

Abstract
Phospholipid remodeling and eicosanoid synthesis are central to lipid-based inflammatory reactions. Studies have revealed that membrane phospholipid remodeling by fatty acids through deacylation/reacylation reactions increases the risk of colorectal cancers (CRC) by allowing the cells to produce excess inflammatory eicosanoids such as prostaglandins, thromboxanes, and leukotrienes. Over the years, efforts have been made to understand the lipid remodeling pathways and to design anti-cancer drugs targeting the enzymes of eicosanoid biosynthesis. Here, we discuss the recent progress in phospholipid remodeling and eicosanoid biosynthesis in CRC.
Keywords: Colon cancer, Arachidonic acid, Lipid remodeling, Cyclooxygenase, Lipooxygenase, Prostaglandins, Leukotrienes
Colorectal cancer (CRC) is prevalent in both developed and developing countries. Epidemiological studies indicate that the consumption of high-fat and high-calorie diets could be linked to CRC. In addition, genetic pre-disposition such as Lynch syndrome (hereditary non-polyposis colorectal cancer) has also been implicated in inducing this deadly disease1,2. In the U.S., however, the overall incidence of colon cancer is on the decline and it has been predicted that this disease will be reduced further if healthier life-styles are followed and anti-cancer medications are made available to the general population3. While the mortality rates for colorectal and other cancers are decreasing in the U.S., for a large population in the developing countries they are on the rise. It is anticipated that the incidence of cancers, including colorectal, breast, lung, and prostate cancer, will increase 60% by 2030 in those countries4. The incidence and survival rates of CRC in India are, however, low compared with those in other Asian countries, and this can probably be attributed to the consumption of a diet richer in vegetables and fruits5.
CRC Genes
Adenocarcinomas are the most common form of CRC. They are the cancerous tumors of the epithelial tissue and are differentiated from adenomas or polyps2. The two subtypes of adenocarcinomas are the signet-ring-cell and mucinous-cell carcinomas. The signet-ring-cell carcinoma is considered to be an aggressive form of CRC, and it can be life threatening6. The mucinous adenocarcinoma is also considered an aggressive form of cancer, and this may be partially due to its ability to spread rapidly7. As far as locations are concerned, the majority of colonic tumors are formed in the distal part of the colon8. The deficient-in-mismatch-repair (dMMR) colonic tumors preferentially localize in the proximal section9. Although the actual reason for this preferential localization is not well understood, it is hypothesized that this could be due to the presence of typical mismatch-repair (MMR) characteristics such as chromosomal instability, loss of heterozygosity and Wnt/β-catenin signaling defects in colorectal cancers. β-Catenin is a transcriptional co-activator that interacts with Wnt/β signaling pathway to regulate embryonic development and cellular homeostasis10. Studies indicate that β-catenin level varies along the colorectal tract. While moderate level of β-catenin supports proximal tumor location, higher concentration prefers distal location9. Alteration of Wnt signaling pathway and mutations in the β-catenin gene affect the regulation of the adenomatous polyposis coli gene (APC), which may lead to CRC11,12.
The major CRC genes identified so far are APC (syndrome: familial adenomatous polyposis or FAP and attenuated FAP), MUTYH MAP (syndrome: MUTYH-associated polyposis, MAP), MLH1, MSH2, MSH6, PMS2, and TACSTD1 (genes associated with the Lynch syndrome); STK11 PJS (the Peutz-Jeghers syndrome), SMAD4 (DPC4), and BMPR1A (juvenile polyposis syndrome), and PTEN (the Cowden Syndrome)13. Three major types of CRC groups exist as follows: (i) the chromosomal instability group, (ii) the microsatellite unstable group, and (iii) the CpG island methylation phenotypic group14. In chromosomal instability (CIN), mis-segregation of the 18q chromosome results in the loss of heterozygosity and aneuploidy, which can lead to CRC13. In addition to 18q, several other genes or loci could be linked to CIN and those include 8q23.3, 8q24, 10p14, 11q23, 15q13, and 18q215,16. In addition, a recent genome-wide screening (GWS) identified 14–15 genes/loci that could also be linked to CIN13.
It is reported that ~15–20% of sporadic and 2–5% of Lynch syndrome CRCs are associated with microsatellite instability (MSI)17. Microsatellites are small DNA segments consisting of short nucleotides (also known as short-sequence repeats or short tandem repeats) that can undergo mutations during replication and genetic recombination. It has been proposed that dMMR-carrying colonic tumors exhibit altered expression of genes and proteins that could be responsible for triggering the MSI. Thus, the mutations and/or alterations of the functions of MMR genes/proteins (i.e., MLH1, MSH2, MSH6, and PMS2) could lead to the MSI17. The phenomenon of DNA methylation of CpG islands (CpG island is a GC-rich region of the genome) is common in sporadic MSI colonic tumors with CpG island methylation phenotype (CIMP). Abnormal DNA methylation of CpG islands affects the function and expression of tumor-suppressor genes18. Interestingly, CIMP is not common in Lynch syndrome and it is more frequently localized in the proximal area rather than the distal region of the colon, where it is linked to serine/threonine-protein kinase B-Raf (BRAF)18. CpG island methylation is an epigenetic phenomenon and is involved in the modulation of gene transcription in normal cells. However, in neoplasia hypermethylation, CpG islands induce unregulated transcription and alter the chromatin structure19. Thus, it appears that MSI, CIMP and BRAF mutation and epigenetic events, although interlinked, do not belong to a consolidated pathway, which makes the entire process incredibly complex. More research is needed to provide understanding of this process.
