% (w/v) glucose mix as a sole carbon source. The 2% (w/v) galactose-containing medium was used to induce the expression of GFP or α-synuclein. For growth assays, cells were pre-grown in 2% (v/v) glycerol/(w/v) lactate/glucose SC medium for 12 h at 28 °C, serially diluted and dropped onto solid YP plates containing 2% (w/v) raffinose or 2% (w/v) galactose (Sigma/Aldrich). Plates were then incubated for 2–3 days at 28 °C.
Protein extraction, membrane fraction isolation, electrophoresis and western immunoblot
Isolation of membrane fractions was performed as explained in (Lee et al., 2002) with slight modifications. Whole cell lysates were washed with PBS and resuspended in homogenization buffer (10 mM Tris-HCl [pH 7.4], 250 mM Sucrose, 2 mM EDTA, Halt Protease Inhibitor Cocktail [Thermo Fisher/Pierce]). Cells were homogenized with a 23-gauge needle (15 passages) and a Dounce homogenizer (50 strokes with a pre-chilled B-type pestle). Cell homogenates were centrifuged at 9600 g for 2 min at 4 °C and supernatants were collected and centrifuged at 100,000×g for 30 min. The supernatant (cytosolic fraction) was collected and the pellet (membrane fraction) was resuspended in buffer (10 mM Tris (pH 7.4), 2 mMEDTA, 1% [v/v] Triton X-100) at a final concentration of less than 1 mg of protein/ml, and incubated for 30 min on ice. The membrane fraction suspensions were again centrifuged at 100,000×g for 30 min at 4 °C. The supernatant was collected and the pellet was discarded. The supernatant (containing remaining soluble and detached peripheral membrane proteins) and cytosolic fractions were precipitated in TCA (final concentration of 25% [v/v]). The precipitates were washed twice with ice-cold acetone and resuspended in lysis buffer.
Isolation of whole cell lysates, western-immunoblotting (WB) and SDS-PAGE of SK-N-SH were performed as explained previously (Rodriguez-Rocha et al., 2012). Blots were incubated with the corresponding primary antibodies overnight (1:1000): p-AMPK1α (Thr172, 2535), cleaved-caspase 3 (Asp 175, 9661), α-synuclein (2642, carboxy-terminal sequence), ubiquitin (3936), and p-Ulk1 (Ser555, 5869) (Cell Signaling); AMPK1α (Ab32047), ATP7A (Ab42486), Nrf2 (Ab62352), and Ulk1 (Ab65050) (Abcam); LC3B (L7543, SIGMA); and Ctr1 (sc66847, Santa Cruz Biotechnology). Blots were reprobed with β-actin (A2228, SIGMA) or GAPDH (2118, Cell Signaling) to verify equal protein loading.
For analysis of α-synuclein oligomers in soluble and insoluble fractions, cells were initially lysed with 10 mM Tris/HCl pH 7.4, 105 mM NaCl, 3 mM MgCl2, 0.1% (v/v) Triton X-100 and Halt protease inhibitor cocktail containing buffer. Samples were centrifuged at 9,600 g. Supernatants (soluble fraction) were analyzed by Blue Native-polyacrylamide gel electrophoresis (BN-PAGE). Pellets were lysed in RIPA buffer (0.1% [w/v] sodium dodecyl sulfate [SDS], 50 mM Tris-base, 150 mM NaCl, 1% [v/v] Triton X-100. 0.5% [w/v] sodium deoxycholate, pH 8.0) and extracts (insoluble fractions) were analyzed by NUPAGE/SDS-PAGE (Life Sciences/Invitrogen).
Filter trap assay for ubiquitinated protein aggregates
Cells were harvested in RIPA buffer. Samples were sonicated, centrifuged, and pellets discarded. Proteins were denatured in LDS-sample buffer (NUPAGE). 25–50 μg of protein were filtered in a nitrocellulose membrane previously equilibrated in NUPAGE transfer buffer using a dot blotter (Scie-Plas). Membrane was washed twice (2% [w/v] SDS, 10 mM Tris-EDTA, pH 7.5) and ubiquitin aggregates were detected by immunoblotting.
Confocal microscopy
Confocal microscopy of GFP-LC3-labeled autophagosomes and MitoTracker Deep Red-labeled mitochondria in SK-N-SH cells was performed as described previously (Garcia-Garcia et al., 2013). For visualization of GFP-tagged α-Synuclein in yeast, pre-cultures of synchronized S. cerevisiae cells were grown in glycerol/lactate medium overnight and transferred into the medium containing 2% [w/v] raffinose or 2% [w/v] galactose with or without CuCl2. Live cells and GFP were then visualized with Olympus IX81 inverted confocal laser scanning microscope (Olympus America) at 488 nm under oil immersion. Images were acquired using Fluoview 500 Software (Olympus America).
Statistical analysis
Experiment replicas were independent and performed on separate days. Collected data were analyzed by using one-way, two-way or three-way ANOVA, and the appropriate post-hoc test using SIGMA-PLOT/STAT package. When ANOVA assumptions were not met (normality or equal variance), Kruskal-Wallis One Way ANOVA on Ranks or data transformation (two-way ANOVA) were performed on collected data. Data were plotted as mean ± standard error of the mean (SE) using the same statistical package for data analysis. Flow cytometry plots and western blots presented show the results of representative experiments. Densitometry analyses of WB were performed in Image J (NIH).
Results
Copper induces caspase-dependent apoptosis, which is modulated by Cu content and transport
CuCl2 treatment for 48 h induced death in dopaminergic neuroblastoma cells in a dose-dependent manner (Figs. 1A–B). Cell death was identified by the loss of plasma membrane integrity (PI uptake) and the depletion in intracellular GSH content (mBCl) (Figs. 1A–B). Cell death was also evaluated by Calcein retention (Fig. 1C). CuCl2-induced cell death was significantly reduced in the presence of the irreversible pan-caspase inhibitor z-VAD-FMK (Figs. 1A–C). CuCl2 also induced the cleavage/activation of caspase 3 (Fig. 1D). However, inhibition of caspase 3 with Ac-DMQD-CHO had no effect on CuCl2 toxicity (Fig. 1C), which might be explained by the compensatory role of other executioner caspases (Zheng et al., 2000). The difference between the sensitivity of PI uptake/GSH depletion (Figs. 1A–B) and Calcein retention assays (Fig. 1C), to detect the loss in cell viability induced byCuCl2, might be related to the fact that Calcein uptake is affected by cell number and proliferation. To confirm the occurrence of apoptosis, we evaluated the externalization of phosphatidylserine using Annexin V binding to phosphatidylserine. Supplementary Fig. 2A–B shows that CuCl2 induces a significant increase in early apoptotic (Annexin V+/PI− cells, Supplementary Fig. 2A contour plots quadrant 3 [Q3] in broken regions) and late necrotic (Annexin V+/PI+ cells, Supplementary Fig. 2A Q1–2). However, the increase in early apoptotic cells is no more than 15% of the total population even when high extracellular [CuCl2] were used (1 mM), while at this concentration, a reduction of 90% in the normal (healthy) population is observed (Annexin V−/PI − cells, Supplementary Fig. 2A–B, Q4). This can be explained by the transition of early apoptotic cells (Annexin V+/PI−) to a late apoptotic stage (Annexin V+/PI+) (Wlodkowic et al., 2011). Indeed, it has been previously demonstrated that in the absence of phagocytes, in charge of the clearance of apoptotic cells/bodies, cells undergoing apoptosis will progress into a late stage where cell membrane becomes permeabilized (i.e. late apoptotic or secondary necrotic cells) (Brauchle et al., 2014; Silva, 2010).
Fig. 1.

Copper exposure induces apoptosis in dopaminergic cells. In A–B, cell survival after CuCl2 treatment (48h) was determined in cells co-stained with PI and mBCl. Two-dimensional 5% probability contour plots in A display cell death (PI uptake) vs GSH content (mBCl). Healthy cells were identified as PI- and high mBCl fluorescence (broken line regions in A) and quantified in the corresponding bar graphs in B. Numbers in quadrants (A) reflect number of cells in %. In C, well death was evaluated via Calcein retention assay and data was normalized vs control (DMSO). When indicated, cells were pre-treated with 50 μM pan-caspase (Z-VAD-FMK) or caspase 3 inhibitor (DMQD-CHO) for 1 h and inhibitors remained present throughout the experiment. In D, WB analysis of CuCl2-induced caspase 3 cleavage/activation. Numbers (italics) represent the densitometry analysis of cleaved caspase 3 normalized to β-actin signal with respect to control. The WB is cropped for simplification, but the unmodified version is found in Supplementary Fig. 1. Data in graphs represent means ± SE of at least n = 3. Two-way ANOVA, Holm-Sidak post hoc test, ap < 0.05 vs DMSO only; bp < 0.05 zVAD or Ac-DMQD vs DMSO within the corresponding [CuCl2] category.
Externalized phosphatidylserine labeling with Annexin V provides an accurate estimation of apoptosis (Wlodkowic et al., 2011), but because it usually labels cells at the very early stages of apoptosis, it cannot be used as an accurate measurement of the final outcome of apoptotic signaling, which is cell death. Indeed, early apoptotic cells with externalized phosphatidylserine can be rescued if the apoptotic stimulus is removed, while loss of plasma membrane integrityis anirreversible feature of cell death (Geske et al., 2001). According to the Nomenclature Committee on Cell Death the loss of plasma membrane integrity is one of the main criteria to define cell death (Kroemer et al., 2009). In addition, several reports demonstrated that: 1) programmed cell death can occur without exposure of PS (Fadeel et al., 1999; King et al., 2000; Lee et al., 2013); 2) that phosphatidylserine externalization is reversible and might be associated to non-cell death processes (Schlegel and Williamson, 2001; Stowell et al., 2009; Yang et al., 2002); and 3) that phosphatidylserine externalization can be regulated in a caspase-independent and -dependent manner (Ferraro-Peyret et al., 2002; Segawa et al., 2014). The protective effect of the pan-caspase inhibitor z-VAD against CuCl2-induced cell death (Figs. 1A–C) clearly points to the occurrence of apoptosis upon CuCl2 treatment. While z-VAD cannot fully protect against CuCl2 (Figs. 1A–C), it has been demonstrated that distinct cell death mechanisms can be activated simultaneously, and that inhibition of one cell death pathway may switch it to a different one (Lemaire et al., 1998; McCall, 2010; Yu et al., 2006). Thus, we decided for our further experiments to evaluate cell death by assessing the loss of plasma membrane integrity (increased PI uptake and reduced Calcein retention) in combination with loss of intracellular GSH (mBCl) to determine overall cell death under our experimental conditions. We and others have previously demonstrated that GSH depletion is a hallmark of apoptotic cell death (Circu and Aw, 2012; Franco and Cidlowski, 2012).
