

Abstract
In many cases congenital heart disease (CHD) is represented by a complex phenotype and an array of several functional and morphological cardiac disorders. These malformations will be briefly summarized in the first part focusing on two severe CHD phenotypes, hypoplastic left heart syndrome (HLHS) and tetralogy of Fallot (TOF). In most cases of CHD the genetic origin remains largely unknown, though the complexity of the clinical picture strongly argues against a dysregulation which can be attributed to a single candidate gene but rather suggests a multifaceted polygenetic origin with elaborate interactions. Consistent with this idea, genome-wide approaches using whole exome sequencing, comparative sequence analysis of multiplex families to identify de novo mutations and global technologies to identify single nucleotide polymorphisms, copy number variants, dysregulation of the transcriptome and epigenetic variations have been conducted to obtain information about genetic alterations and potential predispositions possibly linked to the occurrence of a CHD phenotype. In the second part of this review we will summarize and discuss the available literature on identified genetic alterations linked to TOF and HLHS.
Keywords: Congenital heart disease, Copy number variants, de novo mutations, Epigenetics, Genome-wide association study, Hypoplastic left heart syndrome, Tetralogy of Fallot.
CONGENITAL HEART DISEASE – THE CLINICAL PICTURE
The heart is the first functional organ during embryogenesis. After birth, desaturated blood is received through the right atrium, forwarded to the right ventricle and pumped into the lungs. Oxygenated blood returns through the left atrium and leaves the heart via the left ventricle through the aorta and systemic arteries to supply all organs with oxygen (Fig. 1A). Unfortunately, approximately 4 to 14 per 1.000 live births are diagnosed with congenital heart disease (CHD) [1, 2] where this well controlled circuit is disturbed. The underlying malformations are manifold ranging from small atrial or ventricular septal defects to highly complex malformations for example an abnormal spatial arrangement of the great arteries resulting in serious hemodynamic changes. As most genome-wide analyses have been conducted almost exclusively in patients with hypoplastic left heart syndrome (HLHS) and tetralogy of Fallot (TOF) we will briefly summarize the major clinical malformations and surgical interventions of those two phenotypes only.
Fig. (1).
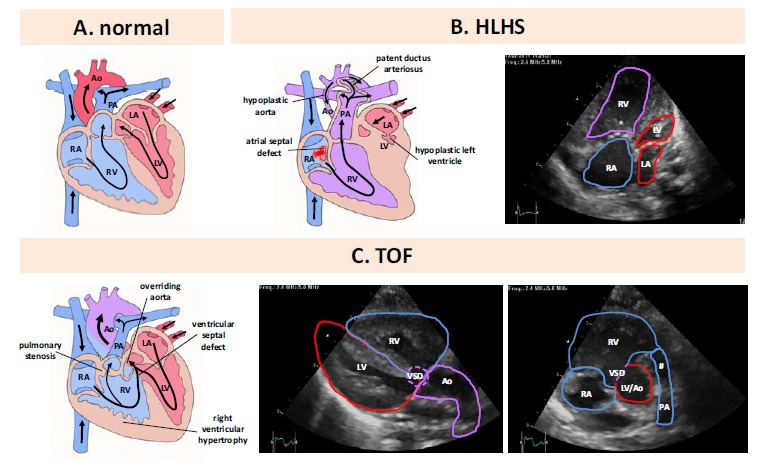
Comparative illustration of morphology and blood flow in normal heart and CHD. A. Morphology and blood flow in a normal heart. B. Left panel. Morphology and blood flow in hypoplastic left heart syndrome. B. right panel. Transthoracic echocardiography, apical four chamber view. C. left panel. Morphology and blood flow in Tetralogy of Fallot. C. middle panel. Transthoracic echocardiography, parasternal long axis view. C. right panel. Transthoracic echocardiography, parasternal short axis view. Ao: aorta, LA: left atrium, LV: left ventricle, RA: right atrium, RV: right ventricle, PA: pulmonary artery, VSD: ventricular septal defect, #: narrow right ventricular outflow tract.