Fatty acid metabolism and CRC
Another important aspect of CRC is elevated fatty acid metabolism and eicosanoid biosynthesis. It has been demonstrated20 that the levels of stearic acid (C18:0), arachidonic acid (C20:4), and decosahexanoic acid (C22:6) increase significantly in CRC patients, although the study has a small sample but results have been interesting and suggested that fatty acids play important roles in CRC. Furthermore, polymorphisms in fatty acid metabolic genes have been linked to CRC21. In 2003, we reviewed the importance of arachidonic acid (AA) in CRCs2. Since then, hundreds of papers have been published underscoring the involvement of fatty acids, diets and CRC. It is hypothesized that while long-chain ω-6 polyunsaturated fatty acids (PUFA; AA for example) are potential inducers of CRCs, ω-3 PUFA suppresses tumorigenesis by interacting with protein kinase CβII signaling pathways2,22. However, a recent epidemiological study suggests that the relationship between ω-6 and ω-3 fatty acid levels and CRC induction/prevention may dependent on gender, locations of tumors (proximal or distal colon), and time of the follow-up study. The study has found that ω-3 PUFA could be linked with the formation of tumors in the distal colon in both men and women in the U.S23. However, more follow-up studies are required to confirm this interesting finding. A large-scale genome-wide study in East Asian population has identified new sets of CRC-linked genetic loci, including fatty acid-metabolism loci, indicating that fatty-acid metabolism could be key to CRC induction24. Therefore, it is conceivable that the association between ω-6 and ω-3 PUFA is more complex than was thought previously and that it may work in concert with several other gene products and pathways of CRC described above.
Phospholipid remodeling and eicosanoid synthesis
Fatty acid metabolism and the synthesis of downstream inflammatory molecules are regulated in part by phospholipid remodeling reactions. In this reaction, newly acquired fatty acids are incorporated into phospholipids (PLs) or lysophospholipids (LPLs) of plasma membranes to generate a phosphoglyceride with a new acyl chain at the SN2 position of the glycerol (Fig. 1, compartment A). The enzymes, such as phospholipase A2s (PLA2s) and acyltransferases (ATs) are important for carrying out the remodeling reaction25. Lysophosphatidylcholine acyltransferase 1 (LPCAT1), the enzyme that transacylases lysophosphatidylcholine (LPC) into phosphatidylcholine (PC) is overexpressed significantly in CRCs, indicating a direct correlation between the transacylation reaction and tumorigenesis/malignancy26. It is shown that the rate of AA incorporation in various PLs and its simultaneous release from arachidonoyl-PL increases significantly in activated human T cells27.
Fig. 1.
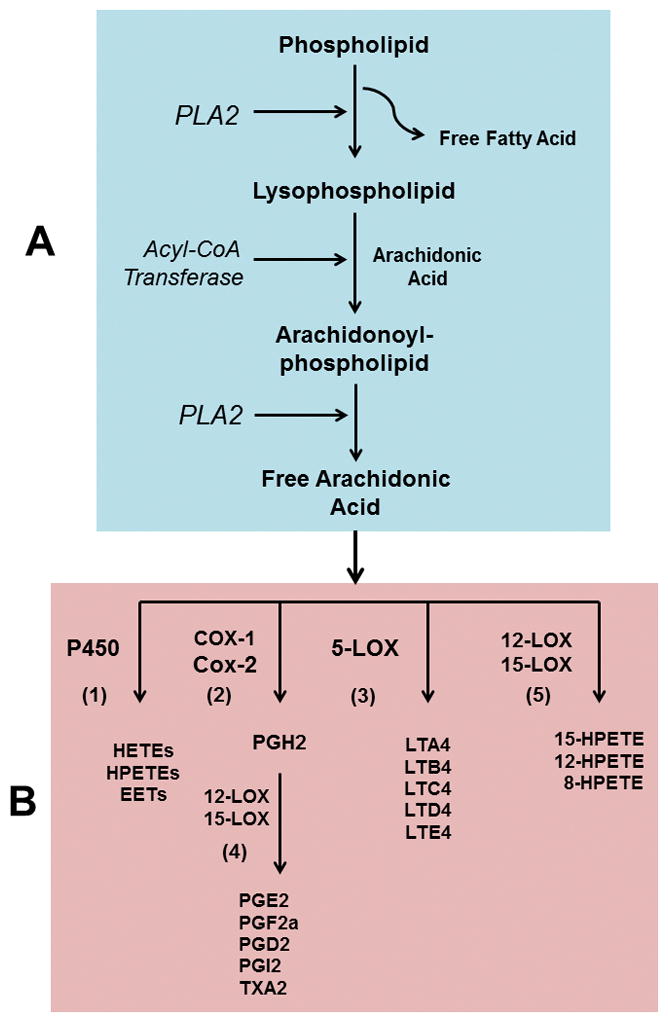
Fatty acid remodeling of membrane phospholipid (compartment A). The existing fatty acids at the SN2 position of a phospholipid is hydrolyzed by PLA2, generating a lysophospholipid molecule. Lysophospholipid serves as an acceptor of a new PUFA (usually arachidonic acid) to synthesize remodeled arachidonoyl-phospholipid catalyzed by acyl-CoA transferase enzymes. In the next step, arachidonoyl-phospholipid undergoes deacylation reaction (by PLA2) releasing free arachidonic acid. Free arachidonic acid acts as a substrate of COX and LOX enzymes of eicosanoid pathway (compartment B) and synthesizes PGs, TXA2, LTs and HETE/HPETE molecules. Compartment B: (1) arachidonic acid can be