Intracellular Cu concentrations cannot be dramatically increased in cells treated with high extracellular Cu due to the high chelating capacity of components present in the medium that are known to form complexes with Cu (Barnea et al., 1989). The requirement for high extracellular [CuCl2] to induce cell death might suggest interference from culture media components. Albumin in FBS can potently bind copper (Masuoka and Saltman, 1994). Thus, it is likely that serum components, including albumin, scavenge copper and reduce its availability for uptake by cells. However, a reduction in FBS content in the media had no effect on CuCl2-induced cell death (Supplementary Fig. 3A). We next correlated the exposure of cells to high [CuCl2] with changes in intracellular Cu content. Cu uptake was evaluated by ICP-MS. Supplementary Fig. 3B shows the increase in intracellular Cu induced by 1 mM CuCl2. A maximum increase was detected at 24 h that was paralleled by significant cell death (> 80%). Thus, we evaluated changes in Cu content after 12 h of exposure to increasing concentrations of CuCl2. Supplementary Fig. 3C shows the changes in intracellular Cu levels induced by CuCl2. We found that an increase of at least 64 μmole/g of protein that corresponded to an exposure of 0.5 mM CuCl2 for 12 h was required to induce dopaminergic cell death. Copper chaperone for superoxide dismutase 1 (CCS1) is a very sensitive biomarker of copper overload. A significant reduction in CCS, but not metallothioneins has been observed after copper supplementation (Suazo et al., 2008). Only high extracellular [CuCl2] (>0.5 mM) induced a decrease in CCS1 levels in dopaminergic cells (Supplementary Fig. 3D).
To corroborate whether the toxic effects of high extracellular Cu are mediated by changes in its intracellular levels, we examined the role of Cu transport systems. In eukaryotic cells, high-affinity Cu1+ import is mediated by Ctr1/SLC31A1 (Kim et al., 2008, 2013; Lee et al., 2002). Ctr1 was found localized in the membrane fraction of two distinct cell clones generated “in house” to stably overexpress hCtr1 (Fig. 2A). Transduction of cells with Ad-Ctr1 significantly increased Cu-induced cell death (Figs. 2C–D). P-type ATPase transporters, ATP7A and ATP7B transport Cu ions from the cytosol across cellular membranes (Kim et al., 2008). Overexpression of ATP7A (Fig. 2B) did not significantly altered CuCl2-induced cell death (Fig. 2E), which might be due to the constant presence of high extracellular Cu levels that could surpass the ability of ATP7A to detoxify cells from Cu. These results demonstrate that the toxic effects of Cu are linked to alterations in its intracellular homeostasis.
Fig. 2.
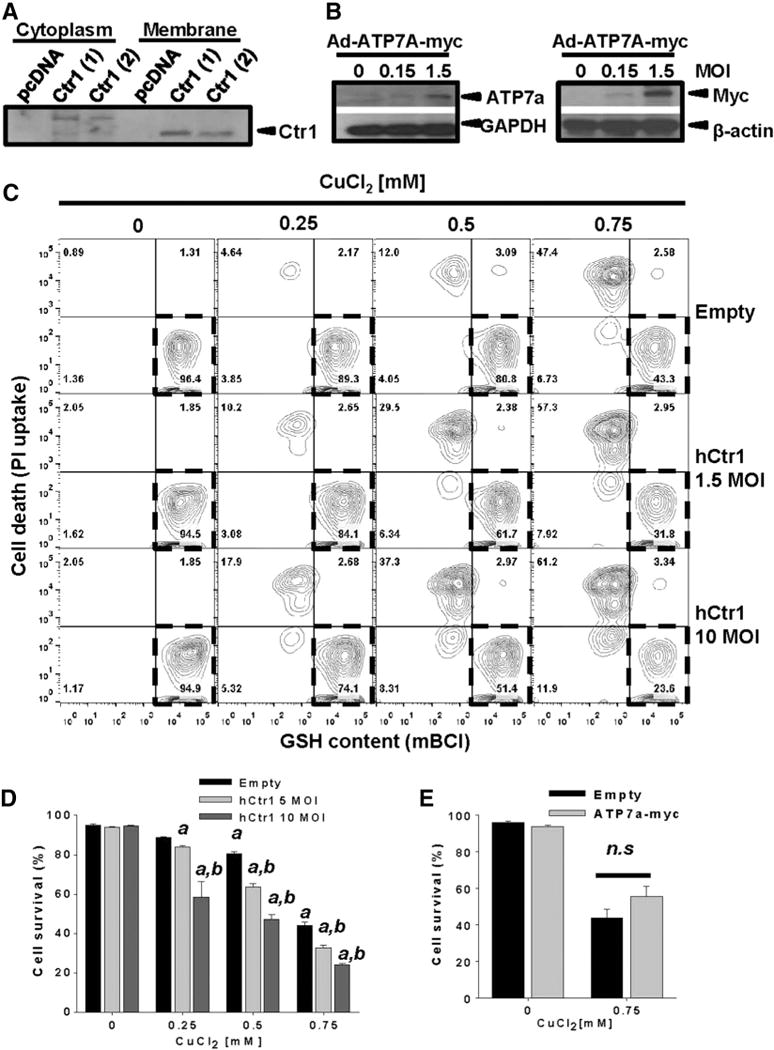
Copper transport regulates cell death. In A, cytosolic and membrane fractions were extracted from two stable cell clones overexpressing hCtr1, and its expression levels were evaluated by WB. In B, whole cell lysates of cells transduced at the indicated MOI for 24 h with Ad-Empty or Ad-ATPA-myc were isolated, and the expression levelsof ATP7A were confirmed with anti-ATP7A (left panel) or anti-myc (right panel) antibodies. In C–E, cells were transduced with Ad-Empty, Ad-hCtr1 or Ad-ATPA-myc at the indicated MOI for 24 h. Then, cells were washed and treated with CuCl2 for 48 h. Cell death was determined by the PI uptake and the loss of in intracellular GSH as explained in Fig. 1A. In C, results were represented in two-dimensional 5%probability contour plots displaying cell death (PI uptake) vs GSH content (mBCl). Healthy cells were identified asPI- and high mBCl fluorescence (broken line regions). Numbers in quadrants reflect %s. Data in D and E is expressed as cell survival (i.e. % of cells with normal GSH content and PI- after treatments). Two-way ANOVA, Holm-Sidak post hoc test, ap < 0.05 vs Empty without CuCl2 supplementation, bp < 0.05 vs Empty within the corresponding [CuCl2] category. n.s. not significant.
Overexpression of WT or A53T α-synuclein increases Cu toxicity independent from oxidative stress or α-synuclein aggregation
We next evaluated the toxicity of the α-synuclein missense mutation A53T and the overexpression of the WT form as a model for gene multiplication using adenoviral vectors (Fig. 3A). No changes in cell survival were detected by short-term (2–3 days) overexpression of WT or A53T α-synuclein (Figs. 3B–D). Interestingly, WT or A53T α-synuclein increased the toxicity of CuCl2 determined by the loss of plasma membrane integrity (PI uptake, Fig. 3C and Calcein retention, Fig. 3D) and cell shrinkage, a marker for apoptotic cell death (Fig. 3C) (Bortner and Cidlowski, 2004). The synergistic toxic effect of Cu exposure and α-synuclein was reproduced in primary midbrain rat dopaminergic cultures supplemented with CuSO4 at significantly lower concentrations (10 μM) than those used in the immortalized dopaminergic neuroblastoma cells (SK-N-SH) (Fig. 3E), demonstrating its reproducibility in a more physiological setting, and that the synergistic effect is independent of the Cu salt used.
Fig. 3.
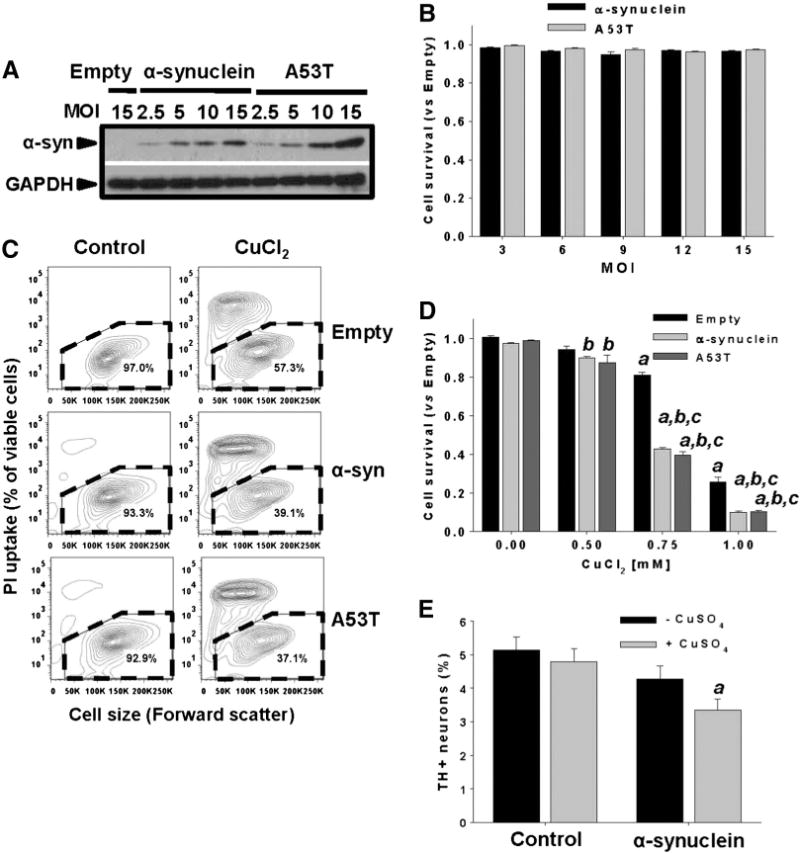
Non-toxic overexpression of α-synuclein sensitizes dopaminergic neuroblastoma cells and primary midbrain cultures to Cu toxicity. In A and B, neuroblastoma cells were transduced with Ad-Empty, Ad-α-synuclein or Ad-α-synuclein A53T mutant (A53T) at the indicated MOI for 24 h. Cells were washed, and the expression levels of WT or A53T α-synuclein were measured by WB after 48 h of transduction (A), while cell survival was evaluated by Calcein retention (B). In C and D, cell death induced by CuCl2 treatment (48 h, 0.75 mM in C) was determined in neuroblastoma cells previously transduced with WT or A53T α-synuclein using PI uptake (C) or Calcein retention assay (D). In C, cell death is represented in two-dimensional 5% probability contour plots of PI fluorescence vs cell size (forward scatter) to depict cells with both compromised plasma membrane integrity (PI uptake) and decreased cell size. %s in contour plots represent the number of viable cells (PI- and normal cell size, broken line regions). Data in B and D represent means ± SE of n = 3. In E, primary midbrain cultures transduced, or not, with WT α-synuclein (MOI = 10, 72 h) were incubated in fresh media with or without CuSO4 (10 μM) for 24 h. Cells were stained with antibodies specific for MAP2 and TH and scored for dopaminergic cell viability. The data are presented as the mean ± SE of n = 7. Two-way ANOVA, Holm-Sidak post hoc test, ap < 0.05 vs Empty without CuCl2 or CuSO4 supplementation; bp < 0.05 vs 0 mM CuCl2 within the corresponding category of α-synuclein or A53T; cp < 0.05 vs Empty within the corresponding [CuCl2] tested.