Hypoplastic Left Heart Syndrome (HLHS)
HLHS is characterized by a stenotic or atretic aortic and/or mitral valve, a high-grade hypoplasia of the aorta ascendens and a highly hypoplastic or even completely missing left ventricle (Fig. 1B). Furthermore, a patent ductus arteriosus must be maintained after birth to achieve sufficient coronary (retrograde via the hypoplastic aorta ascendens) and systemic blood flow. Beside the patent ductus arteriosus an interatrial shunt between the left and right atrium of different sizes ranging from a small foramen ovale to a wide atrial septal defect guarantees shunting of returning pulmonary venous blood from the left to the right atrium. Thereafter the blood is able to pass the tricuspid and pulmonary valve for pulmonary as well as coronary and systemic perfusion via the patent ductus arteriosus.
The HLHS is a rarely occurring CHD, with a prevalence of only 0.15 per 1,000 live births in Germany as reported in the PAN study [2] and the majority of the cases are sporadic. Furthermore, 70% of the affected patients are male, reflecting a strong gender aspect in the disease development [3]. Until recently, HLHS was a uniformly fatal pathologic condition. No definitive treatment existed until in the 1980s the use of the right ventricle to support systemic circulation was first proposed. A tremendous progress in the management of HLHS including prenatal diagnostics, operative procedures and an optimized perioperative intensive care unit treatment has increased patient survival to around 65% at 5- and 55% at 10-years of age [4-6]. At present, the classical surgical strategy encompasses a 3-staged procedure resulting in an univentricular “Fontan” circulation. This palliative treatment ends up with a right ventricle that supports the systemic circulation and a completely passive pulmonary blood flow.
Tetralogy of Fallot (TOF)
One of the most relevant right heart CHDs is TOF first described in 1888 by the French pathologist Étienne-Louis Artur Fallot. TOF has a prevalence of 0.27 per 1.000 live births in Germany [2]. The morphological phenotype is characterized by four malformations: a narrowing of the right outflow tract (pulmonary stenosis), a hypertrophy of the right ventricle, a ventricular septal defect (VSD) and a displacement of the aorta to the right side over the VSD (an overriding aorta) (Fig. 1C). The TOF is a classical cyanotic CHD due to a subpulmonary obstruction with shunting mainly from the right to the left ventricle via the VSD leading to an ejection of deoxygenated blood into the aorta. At present, surgical repair is performed in the first month of life where the stenosis of the right outflow tract is relieved and the VSD is closed enabling an exclusive ejection of oxygenated blood via the left ventricle. Cumulative survival and event-free survival were 72% and 25%, respectively, after 40 years [7].
CONGENITAL HEART DEFECT – A DISEASE WITH MULTIPLE GENETIC ORIGINS
Severe CHDs are characterized by complex clinical phenotypes with numerous malformations. The multi-factor background of CHD with genetic components was first proposed almost half a century ago [8]. Genetic causes have first been mentioned in the early 80-ies [9] and were confirmed by numerous subsequent epidemiological data [10, 11] and family studies [12-14].
The majority of children born with CHD do not have other physical defects. Nevertheless, it has been noted that major chromosomal abnormalities occur in such children [15, 16]. These obvious chromosomal defects can be detected by conventional karyotyping and G-band analysis and about 10% of CHD in children with multiple congenital defects and/or intellectual disability is caused by trisomies 21, 18 and 13 [17]. However, none of these aberrations can be attributed to a single CHD phenotype and they are associated with different CHDs at variable frequencies (Table 1). Being the long-time gold standard the resolution of this traditional method is limited to identify chromosomal aberrations of 5 Mb or larger. The frequent 22q11 deletion causing DiGeorge syndrome, a large 3 Mb deletion encompassing some 60 genes, was first discovered by applying quantitative Southern blot hybridization [18].