utilized by cytochrome P450 enzymes to synthesize HETEs, HPETEs and epoxyeicosatrienoic acids (EETs). (2) COX-1/COX-2 enzymes converts arachidonic acid to PGH2 and then to other PGs facilitated by 12-LOX and 15-LOX as shown in step 4; (3) 5-LOX converts arachidonic acid to various LTs. (5) 15-, 12- and 8-HPETEs are synthesized from arachidonic acid by the help of 12- and 15-LOX enzymes. PLA2, phospholipase A2; PUFA, polyunsaturated fatty acid; PG, prostaglandin; TXA2, thromboxane A2, LT, leukotriene; HETE, hydroxyeicosatetraenoic acids; HPETE, hydroperoxyeicosatetraenoic acids; COX, cycooxygenase; LOX, lipooxygenase.
AA is also distributed in various cellular PLs and this redistribution is proposed as being associated with CoA-dependent and -independent transacylation reactions. The transfer of AA from archidonoyl-PL to other PLs, especially phosphatidylethanolamine has been observed in activated T cells, which could be connected with the cell division and proliferation28. There are numerous reports in the literature that suggest the possible participation of PUFA and PUFA metabolic enzymes in the initiation and progression of CRC, and many of these studies indicate the direct involvement of PL remodeling reactions29.
It can be assumed that PLA2 serves as a key regulator of PL remodeling. PLA2 cleaves fatty acid (usually AA) from the SN2 position of a PL which then functions as a precursor for various types of eicosanoids (illustrated in Fig. 1, compartment B). Based on the substrate preferences, co-factor requirements and genetic/structural uniqueness, PLA2 families can be divided into three major classes-(i) small secreted PLA2 (sPLA2-Ca2+-dependent), (ii) large cytosolic PLA2 (cPLA2-Ca2+-dependent), and (iii) Ca2+-independent intracellular PLA2 (iPLA2) and platelet activating factor (PAF) secreted by PLA2 (PLA2G7)30. sPLA2, cPLA2, and PAF-PLA2 are shown to be associated with CRC. For example, sPLA2 (PLA2G10), which preferably catalyzes phosphatidylcholine (PC), plays an important role in forming tumors in the colon by releasing various small-molecule lipid mediators31. It is demonstrated earlier that AA released from arachidonoyl-PC by PLA2G10 undergoes cyclization by cyclooxygenase (COX) and lipooxygenase (LOX) pathways to produce small, inflammatory lipid molecules32. Likewise, genetic polymorphisms of group 4A cPLA2αs result in phenotypic features similar to those seen in patients with FAP33. Thus cPLA2α may act as a modulator of the disease process. The deletion of cPLA2 locus (pla2G4) in APC Min mice is shown to reduce the formation of new tumors, when compared with APCMincPLA2+ mice34.
The activation of colonic carcinoma cells HT29 by epidermal growth factor (EGF) upregulates cPLA2α activity and produces a significant increase in cell proliferation, indicating that this enzyme is responsible for the growth and proliferation of HT29 cells35. It is also demonstrated that cPLA2α-mediated HT29 proliferation and the concomitant increase in prostaglandin E2 (PGE2) production involves PKA and PKB/Akt pathways35. Similar to groups 4 and 10 PLA2s, PAF-PLA2 (PLA2G7) is also linked with CRC. Based on cellular and genetic studies, it is observed that the deletion of the pla2g7 gene reduces intestinal polyps through a mechanism that involves β-arrestin, Akt phosphatase, and intrinsic apoptotic pathway36. In contrast to these three groups of PLA2s, the role of iPLA2 in CRC is still not clear, and further studies are required to elucidate its role in the initiation and progression of colon cancers37.