We next sought to evaluate if the augmented sensitivity to Cu of cells overexpressing WT or A53T α-synuclein was associated with increased oxidative stress. CuCl2 induced oxidative stress as evidenced by an increase in ROS formation detected by the oxidation of DHE (Fig. 4A), which paralleled intracellular GSH loss (Figs. 1A, 2C and 4B) and the up-regulation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcription factor that mediates antioxidant response (Fig. 4C). Inhibition of de novo GSH synthesis with BSO (an inhibitor of γ-glutamylcysteine ligase) potentiated CuCl2-induced cell death (Supplementary Fig. 4A–B). To ascertain the contribution of mitochondrial vs cytosolic oxidative stress in Cu toxicity and its potentiation by α-synuclein, we used the genetically encoded redox sensitive proteins roGFP and its mitochondria-targeted version (Mito-roGFP) (Hanson et al., 2004; Rodriguez-Rocha et al., 2013). Cu-induced oxidative stress was primarily localized to the cytosolic compartment (Figs. 4D–E). However, overexpression of WT or A53T α-synuclein had no significant effect on oxidative stress in either the cytosol or mitochondrial compartments indicating that the stimulatory effect of α-synuclein on Cu-induced cell death is unlikely to arise from oxidative stress.
Fig. 4.
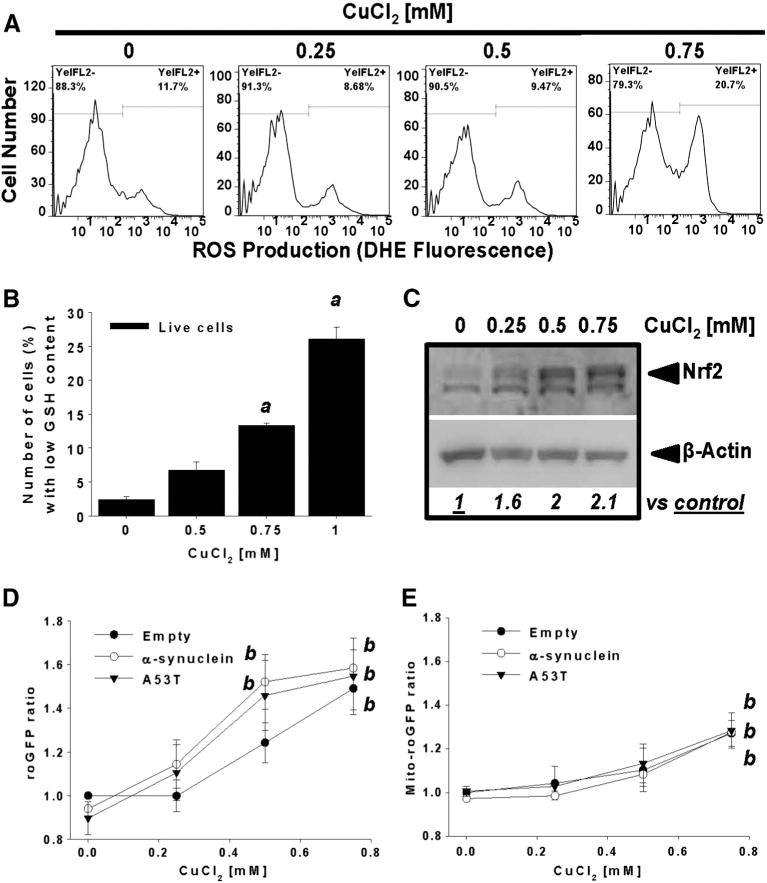
The synergistic toxic effect of α-synuclein and CuCl2 is not mediated by increased oxidative stress. Cells were treated with increasing concentrations of CuCl2 for 48 h. ROS formation and GSH depletion were determined by DHE oxidation (A) and changesin mBCl fluorescence (B), respectively. InC, whole cell lysates of cells treated with CuCl2 were isolated to determine changes in Nrf2 levels by WB. Numbers (italics) represent the densitometry analysis normalized to β-actin with respect to control. In D–E, stable cells overexpressing roGFP and Mito-roGFP were transduced with Ad-Empty, Ad-α-synuclein or Ad-A53T for 24 h (3 MOI). Then, cells were washed and treated with CuCl2 (48 h) as indicated. Cells were stained with PI and only viable cells (PI−) were analyzed to determine alterations in the cytosolic (roGFP) or mitochondrial (Mito-roGFP) redox state by ratiometric analysis. Data in graphs were normalized with respect to Empty control values. Data in graphs represent means ± SE of n = 3 independent experiments. One-way ANOVA, Holm-Sidak post hoc test, ap < 0.05 vs 0 mM CuCl2. Two-way ANOVA, Holm-Sidak post hoc test, bp < 0.05 vs 0 mM CuCl2 within the corresponding virus category.
The toxic effects of α-synuclein are largely ascribed to its aggregation. Cu interacts with α-synuclein accelerating aggregation and fibril formation, as well as its membrane localization (Rasia et al., 2005; Uversky et al., 2001a; X. Wang et al., 2010). We used native and denaturing electrophoresis to determine the formation of 1) high molecular weight (HMW) Triton X-100 (TX-100)-insoluble and -soluble aggregates of α-synuclein (SDS-PAGE), and 2) changes in the native conformation of α-synuclein (BN-PAGE) derived from TX-100-soluble fractions. Overexpression of α-synuclein (WT or A53T mutant) for 72 h, in the presence or absence of CuCl2 supplementation did not induce the accumulation of HMW aggregates of α-synuclein (Supplementary Fig. 5A [soluble and insoluble fractions] and 5B [insoluble fractions]). Under BN-PAGE conditions α-synuclein is reported to exist predominantly as stable unfolded monomers that migrate as 57–60 kDa proteins (Fig. 5A, upper panel) but in denaturing gels, these monomers migrate as 14 kDa proteins (Fig. 5A, lower panel). The larger than expected size of the bands has been proposed to be mainly due to the monomers adopting an unfolded extended conformation, which results in a larger than expected hydrodynamic radius (Fauvet et al., 2012; Lashuel et al., 2013; Weinreb et al., 1996). Other research groups proposed that this migration pattern is also a result of a fraction of α-synuclein existing as a stable tetramer (Bartels et al., 2011; Dettmer et al., 2013). CuCl2 treatment caused a decrease in the abundance of this monomer and the appearance of a band with enhanced migration (Fig. 5A marked with *), which correlated with a decrease in the levels of the monomer determined in TX-100-insoluble fractions (Fig. 5A, lower panel). These results suggest that Cu does not increase the aggregation of α-synuclein when this protein is overexpressed only for 72 h. In contrast, we found that Cu might mediate changes in the endogenous folded state or stability of α-synuclein (monomer or tetramer).
Fig. 5.
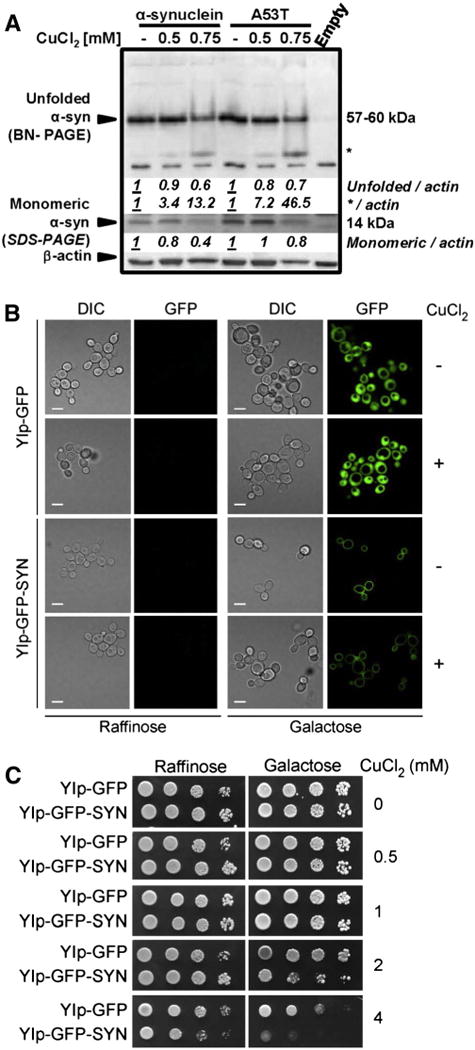
The synergistic toxic effect of α-synuclein and CuCl2 is not associated with changes in α-synuclein aggregation. In A, cells were transduced with Ad-Empty, Ad-α-synuclein or Ad-A53T for 24 h (3 MOI), washed and then treated with or without CuCl2 (48 h). TX-100-soluble (upper panel) and insoluble (lower panel) fractions were analyzed by BN- and SDS-PAGE, respectively, and α-synuclein was visualized by WB. Numbers (italics) represent the densitometry analysis normalized to β-actin with respect to the corresponding control. In B–C, W303 wild type S. cerevisiae strains containing genome-integrated, GAL1 promoter-driven GFP or α-synuclein-GFP expression cassettes were pre-grown in 2% glycerol (v/v)/ lactate (w/v) medium, serially diluted and spotted onto plates containing either 2% raffinose or 2% galactose (w/v) as a sole carbon source, and supplemented with the indicated amounts of CuCl2. Cells were incubated for 2 days at 28 °C. In B, visualization of GFP and α-synuclein-GFP distribution was assessed by confocal microscopy performed after 12 hours of cell growth in the indicated medium with or without 4 mM CuCl2.
α-synuclein has been reported to interact to a different extent with membrane compartments (Auluck et al., 2010), while its A53T mutant has been shown to accumulate in neuronal membrane fractions and induce membrane disruption/permeabilization to a greater extent as compared to the WT form (Tsigelny et al., 2012; Volles and Lansbury, 2002). To further interrogate whether the synergistic toxic effects of Cu and α-synuclein overexpression are linked to changes in α-synuclein aggregation or association with the plasma membrane we used the budding yeast S. cerevisiae model overexpressing an inducible promoter-driven fusion of α-synuclein-GFP. As reviewed in (Khurana and Lindquist, 2010), yeast provides a valid experimental platform to explore the fundamental mechanisms associated with neurodegenerative diseases. In this model, the inducible and regulatable expression of α-synuclein can result in no toxicity, intermediate toxicity, or high toxicity in relation to the levels of α-synuclein being expressed. α-synuclein–GFP overexpressed in yeast at non-toxic levels localizes to the plasma membrane, consistent with its known affinity with phospholipids (Fig. 5B). This is the expected localization of a protein that localizes to synaptic vesicles in neurons, when considering that yeast has constitutive vesicular secretion (Khurana and Lindquist, 2010). In contrast, GFP overexpression alone results in a diffuse localization throughout the cytoplasm (Fig. 5B). Doubling the expression levels of α-synuclein changes its localization tointracytoplasmic inclusions leading to cytotoxicity, consistent with a toxic gain of function (data not shown) (Khurana and Lindquist, 2010). Similar to what was observed in the dopaminergic neuroblastoma cell line, the dose-dependent toxicity induced by Cu in yeast (as reflected by growth inhibition and cell death) was stimulated by overexpression of α-synuclein–GFP (Fig. 5C), without any effect on the plasma membrane localization of the protein (Fig. 5B). Collectively, these results demonstrate that the synergistic toxic effects of α-synuclein overexpression and Cu are not associated with the aggregation of the former component.