It has already been discussed above that PL remodeling by deacylation/reacylation reactions serves as a regulator of eicosanoid biosynthesis. AA or other PUFAs released from PLs (or PL-like molecules) by the action of a specific PLA2 serve as precursors of COX, LOX, and cytochrome P450 enzymes to produce eicosanoids and other bioactive lipid-mediators like hydroxyeicosatetraenoic acids (HETEs) and hydroperoxyeicosatetraenoic acids (HPETEs) (Fig. 1, compartment B). In addition to genetic predisposition, frequent inflammation in the colon is considered as a risk factor of CRC. Molecules like prostaglandins G2 (PGG2), H2 (PGH2), E2 (PGE2), D2 (PGD2) and F2α (PGF2α), prostaglandin I2 (PGI2) and thromoboxaneA2 (TXA2) are the products of two separate COX enzymes, i.e. COX-1 and -2 (Fig. 1B).
While COX-1 is expressed constitutively, COX-2 is an inducible enzyme. The expression of COX-2 increases in inflammatory, mitogenic and cancerous cells38. PGs are secreted by the cells and act through autocrine and paracrine mechanisms via G-protein-coupled prostaglandin receptors (EPs). Eight membrane receptors have been identified so far, including EP1, EP2, EP3, and EP4 for PGE2, DP for PGD2, FP for PGF2, IP for PGI2, and TP for thromboxane A2. Reports suggest that expression and function of eicosanoid receptors are linked to CRC and that they play roles in polyp formation in the colon39. COX-2 expression increases dramatically in CRC and acts through PKC and Ras-signaling pathway40,41.
Using genetic knockout mice, it is demonstrated that COX-2 (but not COX-1) induces colorectal tumors and that suppressing its activity either genetically or chemically reduces tumor formation42. The growth and motility of colon cancer cells is also reported to be dependent upon PGE2 production, one of the products of the COX-2 enzyme43. PGE2 exerts its effect by activating phosphatidylinositol 3 kinase/protein kinase B (PI3K/PKB) pathway. Another product of the COX-1/COX-2 pathway is TXA2 and it has been observed that TXA2 can restore the migration of colonic carcinoma cells that have been inhibited by COX-2 inhibitors44.
AA is also metabolized by 5-lipooxygenase (5-LOX) enzyme to synthesize leukotrienes (LTs), and this reaction is facilitated by 5-LOX activating factor or FLAP45. There are several different types of LTs, which include leukotrienes A4 (LTA4), B4 (LTB4), C4 (LTC4), D4 (LTD4) and E4 (LTE4). LTC4, LTD4, and LTE4 are collectively known as cysteinyl leukotrienes (CysLTs) because of the presence of cysteine residues in the molecule. LTE4 is the most stable of all of the CysLTs. Because LTA4 and LTB4 contain no cysteine, they are not considered as CysLTs46. In addition to 5-LOX, there are also 12-LOX and 15-LOX enzymes, which facilitate the synthesis of 15-, 12- and 8-hydroperoxyeicosatetraenoic acids (15-, 12-, and 8-HPETEs). These short-lived, bioactive lipid mediators are quickly converted to LTs by dehydration reactions45. Interestingly, the 15-LOX activity declines in CRC, resulting in a reduction of apoptosis47. Several G-protein coupled receptor (GPCR) families of receptors function as LT or CysLT receptors that include CysLT1R, CysLT2R, BLT1, and BLT2. LOX activities increase several fold during the initiation and progression of colorectal and other cancers48. Increased expression of CysLT1R has been observed in cultured colon cancer cell lines, and the changes in the CysLT1R/CystLT2R ratio are linked to CRC45. Once the CysLT1R is activated by LTD4, CRC-linked proteins such as COX-2, β-Catenin and Bcl-2 are stimulated. In addition, CysLT1R activation also involves the participation of PI3K, glycogen synthase kinase 3β (GSK-3β), cAMP-response-binding protein (CREB), p90 ribosomal S6 kinase and PI3K-Rac pathways. These signaling molecules/pathways are associated with the survival, proliferation and migration of cancer cells45,49.
Drugs targeting the remodeling/eicosanoid pathway
Aspirin (acetylsalicylic acid) is a common analgesic and anti-inflammatory drug. This non-steroidal anti-inflammatory drug (NSAID) was reported to use treat patients suffering from glycosuria and diabetes mellitus50. It has been found that aspirin is effective in reducing the pain of pancreatic and colonic cancer51 and studies conducted over the past forty years have indicated that NSAIDs, such as aspirin, sulindac, ibuprofen, celecoxib and indomethacin minimize the incidence of CRC by 40%52. In recent studies of the effects of aspirin and other NSAIDs on risk and survival were conducted recently, and it was observed that a daily intake of aspirin (75 mg) lowers the incidence of CRC in high-risk subjects. However, NSAID intake prior to the diagnosis of the disease has failed to yield any significant results53. As far as the molecular mechanisms are concerned, aspirin and other NSAIDs can act through COX-dependent and-independent pathways and influence various signaling pathways such as TGF-β-induced apoptotic pathways, sphingosine phosphatase and kinase-mediated angiogenesis, and cytokines/platelet-derived growth-factor-mediated cells54. The COX-independent actions of NSAIDs involve the NFkB family of molecules, which includes p50 and RelA (p65) heterodimer, I-kappaB, Wnt/B catenin signaling pathway, β-catenin destruction complex, tumor-promoting genes and proteins and cell cycle complex to mention a few. Aspirin stimulates the cell cycle checkpoint proteins, allowing the cells to upregulate tumor suppression proteins54.