Cu-induced cell death is regulated by AMPK/Ulk1 signaling and potentiated by inhibition of Atg5-dependent autophagy
Autophagy is essential for the constitutive protein and organelle turnover and for cell survival during conditions of environmental stress (Murrow and Debnath, 2013). Cu-dopamine complex induces mitochondrial autophagy (mitophagy) and apoptotic cell death (Paris et al., 2009). We observed that CuCl2 induced a dose-dependent accumulation of the autophagosome markers GFP-LC3 puncta (Fig. 6A, white arrows) and LC3-II (Fig. 6B). We did not observe any co-localization of mitochondria with autophagosome as an indicator of mitophagy. LC3-II accumulation was associated with an increase in autophagy flux as demonstrated by its enhanced accumulation in the presence of the lysosomal cargo degradation (autophagy flux) inhibitor chloroquine (CQ, Figs. 6C–D) (Garcia-Garcia et al., 2013). The p62/sequestosome 1 (SQSTM1), is a scaffold protein that recognizes ubiquitinated protein aggregates and non-ubiquitinated proteins and targets them for degradation by autophagy (Bjorkoy et al., 2009). p62 is another marker to study autophagic flux and we observed that CuCl2 also induced the accumulation of p62 in a dose-dependent manner corroborating the increase in autophagy flux (Fig. 6C).
Fig. 6.
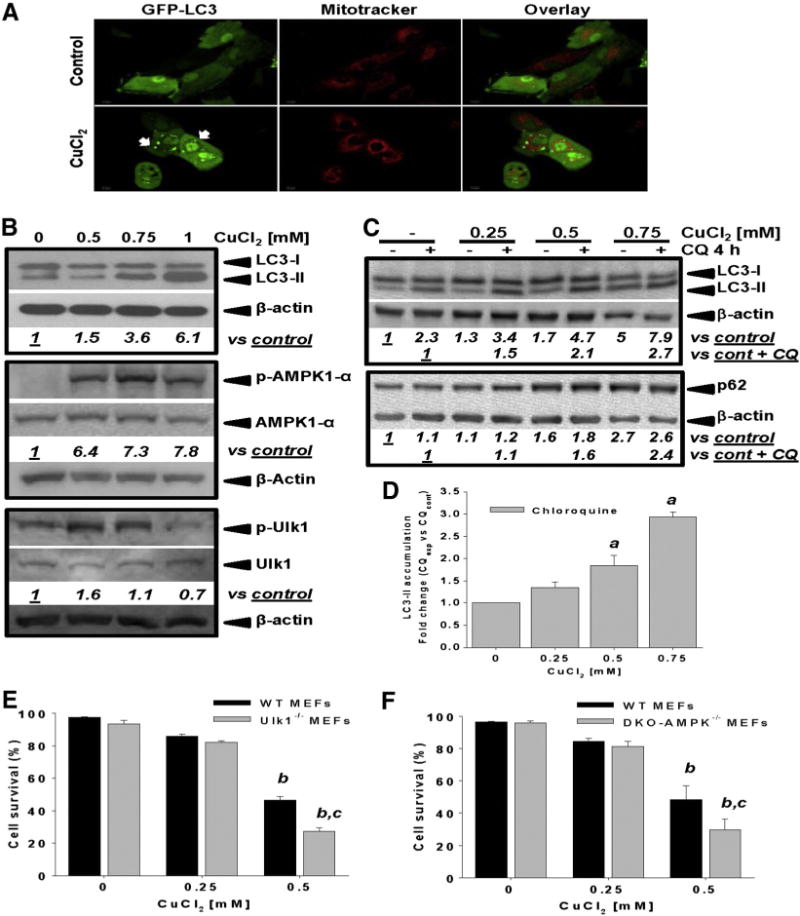
Copper increases autophagy flux and its toxicity is regulated by AMPK-Ulk1 signaling. In A, autophagosome formation induced by 0.75 mM CuCl2 (48 h) was analyzed by confocal microscopy in cells transduced with Ad-GFP-LC3 (3 MOI, 24 h prior CuCl2 treatment). Mitochondria were labeled with Mitotracker. In B, accumulation of LC3-II, and phosphorylation of AMPKα1 and Ulk1 (Ser555) in response to increasing concentrations of CuCl2 (48 h) was evaluated by WB. In C and D, accumulation of LC3-II and p62 induced by CuCl2 (48 h) was determined in the presence or absence of chloroquine (CQ, 40 μM, 4 h prior sample harvesting). Numbers in B, C (italics) represent the densitometry analysis with respect to total AMPKα1, Ulk 1 or β-actin (for LC3-II) and normalized to the corresponding control ± CQ. Data in D represent the densitometry analysis of LC3-II with respect to control + CQ and are means ± SE of n = 3 independent experiments. Controls contained DMSO used as vehicle for CQ. In E–F, Ulk1−/−, DKO AMPK−/− or the corresponding WT MEFs were exposed to CuCl2 at the concentrations indicated (48 h). Cell viability was determined by simultaneous analysis of both PI uptake and changes in intracellular GSH (mBCl) by flow cytometry as exemplified in Fig. 1A. Viable cells were defined as cells with high intracellular glutathione levels (GSH) and PI-. %s represent means ± SE of n = 3 independent experiments. One-way ANOVA, Holm-Sidak post hoc test, ap < 0.05 vs control 0 mM CuCl2. Two-way ANOVA, Holm-Sidak post hoc test, bp < 0.05 vs WT 0 mM CuCl2 cp < 0.05 vs WT within the corresponding [CuCl2] tested.
Collectively, adenosine monophosphate (AMP)-activated protein kinase (AMPK) together with mTOR (mammalianor mechanistic target of rapamycin) and Ulk1 (Unc-51-like kinase1) sense the energy and nutrient status of the cell, and dictate the activation of energy- and amino-acid-generating catabolic processes such as autophagy (Cardaci et al., 2012; Hardie et al., 2012). CuCl2 exposure increases the phosphorylation of AMPK-α1 in a dose-dependent manner (Fig. 6B). Phosphorylation of Ulk1 at Ser555 by AMPK is critical for starvation-induced autophagy and cell survival under conditions of low nutrient and energy availability, as well as upon disruption in mitochondrial homeostasis (Egan et al., 2011). We found that CuCl2 treatment induced a transient increase followed by a decrease in Ulk1 phosphorylation (Fig. 6C). Ulk1−/− and DKO -AMPK (α1 and α2)−/− MEFs were more sensitive to CuCl2-induced toxicity (Figs. 6E–F). These results suggested that Cu-induced AMPK-Ulk1 signaling protects against cell death.
We next sought to understand the role of autophagy in the synergistic toxicity induced by Cu and α-synuclein overexpression. The Atg12-Atg5 complex conjugation is essential for autophagosome formation, which functions as an E3-like ligase for LC3 lipidation (Hanada et al., 2007). Hence overexpression of a dominant negative (dn) form of Atg5 (dnAtg5) is an effective approach to inhibit autophagy (Garcia-Garcia et al., 2013; Klionsky et al., 2012; Yang et al., 2010). Overexpression of dnAtg5 inhibited autophagy induced by CuCl2 (Fig. 7A). dnAtg5 increased CuCl2-induced cell death without affecting the survival of cells overexpressing WT or A53T α-synuclein treated or not with CuCl2 (Figs. 7B–C). α-synuclein is known to interfere with autophagic pathways (Martinez-Vicente et al., 2008; Winslow et al., 2010). However, we observed that overexpression of WT or A53T α-synuclein did not affect autophagy flux irrespective of CuCl2 treatment (Figs. 7D–E, n = 3 p < 0.05, One-way ANOVA).
Fig. 7.
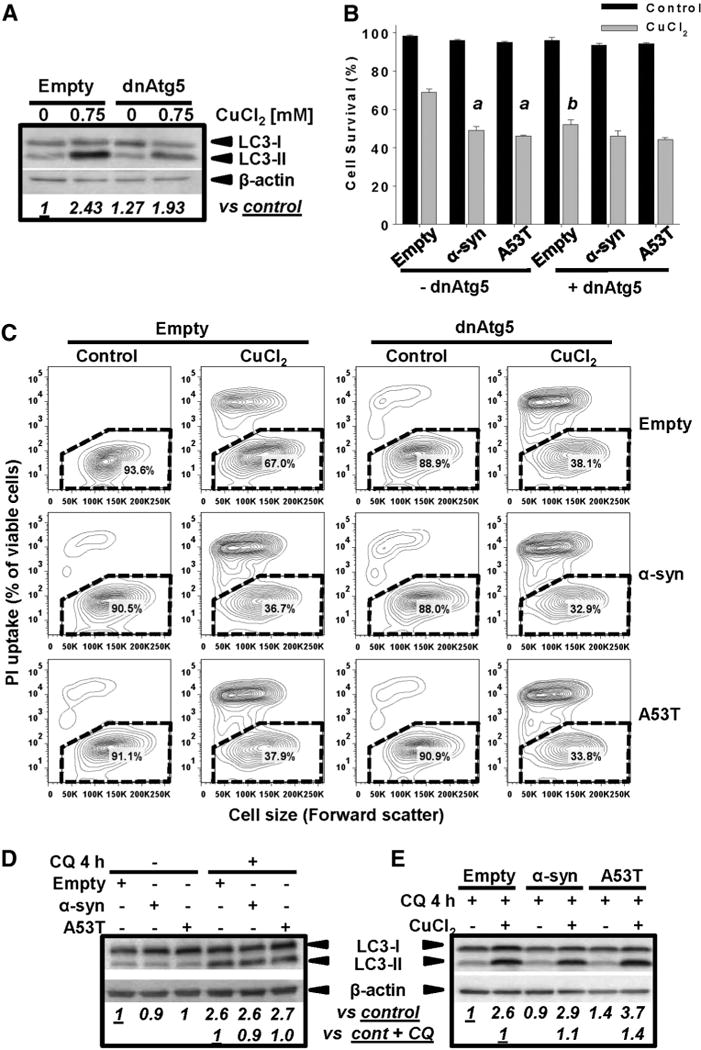
Inhibition of Atg5-dependent autophagy increases Cu toxicity but not the synergistic toxicity of Cu/α-synuclein. Cells were transduced with Ad-dnAtg5 (1.5 MOI) and/or Ad-α-synuclein or Ad-A53T (3 MOI) for 24 h. Ad-Empty was used as control at the MOI required to normalized the MOI used for other treatments (4.5 MOI total), In A, D and E, autophagy (LC3-II accumulation) induced by 0.75 mM CuCl2 (48 h) was evaluated by WB. In D–E, autophagy flux was determined by incubation with CQ (40 μM) 4 h prior to sample isolation. Controls contained DMSO usedas vehicle for CQ. Numbersin WBs (italics) represent the densitometry analysis normalizedto β-actin with respect to controls± CQ. In C, cell death induced by 0.75 mM CuCl2 (48 h) is represented in two-dimensional 5% probability contour plots of PI fluorescence vs cell size (forward scatter) to depict cells with both compromised plasma membrane integrity (PI uptake) and decreased cell size. %s in the contour plots represent the population of viable cells (PI- and normal cell size, broken line regions). B: Two-way ANOVA, Holm-Sidak post hoc test was performed only on CuCl2 data, ap < 0.05 vs Empty + CuCl2; (with or without dnAtg5 transduction); bp < 0.05 vs -dnAtg5 within the corresponding virus category (empty, α-synuclein or A53T).
Copper induces an increase in protein ubiquitination and inhibition of the proteasome stimulates Cu and α-synuclein toxicity
Autophagy and the UPS are major protein quality control mechanisms disrupted in PD (Cook et al., 2012). MG132, an inhibitor of the chymotrypsin-like proteasomal activity, induces a dose-dependent accumulation of ubiquitinated aggregates in dopaminergic cells (Fig. 8A, upper dot blot panel). The half maximal effective concentration (EC50) for the accumulation of ubiquitin bound proteins was estimated at 172.8 nM (Supplementary Fig. 6A), very close to the reported IC50 (100 nM). The levels of ubiquitinated proteins decrease 48 h after MG132 treatment (Fig. 8A, lower dot blot and WB), which is explained by the reported reduction of free ubiquitin levels upon chronic proteasome inhibition (Xu et al., 2004). Changes in the HMW ubiquitinbound proteins in cells treated with CuCl2 were evaluated in the presence of MG132, to distinguish whether alterations were linked to changes in proteasomal activity or protein ubiquitination. CuCl2 induced an increase in ubiquitinated proteins in the presence or absence of MG132, demonstrating that their accumulation is linked to increased ubiquitination. (Fig. 8B WB and dot blot, and Supplementary Fig. 6B). MG132 enhanced CuCl2 toxicity whether WT or A53T α-synuclein were overexpressed or not (Supplementary Fig. 6C and Fig. 8C). Neither WT nor A53T α-synuclein overexpression had an effect on the accumulation of ubiquitinated proteins (Fig. 8D, WB and dot blot, n = 3 p < 0.05 One-way ANOVA). Interestingly, in the presence of the proteasome inhibitor MG132, WT or A53T α-synuclein markedly increased ubiquitinated-protein levels (Fig. 8E and Supplementary Fig. 6D). These results demonstrate that while impairment of autophagy only enhanced Cu toxicity, inhibition of proteasomal activity enhanced both Cu and Cu + α-synuclein toxicity. In addition, WT or A53T α-synuclein stimulated protein ubiquitination but did not affect autophagy flux.
Fig. 8.
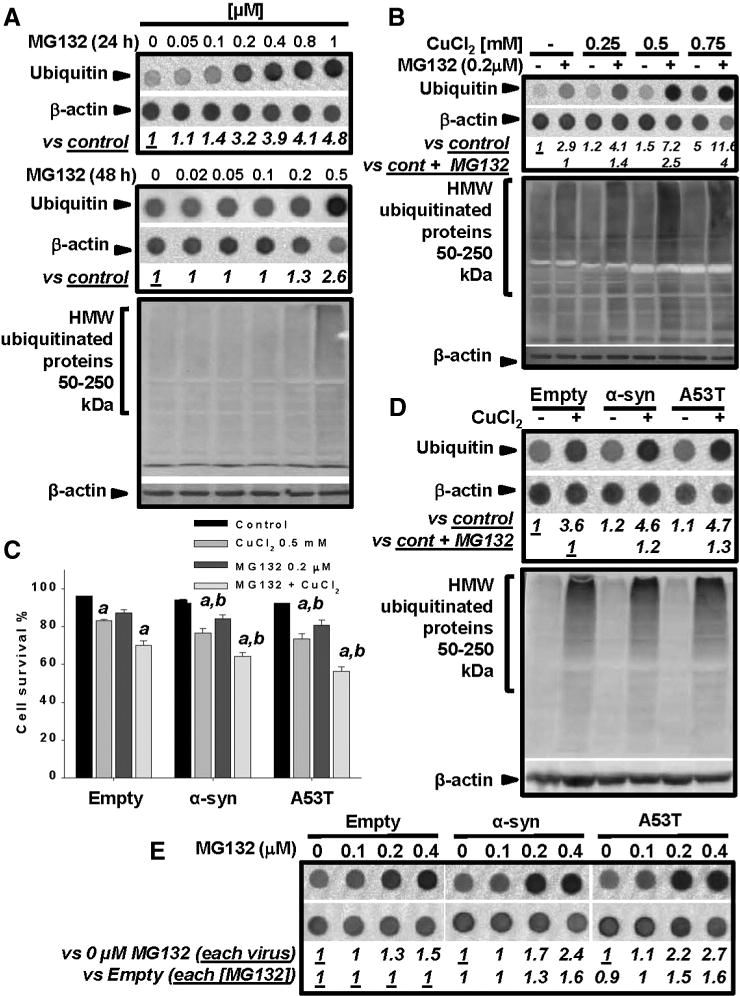
Inhibition of proteasomal activity increases Cu and α-synuclein toxicity. Accumulation of ubiquitin-bound proteins was evaluated by WB or dot blot. Numbers in dot blots (italics) represent the densitometry analysis normalized to β-actin with respect to the indicated controls. In A, cells were treated with MG132 as indicated. In B, cells were treated initially with CuCl2 for 24 h and then MG132 was added for 24 h (CuCl2 remained throughout the experiment). In C, cells were transduced with the indicated viral vectors for 24 h. Then cells were washed and treated with CuCl2 ± MG132 for 48 h. Cell viability was determined by simultaneous analysis of both PI uptake and changes in intracellular GSH (mBCl) by flow cytometry as exemplified in Fig. 1A. Viable cells were defined as cells with high intracellular GSH levels and PI−. %s represent means ± SE of n = 3 independent experiments. Three-way ANOVA, Holm-Sidak post hoc test, ap < 0.05 vs Control within the corresponding category of virus ± MG132;bp < 0.05 vs Empty within the corresponding category ± CuCl2 ± MG132. In D, cells were transduced with the indicated viral vectors for 24 h, washed, and then treated with CuCl2 for 48 h. In E, cells were transduced with the indicated viral vectors for 24 h, washed, and after 24 h, treated with MG132 for additional 24 h (72 h total). Controls contained DMSO used as vehicle for MG132.
Both autophagy and the UPS may act simultaneously in the degradation of α-synuclein. We next decided to evaluate the contribution of both degradation pathways to α-synuclein accumulation. In contrast to CuCl2 treatment (Supplementary Fig. 5), inhibition of proteasomal activity increased the accumulation of HMW α-synuclein aggregates isolated from total cell lysates (Supplementary Fig. 7A), and TX-100-insoluble fractions (Supplementary Fig. 7B) Inhibition of autophagy per se had no effect on the accumulation of HMW (Supplementary Fig. 7A–B). Simultaneous inhibition of autophagy and the proteasome only potentiated the accumulation of HMW aggregates in cells expressing WT α-synuclein in triton soluble fractions, but not A53T (Supplementary Fig. 7B). Similar to CuCl2 treatment, inhibition of the proteasome but not autophagy induced an accumulation of a protein band with a lower migration profile in cells overexpressing either WT or A53T forms (Supplementary Fig. 7C, highlighted by *). This effect was potentiated by expression of dnAtg5. Impairment of both autophagy and the proteasome had no additive effect on the toxicity of CuCl2 in the presence or absence of either α-synuclein or A53T (Supplementary Fig. 8). Our results suggest then that Cu can exert a synergistic toxic effect with WT or A53T α-synuclein independent from α-synuclein aggregation. This synergistic effect seems to be mediated by an increase “stress load” to protein degradation mechanisms, primarily the ubiquitin/proteasome system and, to a lesser extent, autophagy (Supplementary Fig. 9).
Discussion
Mutations or multiplications in SNCA and the oligomerization, aggregation and accumulation of its protein product (α-synuclein) have been linked to PD. However, the mechanisms by which α-synuclein contributes to the loss of dopaminergic neurons are unclear. Dysregulation of intracellular metal homeostasis has been suggested as a likely contributor to PD (Ayton et al., 2012; Bharucha et al., 2008). In the present study, we demonstrated that non-toxic α-synuclein overexpression can increase the sensitivity of cells to Cu. While previous studies have demonstrated that Cu can promote α-synuclein aggregation, we found that the increased sensitivity to Cu of cells overexpressing α-synuclein is independent from its aggregation or oxidative stress. Interestingly, impairment of autophagy flux only increased Cu toxicity, while inhibition of the UPS enhanced the toxicity induced by Cu and Cu + α-synuclein. Our results demonstrate that α-synuclein enhances Cu toxicity in dopaminergic cells via modulation of protein degradation pathways (Supplementary Fig. 9).
Cu mediates oxidation-reduction reactions participating in the generation of ROS and subsequent oxidative damage. Indeed, we observed that Cu induced caspase-dependent apoptotic cell death in dopaminergic cells was associated with increased oxidative stress. Chronic intracellular accumulation of misfolded α-synuclein also promotes oxidative stress and mitochondrial dysfunction (Hsu et al., 2000), and α-synuclein-Cu complexes trigger ROS formation (Meloni and Vasak, 2011; C. Wang et al., 2010). Moreover, α-synuclein also potentiates oxidative stress induced by pesticides (Kanda et al., 2000; Ko et al., 2000). However, by using redox sensitive reporters we observed that WT or A53T α-synuclein overexpression had no significant effect on cytosolic or mitochondrial oxidative stress induced by Cu.
The extracellular Cu concentrations used in this work to induce cell death in dopaminergic neuroblastoma cells are high (500–750 μM) compared to those found in the CSF of PD patients (0.3 μM). Cu-induced toxicity has been shown to be potentiated by dopamine (Paris et al., 2001, 2009). The human SK-N-SH neuroblastoma cell line used here has been reported to have low levels of tyrosine hydroxylase activity (Klongpanichapak et al., 2008; West et al., 1977). Tyrosine hydroxylase levels in human neuroblastoma cells can be increased by their differentiation with retinoic acid, but their differentiation can makethem resistant to cell death by upregulation of anti-apoptotic signaling mechanisms (Cheung et al., 2009). Dopamine increases the sensitivity of dopaminergic cells to PD-related insults. Thus, the reduced sensitivity of the neuroblastoma cells to Cu + α-synuclein might be due to reduced dopamine content. Dopamine and/or its metabolites promote α-synuclein aggregation and toxicity and concomitantly, α-synuclein deregulates dopamine homeostasis (Conway et al., 2001 ; Lotharius and Brundin, 2002; Xu et al., 2002). The synergistic toxic interaction found in our experimental paradigm seems to be independent from α-synuclein aggregation. However, the pro-oxidant properties of dopamine might promote the synergistic toxicity induced by Cu + α-synuclein independent from α-synuclein oligomerization or aggregation. As such, we could reproduce the synergistic toxicity of Cu and α-synuclein in primary midbrain neuronal cultures using significantly lower concentrations of Cu (10 μM). Interestingly, localized Cu release from synaptic clefts was estimated to reach ~ 10–100 μM during neuronal activity (Hopt et al., 2003; Kardos et al., 1989).
The brain contains the highest Cu content in the body next to the liver (6–20 μg/g dry weight) (Kornhuber et al., 1994; Prohaska, 1987). Primary cortical neurons are reported to contain ten times higher levels of Cu than astrocytes (200 μg/g of protein) (Hare et al., 2013). The substantia nigra (59 μg/g dry weight) and the locus coeruleus (201 μg/g dry weight), where neuronal loss in PD is reported to be greater than in the substantia nigra (Zarow et al., 2003), contain higher concentrations of Cu compared to other brain regions (<35 μg/g dry weight) (Davies et al., 2013b; Lutsenko et al., 2010; Prohaska, 1987). At least 500 μM CuCl2 was required in our study to induce synergistic toxicity in dopaminergic cells when combined with α-synuclein overexpression. High extracellular Cu concentrations can exert nonspecific effects through alterations in membrane function and stability. However, in our experimental model exposure to 500 μM CuCl2 only correlated with an increase in Cu from 1.3 μg (0.02μmole) under normal conditions, to 64 μg/g of protein (1 μmole Cu/g protein). Thus, the intracellular Cu levels required to potentiate cell death in the presence of α-synuclein are similar to the levels found in the substantia nigra.