Similar to COX enzymes, the deactivation of 5-LOX activity also blocks cell proliferation by inducing apoptosis. The 5-LOX activity is associated with polyp formation and may direct myeloid-derived suppressor cells to the polyp site55. The inhibitors of LOX pathways are also useful in treating asthma and arthritis. Curcumin, meclofenamate, auranofin, baicalein, caffeic acid, esculetin, L-655, 238, lycopodiene, NDGA, MK-886, and ziluton target LOX enzymes and have been shown to be effective in treating various cancers48. Anti-LOX drugs such as pranlukast, zafirlukast and montelukast are also available and are useful for treating asthma patients. These drugs are CysT1R antagonists and are effective in reducing colonic tumor growth in experimental nude mice model45. Cyst LT1R antagonists such as ZM 198 and ZM 615 decrease the growth of colon cancer cells in culture. Interestingly, it is noted that the combined effects of LTR1 antagonists and COX-2 inhibitors are more effective in blocking cancer cell proliferation and inducing apoptosis56.
Future studies
Future studies should be focused on identifying common genes and proteins that connect the remodeling network with other pathways. In this context, the recent development of genome-wide screening (GWS) and metabolomic study should yield conclusive results. In addition, the screening of small molecules and the development of non-toxic therapies need to be considered. Currently, there are many new drugs to treat CRCs, but they are not designed to target the COX/LOX pathways. It will be interesting to see, if NSAID molecules can be coupled to these new drugs to make cancer therapy more effective. For example, in the past, attempts were made to design nitric oxide (NO)-donating NSAIDs (NO-NSAID), which showed promising results in reducing the growth of cancer cells57.
Acknowledgments
Dr. Siddhartha Das was supported by a grant (R01AI096667) from the National Institutes of Allergy and Infectious Diseases (NIAID, NIH). Dr. Leobarda Robles Martinez was supported by a post-doctoral fellowship from CONACYT (Mexico). The authors thank Mr. Christiancel Salazar for the art-work.
Abbreviations
- APC
adenomatous polyposis coli
- AA
archidonic acid
- CRC
colorectal cancer
- CIN
chromosomal instability
- COX
cyclooxygenase
- cPLA2
cytoplasmic PLA2
- GWS
genome-wide screening
- iPLA2
intracellular phospholipase A2
- LOX
lipooxygenase
- LT
leukotriene
- MSI
microsatellite instability
- NSAID
non-steroidial anti-inflammatory drugs
- PG
prostaglandin
- PUFA
poly unsaturated fatty acid
- PLA2
phospholipase A2
References
- 1.Barrow E, Hill J, Evans DG. Cancer risk in Lynch Syndrome. Fam Cancer. 2013;12:229–240. doi: 10.1007/s10689-013-9615-1. [DOI] [PubMed] [Google Scholar]
- 2.Jones R, Adel-Alvarez LA, Alvarez OR, Broaddus R, Das S. Arachidonic acid and colorectal carcinogenesis. Mol Cell Biochem. 2003;253:141–149. doi: 10.1023/a:1026060426569. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics. CA Cancer J Clin. 2014;64:104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 4.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 5.Pathy S, Lambert R, Sauvaget C, Sankaranarayanan R. The incidence and survival rates of colorectal cancer in India remain low compared with rising rates in East Asia. Dis Colon Rectum. 2012;55:900–906. doi: 10.1097/DCR.0b013e31825afc4e. [DOI] [PubMed] [Google Scholar]
- 6.Ponz de Leon M, Di Gregorio C. Pathology of colorectal cancer. Dig Liver Dis. 2001;33:372–388. doi: 10.1016/s1590-8658(01)80095-5. [DOI] [PubMed] [Google Scholar]
- 7.Du W, Mah JT, Lee J, Sankila R, Sankaranarayanan R, Chia KS. Incidence and survival of mucinous adenocarcinoma of the colorectum: a population-based study from an Asian country. Dis Colon Rectum. 2004;47:78–85. doi: 10.1007/s10350-003-0014-9. [DOI] [PubMed] [Google Scholar]
- 8.Wu X, Chen VW, Martin J, Roffers S, Groves FD, Correa CN, Hamilton-Byrd E, Jemal A. Subsite-specific colorectal cancer incidence rates and stage distributions among Asians and Pacific Islanders in the United States, 1995 to 1999. Cancer Epidemiol Biomarkers Prev. 2004;13:1215–1222. [PubMed] [Google Scholar]