DMEM:F12 media formulation contains 5.2 nM CuSO4. Thus, a 10,000 fold increase in extracellular Cu (500 μM) is only translated to a 50 fold increase in intracellular Cu levels, suggesting that a limited permeability for translocation of Cu across the plasma membrane or an active efflux mechanism maintain intracellular Cu homeostasis. Importantly, this threshold is based on a dose-response, not a time course analysis. Thus, longer exposures to Cu might be required for lower concentrations of Cu to overcome cellular “defenses” and induce toxicity. Accordingly, we observed that overexpression of Ctr1 increased Cu-induced cell death demonstrating that the toxic effects of high extracellular Cu are mediated by changes in its intracellular levels. Interestingly, a recent report demonstrates that low levels of Ctr1 are found in survival neurons in the substantia nigra of PD patients (Davies et al., 2013a).
Another possibility for the requirement of high extracellular Cu levels to induce toxicity is likely associated with the very effective intracellular chelating systems for Cu, such as methallothioneins, metallochaperones and GSH (Meloni and Vasak, 2011; White et al., 1999). GSH depletion is an early biomarker of PD progression observed prior to any major alterations in metal homeostasis (Dexter et al., 1994; Pearce et al., 1997). GSH depletion/oxidation increases α-synuclein expression and aggregation (Y.H. Kim et al., 2011; Paik et al., 2003). Similarly, Cu-induced α-synuclein aggregation is enhanced by GSH depletion (Cheng and Trombetta, 2004). We found that GSH depletion significantly increased Cu-induced cell death. Metallothioneins exert protective effects against toxicological models of PD including oxidative stress inducedby α-synuclein complexes (Meloni and Vasak, 2011). Cu/metallothionein or Cu/ceruloplasmin ratios in plasma are not indicative of PD disease progression (Arnal et al., 2010). However, variations in ceruloplasmin synthesis and/or its Cu-binding properties have been proposed as essential features of PD (Bharucha et al., 2008; Torsdottir et al., 1999) and mediators of α-synuclein aggregation in vitro (Kim et al., 2002). It is important to note that the pathological consequences Cu are not necessary linked to the total amount of Cu in the cells but rather to the labile Cu pool available.
Dopamine (Gomez-Santos et al., 2003), neuromelanin (Li et al., 2012), environmental toxicants including pesticides (paraquat, dieldrin, rotenone) (Manning-Bog et al., 2002; Mulcahy et al., 2012; Sherer et al., 2003; Sun et al., 2005; Uversky et al., 2001b) and metals (Cu, Fe and Al) (Ostrerova-Golts et al., 2000; VanDuyn et al., 2013; X. Wang et al., 2010), as well as mitochondrial toxicants (MPTP/MPP+) (Fornai et al., 2005) can induce dopaminergic cell death and/or potentiate WT or mutant α-synuclein toxicity by increasing its expression levels or aggregation. α-synuclein oligomerization and fibrillogenesis are also modulated byphosphorylation, C-terminal truncations and interactions with metals or pro-oxidant conditions (Breydo et al., 2012; Stefanis, 2012). A number of studies have reported that Cu interacts with α-synuclein accelerating aggregation and fibril formation and also modulating its membrane localization (Bortolus et al., 2010; Rasia et al., 2005; Rose et al., 2011; Uversky et al., 2001a; X. Wang et al., 2010). The neurotoxicity of α-synuclein was also postulated to require Cu (Wright et al., 2009). Our data show that while inhibition of the proteasome increased the accumulation of HMW aggregates of WT or A53T α-synuclein, Cu had no major effects on either α-synuclein aggregation or membrane localization when the latter was overexpressedin ashort-term/non-toxic manner. These results are explained by the fact that formation of HMW aggregates in cells takes ~7 days of α-synuclein over-expression (Ko et al., 2008). Thus, our results do not necessarily contradict the well-establish stimulatory effect of Cu on α-synuclein aggregation. Instead, we report that overexpression of WT or A53T α-synuclein can also exert synergistic effects with Cu via modulation of protein degradation pathways. Most of the studies so far have associated the toxicity of α-synuclein overexpression and mutation to its aggregation. Interestingly, it was demonstrated that the toxicity of α-synuclein overexpression in a conditional mouse model is independent from filamentous inclusion body formation (Nuber et al., 2008). Very few studies exist to date that report on the effects of non-toxic/low expression levels of α-synuclein and its deleterious effects irrespective of its aggregation. Non-toxic overexpression of A30P α-synuclein lowers proteasomal activity and increases the intrinsic pathway of apoptosis (Tanaka et al., 2001). To the best of our knowledge, this is the first report demonstratingthat non-toxic α-synuclein overexpression can have deleterious effects by synergizing with metal toxicity (Cu) and proteasome dysfunction.
Endogenous α-synuclein is found natively unfolded and soluble in the cytoplasm, with a small fraction associated with lipid membranes. An interesting observation in our work was that both excess Cu and proteasome inhibition seem to alter the native conformation of α-synuclein. It has been demonstrated that α-synuclein exists predominantly as stable unfolded monomer or a tetramer that migrates as 57–60 kDa protein under native conditions. We found that both Cu and inhibition of the proteasomal activity induced the appearance of 14 kDa protein band under native conditions suggesting that Cu may be inducing either: 1) an alteration in the native conformation; 2) changes in the oligomerization (tetramerization) state; or 3) cleavage of α-synuclein. Furthermore, α-synuclein in such “modified state” appears to be more proteasomal degradation-prone, as suggested by its accumulation in the presence of MG132. Native α-synuclein has been proposed to exist in equilibrium between different conformational and/or oligomeric states (Lashuel et al., 2013), and the stable native state may prevent α-synuclein from forming toxic oligomers. Thus, the synergistic effect of Cu and α-synuclein (WT or A53T) overexpression might be linked to alterations (destabilization) in the protein’s non-toxic state. Consistent with this idea, proteases such as calpain, cathepsins or metalloproteases are known to cleave α-synuclein (Dufty et al., 2007), and the resulting C-terminal truncations have been shown to enhance α-synuclein toxicity (Burre et al., 2012). Identifying the nature of this modified/destabilized state and the mechanisms mediating its accumulation by Cu exposure or by inhibition of the proteasome will require more research.
Clearance of WT and/or A53T α-synuclein aggregates has been shown to be mediated by both autophagy and the ubiquitin/proteasome pathway (Cuervo et al., 2004; Ebrahimi-Fakhari et al., 2011 ; Webb et al., 2003). Reciprocally, α-synuclein can interfere with autophagy (Martinez-Vicente et al., 2008; Winslow et al., 2010) and the ubiquitin/proteasome system (Chen et al., 2005; Lindersson et al., 2004; Martin-Clemente et al., 2004; Snyder et al., 2003; Tanaka et al., 2001; Zhang et al., 2008). For example, WT and A53T α-synuclein variants inhibit autophagy via impairment of the cytosolic translocation of the high mobility group box 1 (HMGB1), reducing HMGB1-beclin-1 binding, and increasing Beclin-1-Bcl-2 association (Song et al., 2014), as well as by a decrease in the activity of the transcriptional regulator of the autophagy-lysosome pathway, TFEB (transcriptional factor EB) (Decressac et al., 2013). Oxidation of α-synuclein has been reported to reduce its degradation by the 20S proteasome (Alvarez-Castelao et al., 2013). Cu has been recently shown to induce the aggregation of ubiquitin (Arnesano et al., 2009). We found that Cu induced both an increase in protein ubiquitination and autophagy. The accumulation of ubiquitinated proteins induced by Cu was not associated with impairment in the proteasome pathway as evidenced by its enhancement in the presence of MG132, and its non-saturable kinetic in the presence or absence of the proteasome inhibitor. Overexpression of WT or A53T α-synuclein also stimulated both autophagy and the accumulation of ubiquitinated proteins. Because impairment of autophagy only enhanced Cu toxicity, while inhibition of the proteasome enhanced both Cu and WT/A53T α-synuclein toxicity, our results suggest that α-synuclein exerts its effect primarily via modulation of the UPS.
Autophagy is a catabolic process involved in cellular survival during conditions of environmental stress (Murrow and Debnath, 2013). In very few circumstances there is actual genetic-based evidence for a role of autophagy as a cell death pathway (Shen et al., 2012). We found that the AMPK-Ulk1 signaling and Atg5-dependent autophagy exerted a protective effect against Cu-induced apoptosis. While macroautophagy (here referred to as autophagy) depends on the formation of the Atg12-Atg5 conjugation system for autophagosome formation, chaperone-mediated autophagy (CMA) involves the direct delivery of substrates to the lysosome via recognition of specific chaperones (Kroemer et al., 2010). Both autophagy and CMA have been reported to mediate the clearance of α-synuclein (Cuervo et al., 2004; Malkus and Ischiropoulos, 2012; Vogiatzi et al., 2008). Our data cannot completely rule-out the role of autophagic processes in the synergistic toxic effects of Cu and α-synuclein because previous studies have showed that impaired macroautophagy leads to constitutive activation of CMA (Kaushik et al., 2008).
Overall, our work demonstrates, for the first time, that Cu exposure and α-synuclein (WT or A53T) exert a synergistic toxic effect on dopaminergic cells independent from α-synuclein aggregation. Our data also suggest that this synergistic effect is mediated by an increased “stress burden” to protein degradation mechanisms, primarily the UPS, and, to a lesser extent autophagy (Supplementary Fig. 9). The toxic effects of Cu/proteasome dysfunction and α-synuclein might be associated with alterations in the native state of the monomer.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Grant P20RR17675 Centers of Biomedical Research Excellence (COBRE), ES016337 (J.L.), GM108975 (O.K.) and the Alzheimer’s Association Grant NIRGD-10-172826 (R.F. and J.L.). L.Z.F. received a Post-doctoral Fellowship from the National Council of Science and Technology (CONACYT 211456) of Mexico. We would like to thank Dr. Charles A. Kuszynski and Daniel Shea at the Nebraska Center for Virology (NIGMS grant number P30 GM103509) for their helpintheflow cytometry analyses and cell sorting, as well as You Zhou and Terri Fangman at the Center for Biotechnology Microscopy Core for help with thefluorescent live-cell imaging.
Abbreviations
- A53T
mutant A53T α-synuclein
- AMPK
adenosine monophosphate (AMP)-activated protein kinase
- Atg5
autophagy protein 5
- ATP7A
Cu-transporting ATPase 1
- BSO
L-buthionine-sulfoximide
- Ctr1
Cu transporter protein 1
- Cu
copper
- CuCl2
copper chloride
- CuSO4
copper sulfate
- CMA
chaperone-mediated autophagy
- CQ
chloroquine
- DHE
dihydroethidine
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
- FRE
ferric reductase
- GSH
glutathione
- HMW
high molecular weight
- ICP-MS
Inductively Coupled Plasma Mass Spectrometry
- LC3
microtubule-associated protein 1A/1B-light chain 3
- mBCl
monochlorobimane
- MEFs
mouse embryonic fibroblasts
- MOI
multiplicity of infection
- MR
metalloreductasesNrf2, nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcription factor
- PD
Parkinson’s disease
- PI
propidium iodide
- roGFP
reduction-oxidation sensitive green fluorescent protein
- ROS
reactive oxygen species
- SNCA/PARK1
synuclein, alpha (non A4 component of amyloid precursor)/Parkinson disease, familial, type 1 gene
- STEAP
six-transmembrane epithelial antigen protein
- TBS
tris-buffered saline
- Ulk1
Unc-51-like kinase 1
- UPS
ubiquitin-proteasome system
- WT
wild type
- z-VAD-FMK
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2014.11.018.