- 9.Albuquerque C, Bakker ER, van Veelen W, Smits R. Colorectal cancers choosing sides. Colorectal cancers choosing sides. Biochim Biophys Acta. 2011;1816:219–231. doi: 10.1016/j.bbcan.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 10.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 12.Schneikert J, Vijaya Chandra SH, Ruppert JG, Ray S, Wenzel EM, Behrens J. Functional comparison of human adenomatous polyposis coli (APC) and APC-like in targeting beta-catenin for degradation. PLoS One. 2013;8:e68072. doi: 10.1371/journal.pone.0068072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Migliore L, Migheli F, Spisni R, Coppede F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J Biomed Biotechnol. 2011;2011:792362. doi: 10.1155/2011/792362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol. 2014;27:9–14. [PMC free article] [PubMed] [Google Scholar]
- 15.Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, Semple C, Clark AJ, Reid FJ, Smith LA, Kavoussanakis K, Koessler T, Pharoah PD, Buch S, Schafmayer C, Tepel J, Schreiber S, Volzke H, Schmidt CO, Hampe J, Chang-Claude J, Hoffmeister M, Brenner H, Wilkening S, Canzian F, Capella G, Moreno V, Deary IJ, Starr JM, Tomlinson IP, Kemp Z, Howarth K, Carvajal-Carmona L, Webb E, Broderick P, Vijayakrishnan J, Houlston RS, Rennert G, Ballinger D, Rozek L, Gruber SB, Matsuda K, Kidokoro T, Nakamura Y, Zanke BW, Greenwood CM, Rangrej J, Kustra R, Montpetit A, Hudson TJ, Gallinger S, Campbell H, Dunlop MG. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40:631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, Jaeger E, Fielding S, Rowan A, Vijayakrishnan J, Domingo E, Chandler I, Kemp Z, Qureshi M, Farrington SM, Tenesa A, Prendergast JG, Barnetson RA, Penegar S, Barclay E, Wood W, Martin L, Gorman M, Thomas H, Peto J, Bishop DT, Gray R, Maher ER, Lucassen A, Kerr D, Evans DG, Consortium C, Schafmayer C, Buch S, Volzke H, Hampe J, Schreiber S, John U, Koessler T, Pharoah P, van Wezel T, Morreau H, Wijnen JT, Hopper JL, Southey MC, Giles GG, Severi G, Castellvi-Bel S, Ruiz-Ponte C, Carracedo A, Castells A, Consortium E, Forsti A, Hemminki K, Vodicka P, Naccarati A, Lipton L, Ho JW, Cheng KK, Sham PC, Luk J, Agundez JA, Ladero JM, de la Hoya M, Caldes T, Niittymaki I, Tuupanen S, Karhu A, Aaltonen L, Cazier JB, Campbell H, Dunlop MG, Houlston RS. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008;40:623–630. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 17.Buecher B, Cacheux W, Rouleau E, Dieumegard B, Mitry E, Lievre A. Role of microsatellite instability in the management of colorectal cancers. Dig Liver Dis. 2013;45:441–449. doi: 10.1016/j.dld.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Curtin K, Slattery ML, Samowitz WS. CpG island methylation in colorectal cancer: past, present and future. Patholog Res Int. 2011;2011:902674. doi: 10.4061/2011/902674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grady WM. Epigenetic events in the colorectum and in colon cancer. Biochem Soc Trans. 2005;33:684–688. doi: 10.1042/BST0330684. [DOI] [PubMed] [Google Scholar]
- 20*.Neoptolemos JP, Husband D, Imray C, Rowley S, Lawson N. Arachidonic acid and docosahexaenoic acid are increased in human colorectal cancer. Gut. 1991;32:278–281. doi: 10.1136/gut.32.3.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoeft B, Linseisen J, Beckmann L, Muller-Decker K, Canzian F, Husing A, Kaaks R, Vogel U, Jakobsen MU, Overvad K, Hansen RD, Knuppel S, Boeing H, Trichopoulou A, Koumantaki Y, Trichopoulos D, Berrino F, Palli D, Panico S, Tumino R, Bueno-de-Mesquita HB, van Duijnhoven FJ, van Gils CH, Peeters PH, Dumeaux V, Lund E, Huerta Castano JM, Munoz X, Rodriguez L, Barricarte A, Manjer J, Jirstrom K, Van Guelpen B, Hallmans G, Spencer EA, Crowe FL, Khaw KT, Wareham N, Morois S, Boutron-Ruault MC, Clavel-Chapelon F, Chajes V, Jenab M, Boffetta P, Vineis P, Mouw T, Norat T, Riboli E, Nieters A. Polymorphisms in fatty-acid-metabolism-related genes are associated with colorectal cancer risk. Carcinogenesis. 2010;31:466–472. doi: 10.1093/carcin/bgp325. [DOI] [PubMed] [Google Scholar]
- 22.Murray NR, Weems C, Chen L, Leon J, Yu W, Davidson LA, Jamieson L, Chapkin RS, Thompson EA, Fields AP. Protein kinase C betaII and TGFbetaRII in omega-3 fatty acid-mediated inhibition of colon carcinogenesis. J Cell Biol. 2002;157:915–920. doi: 10.1083/jcb.200201127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song M, Chan AT, Fuchs CS, Ogino S, Hu FB, Mozaffarian D, Ma J, Willett WC, Giovannucci EL, Wu K. Dietary intake of fish, ω-3 and ω-6 fatty acids and risk of colorectal cancer: A prospective study in U.S. men and women. Int J Cancer. 2014 doi: 10.1002/ijc.28878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Jia WH, Matsuo K, Shin A, Xiang YB, Matsuda K, Jee SH, Kim DH, Cheah PY, Ren Z, Cai Q, Long J, Shi J, Wen W, Yang G, Ji BT, Pan ZZ, Matsuda F, Gao YT, Oh JH, Ahn YO, Kubo M, Thean LF, Park EJ, Li HL, Park JW, Jo J, Jeong JY, Hosono S, Nakamura Y, Shu XO, Zeng YX, Zheng W. Genome-wide association study identifies a new SMAD7 risk variant associated with colorectal cancer risk in East Asians. Int J Cancer. 2014;135:948–955. doi: 10.1002/ijc.28733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita A, Sugiura T, Waku K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J Biochem. 1997;122:1–16. doi: 10.1093/oxfordjournals.jbchem.a021715. [DOI] [PubMed] [Google Scholar]
- 26.Mansilla F, da Costa KA, Wang S, Kruhoffer M, Lewin TM, Orntoft TF, Coleman RA, Birkenkamp-Demtroder K. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) overexpression in human colorectal cancer. J Mol Med (Berl) 2009;87:85–97. doi: 10.1007/s00109-008-0409-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robichaud PP, Boulay K, Munganyiki JE, Surette ME. Fatty acid remodeling in cellular glycerophospholipids following the activation of human T cells. J Lipid Res. 2013;54:2665–2677. doi: 10.1194/jlr.M037044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomita M, Baker RC, Ando S, Santoro TJ. Arachidonoyl-phospholipid remodeling in proliferating murine T cells. Lipids Health Dis. 2004;3:1. doi: 10.1186/1476-511X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen-Petrik MB, McEntee MF, Johnson BT, Obukowicz MG, Masferrer J, Zweifel B, Chiu CH, Whelan J. Selective inhibition of Delta-6 desaturase impedes intestinal tumorigenesis. Cancer Lett. 2002;175:157–163. doi: 10.1016/s0304-3835(01)00715-7. [DOI] [PubMed] [Google Scholar]
- 30.Dennis EA. Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994;269:13057–13060. [PubMed] [Google Scholar]
- 31.Surrel F, Jemel I, Boilard E, Bollinger JG, Payre C, Mounier CM, Talvinen KA, Laine VJ, Nevalainen TJ, Gelb MH, Lambeau G. Group X phospholipase A2 stimulates the proliferation of colon cancer cells by producing various lipid mediators. Mol Pharmacol. 2009;76:778–790. doi: 10.1124/mol.108.053371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanasaki K, Ono T, Saiga A, Morioka Y, Ikeda M, Kawamoto K, Higashino K, Nakano K, Yamada K, Ishizaki J, Arita H. Purified group X secretory phospholipase A(2) induced prominent release of arachidonic acid from human myeloid leukemia cells. J Biol Chem. 1999;274:34203–34211. doi: 10.1074/jbc.274.48.34203. [DOI] [PubMed] [Google Scholar]
- 33.Umeno J, Matsumoto T, Esaki M, Kukita Y, Tahira T, Yanaru-Fujisawa R, Nakamura S, Arima H, Hirahashi M, Hayashi K, Iida M. Impact of group IVA cytosolic phospholipase A2 gene polymorphisms on phenotypic features of patients with familial adenomatous polyposis. Int J Colorectal Dis. 2010;25:293–301. doi: 10.1007/s00384-009-0808-x. [DOI] [PubMed] [Google Scholar]
- 34.Hong KH, Bonventre JC, O’Leary E, Bonventre JV, Lander ES. Deletion of cytosolic phospholipase A(2) suppresses Apc(Min)-induced tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:3935–3939. doi: 10.1073/pnas.051635898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kisslov L, Hadad N, Rosengraten M, Levy R. HT-29 human colon cancer cell proliferation is regulated by cytosolic phospholipase A(2)α dependent PGE(2)via both PKA and PKB pathways. Biochim Biophys Acta. 2012;1821:1224–1234. doi: 10.1016/j.bbalip.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Xu C, Reichert EC, Nakano T, Lohse M, Gardner AA, Revelo MP, Topham MK, Stafforini DM. Deficiency of phospholipase A2 group 7 decreases intestinal polyposis and colon tumorigenesis in Apc(Min/+) mice. Cancer Res. 2013;73:2806–2816. doi: 10.1158/0008-5472.CAN-12-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liou JY, Aleksic N, Chen SF, Han TJ, Shyue SK, Wu KK. Mitochondrial localization of cyclooxygenase-2 and calcium-independent phospholipase A2 in human cancer cells: implication in apoptosis resistance. Exp Cell Res. 2005;306:75–84. doi: 10.1016/j.yexcr.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 38.Konturek PC, Kania J, Burnat G, Hahn EG, Konturek SJ. Prostaglandins as mediators of COX-2 derived carcinogenesis in gastrointestinal tract. J Physiol Pharmacol. 2005;56(Suppl 5):57–73. [PubMed] [Google Scholar]