Footnotes
Conflict of interest statement
The authors certify that there is no conflict of interest.
Available online on ScienceDirect (www.sciencedirect.com).
References
- Alvarez-Castelao B, et al. Mechanism of cleavage of alpha-synuclein by the 20S proteasome and modulation of its degradation by the RedOx state of the N-terminal methionines. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbamcr.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Arnal N, et al. Clinical utility of copper, ceruloplasmin, and metallothionein plasma determinations in human neurodegenerative patients and their first-degree relatives. Brain Res. 2010;1319:118–130. doi: 10.1016/j.brainres.2009.11.085. [DOI] [PubMed] [Google Scholar]
- Arnesano F, et al. Copper-triggered aggregation of ubiquitin. PLoS One. 2009;4:e7052. doi: 10.1371/journal.pone.0007052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auluck PK, et al. alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Ayton S, et al. Ceruloplasmin dysfunction and therapeutic potential for parkinson disease. Ann Neurol. 2012 doi: 10.1002/ana.23817. [DOI] [PubMed] [Google Scholar]
- Bampton ET, et al. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Barde I, et al. Production and titration of lentiviral vectors. Curr Protoc Neurosci. 2010 doi: 10.1002/0471142301.ns0421s53. (Chapter 4, Unit 4 21) [DOI] [PubMed] [Google Scholar]
- Barnea A, et al. High-affinity uptake of 67Cu into a veratridine-releasable pool in brain tissue. Am J Physiol. 1989;257:C315–C322. doi: 10.1152/ajpcell.1989.257.2.C315. [DOI] [PubMed] [Google Scholar]
- Bartels T, et al. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharucha KJ, et al. Lower serum ceruloplasmin levels correlate with younger age of onset in Parkinson’s disease. J Neurol. 2008;255:1957–1962. doi: 10.1007/s00415-009-0063-7. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. doi: 10.1016/S0076-6879(08)03612-4. [DOI] [PubMed] [Google Scholar]
- Bortner CD, Cidlowski JA. The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch. 2004;448:313–318. doi: 10.1007/s00424-004-1266-5. [DOI] [PubMed] [Google Scholar]
- Bortolus M, et al. Structural characterization of a high affinity mononuclear site in the copper(II)-alpha-synuclein complex. J Am Chem Soc. 2010;132:18057–18066. doi: 10.1021/ja103338n. [DOI] [PubMed] [Google Scholar]
- Brauchle E, et al. Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy. Sci Rep. 2014;4:4698. doi: 10.1038/srep04698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breydo L, et al. Alpha-synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta. 2012;1822:261–285. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Burre J, et al. Systematic mutagenesis of alpha-synuclein reveals distinct sequence requirements for physiological and pathological activities. J Neurosci. 2012;32:15227–15242. doi: 10.1523/JNEUROSCI.3545-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon JR, Greenamyre JT. Gene-environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiol Dis. 2013;57:38–46. doi: 10.1016/j.nbd.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardaci S, et al. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci. 2012;125:2115–2125. doi: 10.1242/jcs.095216. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chen Q, et al. Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J Biol Chem. 2005;280:30009–30017. doi: 10.1074/jbc.M501308200. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Trombetta LD. The induction of amyloid precursor protein and alpha-synuclein in rat hippocampal astrocytes by diethyldithiocarbamate and copper with or without glutathione. Toxicol Lett. 2004;146:139–149. doi: 10.1016/j.toxlet.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Cheung YT, et al. Effects of all-trans-retinoic acid on human SH-SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology. 2009;30:127–135. doi: 10.1016/j.neuro.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Circu ML, Aw TY. Glutathione and modulation of cell apoptosis. Biochim Biophys Acta. 2012;1823:1767–1777. doi: 10.1016/j.bbamcr.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway KA, et al. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- Cook C, et al. Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009423. doi: 10.1101/cshperspect.a009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, et al. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Davies KM, et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol Aging. 2013a doi: 10.1016/j.neurobiolaging.2013.09.034. [DOI] [PubMed] [Google Scholar]
- Davies KM, et al. Localization of copper and copper transporters in the human brain. Metallomics. 2013b;5:43–51. doi: 10.1039/c2mt20151h. [DOI] [PubMed] [Google Scholar]
- Decressac M, et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, et al. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter DT, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- Dexter DT, et al. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol. 1994;35:38–44. doi: 10.1002/ana.410350107. [DOI] [PubMed] [Google Scholar]
- Dufty BM, et al. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci. 2011;31:14508–14520. doi: 10.1523/JNEUROSCI.1560-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadeel B, et al. Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression. Biochem Biophys Res Commun. 1999;266:504–511. doi: 10.1006/bbrc.1999.1820. [DOI] [PubMed] [Google Scholar]
- Fauvet B, et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro-Peyret C, et al. Caspase-independent phosphatidylserine exposure during apoptosis of primary T lymphocytes. J Immunol. 2002;169:4805–4810. doi: 10.4049/jimmunol.169.9.4805. [DOI] [PubMed] [Google Scholar]
- Fornai F, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. Glutathione efflux and cell death. Antioxid Redox Signal. 2012;17:1694–1713. doi: 10.1089/ars.2012.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, et al. Glutathione depletion is necessary for apoptosis in lymphoid cells independent of reactive oxygen species formation. J Biol Chem. 2007;282:30452–30465. doi: 10.1074/jbc.M703091200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, et al. Glutathione depletion and disruption of intracellular ionic homeostasis regulate lymphoid cell apoptosis. J Biol Chem. 2008;283:36071–36087. doi: 10.1074/jbc.M807061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaier ED, et al. Copper signaling in the mammalian nervous system: synaptic effects. J Neurosci Res. 2013;91:2–19. doi: 10.1002/jnr.23143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin JE, et al. Synucleinopathies: clinical and pathological implications. Arch Neurol. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- Garcia-Garcia A, et al. Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP+-induced apoptosis but Not rotenone or 6-hydroxydopamine toxicity. Toxicol Sci. 2013;136:166–182. doi: 10.1093/toxsci/kft188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geske FJ, et al. Early stages of p53-induced apoptosis are reversible. Cell Death Differ. 2001;8:182–191. doi: 10.1038/sj.cdd.4400786. [DOI] [PubMed] [Google Scholar]
- Gomez-Santos C, et al. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- Gorell JM, et al. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology. 1997;48:650–658. doi: 10.1212/wnl.48.3.650. [DOI] [PubMed] [Google Scholar]
- Gorell JM, et al. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- Hanada T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- Hanson GT, et al. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem. 2004;279:13044–13053. doi: 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- Hardie DG, et al. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare DJ, et al. Profiling the iron, copper and zinc content in primary neuron and astrocyte cultures by rapid online quantitative size exclusion chromatography-inductively coupled plasma-mass spectrometry. Metallomics. 2013;5:1656–1662. doi: 10.1039/c3mt00227f. [DOI] [PubMed] [Google Scholar]
- He TC, et al. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopt A, et al. Methods for studying synaptosomal copper release. J Neurosci Methods. 2003;128:159–172. doi: 10.1016/s0165-0270(03)00173-0. [DOI] [PubMed] [Google Scholar]
- Horowitz MP, Greenamyre JT. Gene-environment interactions in Parkinson’s disease: the importance of animal modeling. Clin Pharmacol Ther. 2010;88:467–474. doi: 10.1038/clpt.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hozumi I, et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J Neurol Sci. 2011;303:95–99. doi: 10.1016/j.jns.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Hsu LJ, et al. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Jimenez FJ, et al. Serum levels of zinc and copper in patients with Parkinson’s disease. J Neurol Sci. 1992;112:30–33. doi: 10.1016/0022-510x(92)90127-7. [DOI] [PubMed] [Google Scholar]
- Kanda S, et al. Enhanced vulnerability to oxidative stress by alpha-synuclein mutations and C-terminal truncation. Neuroscience. 2000;97:279–284. doi: 10.1016/s0306-4522(00)00077-4. [DOI] [PubMed] [Google Scholar]
- Kardos J, et al. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett. 1989;103:139–144. doi: 10.1016/0304-3940(89)90565-x. [DOI] [PubMed] [Google Scholar]
- Kaushik S, et al. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008;19:2179–2192. doi: 10.1091/mbc.E07-11-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Lindquist S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker’s yeast? Nat Rev Neurosci. 2010;11:436–449. doi: 10.1038/nrn2809. [DOI] [PubMed] [Google Scholar]
- Kim KS, et al. The ceruloplasmin and hydrogen peroxide system induces alpha-synuclein aggregation in vitro. Biochimie. 2002;84:625–631. doi: 10.1016/s0300-9084(02)01435-9. [DOI] [PubMed] [Google Scholar]
- Kim BE, et al. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011a;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, et al. Inducible dopaminergic glutathione depletion in an alpha-synuclein transgenic mouse model results in age-related olfactory dysfunction. Neuroscience. 2011b;172:379–386. doi: 10.1016/j.neuroscience.2010.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, et al. SLC31 (CTR) family of copper transporters in health and disease. Mol Aspects Med. 2013;34:561–570. doi: 10.1016/j.mam.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MA, et al. There is substantial nuclear and cellular disintegration before detectable phosphatidylserine exposure during the camptothecin-induced apoptosis of HL-60 cells. Cytometry. 2000;40:10–18. [PubMed] [Google Scholar]
- Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klongpanichapak S, et al. Melatonin inhibits amphetamine-induced increase in alpha-synuclein and decrease in phosphorylated tyrosine hydroxylase in SK-N-SH cells. Neurosci Lett. 2008;436:309–313. doi: 10.1016/j.neulet.2008.03.053. [DOI] [PubMed] [Google Scholar]
- Klucken J, et al. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy. 2012;8:754–766. doi: 10.4161/auto.19371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko L, et al. Sensitization of neuronal cells to oxidative stress with mutated human alpha-synuclein. J Neurochem. 2000;75:2546–2554. doi: 10.1046/j.1471-4159.2000.0752546.x. [DOI] [PubMed] [Google Scholar]
- Ko LW, et al. Aggregates assembled from overexpression of wild-type alpha-synuclein are not toxic to human neuronal cells. J Neuropathol Exp Neurol. 2008;67:1084–1096. doi: 10.1097/NEN.0b013e31818c3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, et al. Iron, copper, zinc, magnesium, and calcium in postmortem brain tissue from schizophrenic patients. Biol Psychiatry. 1994;36:31–34. doi: 10.1016/0006-3223(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Kroemer G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, et al. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, et al. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, et al. Biochemical characterization of the human copper transporter Ctr1. J Biol Chem. 2002;277:4380–4387. doi: 10.1074/jbc.M104728200. [DOI] [PubMed] [Google Scholar]
- Lee SH, et al. Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death Differ. 2013;20:64–76. doi: 10.1038/cdd.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire C, et al. Inhibition of caspase activity induces a switch from apoptosis to necrosis. FEBS Lett. 1998;425:266–270. doi: 10.1016/s0014-5793(98)00252-x. [DOI] [PubMed] [Google Scholar]
- Li J, et al. Neuromelanin enhances the toxicity of alpha-synuclein in SK-N-SH cells. J Neural Transm. 2012;119:685–691. doi: 10.1007/s00702-011-0753-z. [DOI] [PubMed] [Google Scholar]
- Lindersson E, et al. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- Liu F, et al. Methionine sulfoxide reductase A protects dopaminergic cells from Parkinson’s disease-related insults. Free Radic Biol Med. 2008a;45:242–255. doi: 10.1016/j.freeradbiomed.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, et al. Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson’s disease. J Neurochem. 2008b;105:2435–2453. doi: 10.1111/j.1471-4159.2008.05333.x. [DOI] [PubMed] [Google Scholar]
- Liu Y, et al. Comparative features of copper ATPases ATP7A and ATP7B heterologously expressed in COS-1 cells. Biochemistry. 2010;49:10006–10012. doi: 10.1021/bi101423j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P. Impaired dopamine storage resulting from alpha-synuclein mutations may contribute to the pathogenesis of Parkinson’s disease. Hum Mol Genet. 2002;11:2395–2407. doi: 10.1093/hmg/11.20.2395. [DOI] [PubMed] [Google Scholar]
- Lutsenko S, et al. Copper handling machinery of the brain. Metallomics. 2010;2:596–608. doi: 10.1039/c0mt00006j. [DOI] [PubMed] [Google Scholar]
- Malkus KA, Ischiropoulos H. Regional deficiencies in chaperone-mediated autophagy underlie alpha-synuclein aggregation and neurodegeneration. Neurobiol Dis. 2012;46:732–744. doi: 10.1016/j.nbd.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Bog AB, et al. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- Mariani S, et al. Fe and Cu do not differ in Parkinson’s disease: a replication study plus meta-analysis. Neurobiol Aging. 2013;34:632–633. doi: 10.1016/j.neurobiolaging.2012.05.015. [DOI] [PubMed] [Google Scholar]
- Martin-Clemente B, et al. alpha-Synuclein expression levels do not significantly affect proteasome function and expression in mice and stably transfected PC12 cell lines. J Biol Chem. 2004;279:52984–52990. doi: 10.1074/jbc.M409028200. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuoka J, Saltman P. Zinc(II) and copper(II) binding to serum albumin. A comparative study of dog, bovine, and human albumin. J Biol Chem. 1994;269:25557–25561. [PubMed] [Google Scholar]
- McCall K. Genetic control of necrosis – another type of programmed cell death. Curr Opin Cell Biol. 2010;22:882–888. doi: 10.1016/j.ceb.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloni G, Vasak M. Redox activity of alpha-synuclein-Cu is silenced by Zn(7)-metallothionein-3. Free Radic Biol Med. 2011;50:1471–1479. doi: 10.1016/j.freeradbiomed.2011.02.003. [DOI] [PubMed] [Google Scholar]
- Messam CA, Pittman RN. Asynchrony and commitment to die during apoptosis. Exp Cell Res. 1998;238:389–398. doi: 10.1006/excr.1997.3845. [DOI] [PubMed] [Google Scholar]
- Mulcahy P, et al. Development and characterisation of a novel rat model of Parkinson’s disease induced by sequential intranigral administration of AAV-alpha-synuclein and the pesticide, rotenone. Neuroscience. 2012;203:170–179. doi: 10.1016/j.neuroscience.2011.12.011. [DOI] [PubMed] [Google Scholar]
- Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol. 2013;8:105–137. doi: 10.1146/annurev-pathol-020712-163918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, et al. Neurodegeneration and motor dysfunction in a conditional model of Parkinson’s disease. J Neurosci. 2008;28:2471–2484. doi: 10.1523/JNEUROSCI.3040-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo A, et al. NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins. Hum Mol Genet. 2013;22:1699–1708. doi: 10.1093/hmg/ddt016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrerova-Golts N, et al. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci. 2000;20:6048–6054. doi: 10.1523/JNEUROSCI.20-16-06048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik SR, et al. Oxidized glutathione stimulated the amyloid formation of alpha-synuclein. FEBS Lett. 2003;537:63–67. doi: 10.1016/s0014-5793(03)00081-4. [DOI] [PubMed] [Google Scholar]
- Pall HS, et al. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet. 1987;2:238–241. doi: 10.1016/s0140-6736(87)90827-0. [DOI] [PubMed] [Google Scholar]
- Paris I, et al. Copper neurotoxicity is dependent on dopamine-mediated copper uptake and one-electron reduction of aminochrome in a rat substantia nigra neuronal cell line. J Neurochem. 2001;77:519–529. doi: 10.1046/j.1471-4159.2001.00243.x. [DOI] [PubMed] [Google Scholar]
- Paris I, et al. Copper dopamine complex induces mitochondrial autophagy preceding caspase-independent apoptotic cell death. J Biol Chem. 2009;284:13306–13315. doi: 10.1074/jbc.M900323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RK, et al. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Prohaska JR. Functions of trace elements in brain metabolism. Physiol Rev. 1987;67:858–901. doi: 10.1152/physrev.1987.67.3.858. [DOI] [PubMed] [Google Scholar]
- Rasia RM, et al. Structural characterization of copper(II) binding to alpha-synuclein: Insights into the bioinorganic chemistry of Parkinson’s disease. Proc Natl Acad Sci U S A. 2005;102:4294–4299. doi: 10.1073/pnas.0407881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochet JC, et al. Molecular insights into Parkinson’s disease. Prog Mol Biol Transl Sci. 2012;107:125–188. doi: 10.1016/B978-0-12-385883-2.00011-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rocha H, et al. Glutaredoxin 1 protects dopaminergic cells by increased protein glutathionylation inexperimental Parkinson’s disease. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rocha H, et al. Compartmentalized oxidative stress in dopaminergic cell death induced by pesticides and complex I inhibitors: Distinct roles of superoxide anion and superoxide dismutases. Free Radic Biol Med. 2013;61C:370–383. doi: 10.1016/j.freeradbiomed.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose F, et al. Mechanism of copper(II)-induced misfolding of Parkinson’s disease protein. Sci Rep. 2011;1:11. doi: 10.1038/srep00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybicki BA, et al. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov Disord. 1993;8:87–92. doi: 10.1002/mds.870080116. [DOI] [PubMed] [Google Scholar]
- Santner A, Uversky VN. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics. 2010;2:378–392. doi: 10.1039/b926659c. [DOI] [PubMed] [Google Scholar]
- Schlegel RA, Williamson P. Phosphatidylserine, a death knell. Cell Death Differ. 2001;8:551–563. doi: 10.1038/sj.cdd.4400817. [DOI] [PubMed] [Google Scholar]
- Segawa K, et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- Shen S, et al. The end of autophagic cell death? Autophagy. 2012;8:1–3. doi: 10.4161/auto.8.1.16618. [DOI] [PubMed] [Google Scholar]
- Sherer TB, et al. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179:9–16. doi: 10.1006/exnr.2002.8072. [DOI] [PubMed] [Google Scholar]
- Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- Snyder H, et al. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- Song JX, et al. HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy. 2014;10:144–154. doi: 10.4161/auto.26751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell SR, et al. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol Biol Cell. 2009;20:1408–1418. doi: 10.1091/mbc.E08-07-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathearn KE, et al. Neuroprotective effects of anthocyanin- and proanthocyanidin-rich extracts in cellular models of Parkinsons disease. Brain Res. 2014;1555:60–77. doi: 10.1016/j.brainres.2014.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suazo M, et al. CCS and SOD1 mRNA are reduced after copper supplementation in peripheral mononuclear cells of individuals with high serum ceruloplasmin concentration. J Nutr Biochem. 2008;19:269–274. doi: 10.1016/j.jnutbio.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Sun F, et al. Dieldrin induces ubiquitin-proteasome dysfunction in alpha-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. J Pharmacol Exp Ther. 2005;315:69–79. doi: 10.1124/jpet.105.084632. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, et al. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Torsdottir G, et al. Copper, ceruloplasmin, superoxide dismutase and iron parameters in Parkinson’s disease. Pharmacol Toxicol. 1999;85:239–243. doi: 10.1111/j.1600-0773.1999.tb02015.x. [DOI] [PubMed] [Google Scholar]
- Tsigelny IF, et al. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012;279:1000–1013. doi: 10.1111/j.1742-4658.2012.08489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, et al. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem. 2001a;276:44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- Uversky VN, et al. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson’s disease. FEBS Lett. 2001b;500:105–108. doi: 10.1016/s0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- Vance JM, et al. Gene-environment interactions in Parkinson’s disease and other forms of parkinsonism. Neurotoxicology. 2010;31:598–602. doi: 10.1016/j.neuro.2010.04.007. [DOI] [PubMed] [Google Scholar]
- VanDuyn N, et al. The metal transporter SMF-3/DMT-1 mediates aluminum-induced dopamine neuron degeneration. J Neurochem. 2013;124:147–157. doi: 10.1111/jnc.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, et al. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a porelike mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- Wach A, et al. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- Wang C, et al. Redox reactions of the alpha-synuclein-Cu(2+) complex and their effects on neuronal cell viability. Biochemistry. 2010a;49:8134–8142. doi: 10.1021/bi1010909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J Neurochem. 2010b;113:704–714. doi: 10.1111/j.1471-4159.2010.06638.x. [DOI] [PubMed] [Google Scholar]
- Webb JL, et al. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, et al. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- West GJ, et al. Adrenergic, cholinergic, and inactive human neuroblastoma cell lines with the action-potential Na + ionophore. Cancer Res. 1977;37:1372–1376. [PubMed] [Google Scholar]
- White AR, et al. Exacerbation of copper toxicity in primary neuronal cultures depleted of cellular glutathione. J Neurochem. 1999;72:2092–2098. doi: 10.1046/j.1471-4159.1999.0722092.x. [DOI] [PubMed] [Google Scholar]
- Winslow AR, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodkowic D, et al. Apoptosis and beyond: cytometry in studies of programmed cell death. Methods Cell Biol. 2011;103:55–98. doi: 10.1016/B978-0-12-385493-3.00004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JA, et al. Unique copper-induced oligomers mediate alpha-synuclein toxicity. FASEB J. 2009;23:2384–2393. doi: 10.1096/fj.09-130039. [DOI] [PubMed] [Google Scholar]
- Xu J, et al. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- Xu Q, et al. Bortezomib rapidly suppresses ubiquitin thiolesterification to ubiquitin-conjugating enzymes and inhibits ubiquitination of histones and type I inositol 1,4,5-trisphosphate receptor. Mol Cancer Ther. 2004;3:1263–1269. [PubMed] [Google Scholar]
- Yang MY, et al. Reversible phosphatidylserine expression on blood granulocytes related to membrane perturbation but not DNA strand breaks. J Leukoc Biol. 2002;71:231–237. [PubMed] [Google Scholar]
- Yang L, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C, et al. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- Zhang NY, et al. alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem. 2008;283:20288–20298. doi: 10.1074/jbc.M710560200. [DOI] [PubMed] [Google Scholar]
- Zheng TS, et al. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.