- 39.Mutoh M, Takahashi M, Wakabayashi K. Roles of prostanoids in colon carcinogenesis and their potential targeting for cancer chemoprevention. Curr Pharm Des. 2006;12:2375–2382. doi: 10.2174/138161206777698972. [DOI] [PubMed] [Google Scholar]
- 40.Glinghammar B, Rafter J. Colonic luminal contents induce cyclooxygenase 2 transcription in human colon carcinoma cells. Gastroenterology. 2001;120:401–410. doi: 10.1053/gast.2001.21188. [DOI] [PubMed] [Google Scholar]
- 41.O’Brian CA, Ward NE, Gravitt KR, Gupta KP. The tumor promoter receptor protein kinase C: a novel target for chemoprevention and therapy of human colon cancer. Prog Clin Biol Res. 1995;391:117–120. [PubMed] [Google Scholar]
- 42.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 43.Sheng H, Shao J, Washington MK, DuBois RN. Akt/PKB activity is required for Ha-Ras-mediated transformation of intestinal epithelial cells. J Biol Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M010093200. [DOI] [PubMed] [Google Scholar]
- 44.Daniel TO, Liu H, Morrow JD, Crews BC, Marnett LJ. Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial migration and angiogenesis. Cancer Res. 1999;59:4574–4577. [PubMed] [Google Scholar]
- 45.Savari S, Vinnakota K, Zhang Y, Sjolander A. Cysteinyl leukotrienes and their receptors: bridging inflammation and colorectal cancer. World J Gastroenterol. 2014;20:968–977. doi: 10.3748/wjg.v20.i4.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 47.Oliver L, Cordel S, Barbieux I, LeCabellec MT, Meflah K, Gregoire M, Vallette FM. Resistance to apoptosis is increased during metastatic dissemination of colon cancer. Clin Exp Metastasis. 2002;19:175–180. doi: 10.1023/a:1014510508664. [DOI] [PubMed] [Google Scholar]
- 48.Bishayee K, Khuda-Bukhsh AR. 5-lipoxygenase antagonist therapy: a new approach towards targeted cancer chemotherapy. Acta Biochim Biophys Sin (Shanghai) 2013;45:709–719. doi: 10.1093/abbs/gmt064. [DOI] [PubMed] [Google Scholar]
- 49.Paruchuri S, Sjolander A. Leukotriene D4 mediates survival and proliferation via separate but parallel pathways in the human intestinal epithelial cell line Int 407. J Biol Chem. 2003;278:45577–45585. doi: 10.1074/jbc.M302881200. [DOI] [PubMed] [Google Scholar]
- 50.Williamson RT. On the treatment of glycosuria and Diabetes mellitus with aspirin. Br Med J. 1902;2:1946–1948. doi: 10.1136/bmj.2.2191.1946-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moertel CG, Ahmann DL, Taylor WF, Schwartau N. Aspirin and pancreatic cancer pain. Gastroenterology. 1971;60:552–553. [PubMed] [Google Scholar]
- 52.Fournier DB, Gordon GB. COX-2 and colon cancer: potential targets for chemoprevention. J Cell Biochem Suppl. 2000;34:97–102. doi: 10.1002/(sici)1097-4644(2000)77:34+<97::aid-jcb16>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 53.Din FV, Theodoratou E, Farrington SM, Tenesa A, Barnetson RA, Cetnarskyj R, Stark L, Porteous ME, Campbell H, Dunlop MG. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut. 2010;59:1670–1679. doi: 10.1136/gut.2009.203000. [DOI] [PubMed] [Google Scholar]
- 54*.Stolfi C, De Simone V, Pallone F, Monteleone G. Mechanisms of action of non-steroidal anti-inflammatory drugs (NSAIDs) and mesalazine in the chemoprevention of colorectal cancer. Int J Mol Sci. 2013;14:17972–17985. doi: 10.3390/ijms140917972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheon EC, Strouch MJ, Krantz SB, Heiferman MJ, Bentrem DJ. Genetic deletion of 5-lipoxygenase increases tumor-infiltrating macrophages in Apc(Δ468) mice. J Gastrointest Surg. 2012;16:389–393. doi: 10.1007/s11605-011-1761-x. [DOI] [PubMed] [Google Scholar]
- 56.Cianchi F, Cortesini C, Magnelli L, Fanti E, Papucci L, Schiavone N, Messerini L, Vannacci A, Capaccioli S, Perna F, Lulli M, Fabbroni V, Perigli G, Bechi P, Masini E. Inhibition of 5-lipoxygenase by MK886 augments the antitumor activity of celecoxib in human colon cancer cells. Mol Cancer Ther. 2006;5:2716–2726. doi: 10.1158/1535-7163.MCT-06-0318. [DOI] [PubMed] [Google Scholar]
- 57.Rigas B. The use of nitric oxide-donating nonsteroidal anti-inflammatory drugs in the chemoprevention of colorectal neoplasia. Curr Opin Gastroenterol. 2007;23:55–59. doi: 10.1097/MOG.0b013e32801145b0. [DOI] [PubMed] [Google Scholar]