Table 1.
Aneuploidity and congenital heart defects.
| Chromosomal Aberration | Frequency a | Association with Congenital Heart Defects | CHD Frequency in |
|---|---|---|---|
| Trisomy 21 (Down Syndrome) |
1 : 700 | ASD, AVSD, TOF, VSD | 50 % |
| Trisomy 13 (Patau Syndrome) |
1 – 2 : 10.000 | ASD, HLHS, PDA, VSD | 80 % |
| Trisomy 18 (Edwards Syndrome) |
1 : 2.500 | ASD, BAV, CoA, DORV, PDA, TOF, VSD |
> 90 % |
| Monosomy X (Turner Syndrome) |
1 : 2.500 | AoS, BAV, CoA, HLHS | 33 % |
| 47, XXY (Klinefelter Syndrome) |
1 – 2 : 1.000 | ASD, MVP, PDA | 50 % |
per live births. Adapted from [31].
aortic stenosis, ASD: atrial septal defect, AVSD: atrioventricular septal defect, BAV: bicuspid aortic valve, CoA: coarctation of the aorta, DORV: double-outlet right ventricle, HLHS: hypoplastic left heart syndrome, MVP: mitral valve prolapse, PDA: patent ductus arteriosus, TOF: tetralogy of Fallot, VSD: ventricular septal defect.
Such syndromes, however, appear only in a minor fraction of CHD. Almost 80% of CHDs occur sporadically and their inheritance does not follow a Mendelian order. Meanwhile a large list of potential candidate genes has been identified [19], though these genes have only exceptionally been shown to be causative for the disease such as TBX5 for the autosomal-dominant inherited Holt-Oram syndrome [20]. Mutations in further essential cardiac transcription factors such as MESP1 [21], NKX2.5 [22-24], GATA4 [25], GATA6 [26] or TBX20 [27] and combinations thereof [28] have been described in patients with CHD but without any convincing proof of their causative function.
Therefore, in most cases of CHD the genetic origin still remains largely unknown though the accumulation of alterations in the genome of patients affecting a large number of causative genes appears a plausible explanation. To identify polygenic networks on a genome-wide basis with increased sensitivity, novel technologies have been developed. Array-based comparative genomic hybridization (CGH) techniques allow the detection of copy number changes in the range of 5 to 10 kb [29] or even down to 200 bp [30] and state-of-the-art chips either facilitate the analysis of several millions of SNPs, a genome-wide detection of methylation sites or the global transcriptome evaluation. Next-generation sequencing technologies replace Sanger sequencing and gradually also genomic micorarrays. These high-throughput approaches require a substantial computational and bioinformatic power but yield far more sequence information in less time and at lower costs which makes them the method of choice to analyze multifactorial and multigenetic diseases such as CHD. In the following we will summarize and discuss such genome-wide approaches which have been conducted to identify genetic alterations and potential candidate genes and network-interactions in CHD populations with a defined clinical phenotype.
SINGLE NUCLEOTIDE VARIANTS (TABLE 2)
Table 2.
Genome-wide association studies in CHD patients.
| Platform(s) | Patients | Genetic Alteration | Impact | Reference |
|---|---|---|---|---|
| Illumina 660W-Quad | 798 TOF | 12q24 (PTPN11) 13q32 (GPC5, NRP1) |
risk allele for TOF risk allele for TOF |
[33] |
| SEQUENOM MALDI-TOF | 362 TOF | rs11066320 (PTPN11) | risk allele for TOF | [34] |
| NimbleGen sequence capture 365K array Genome Sequencer FLX Illumina Genome Analyzer |
26 TOF | deleterious SNPs in sixteen genes | imbalance of functional networks in TOF | [40] |
| Applied Biosystems Linkage Mapping Set (medium-density) |
43 families with LVOT | 2q23, 10q21, 16p12 16p12 10q11 2p15 |
risk loci for LVOT risk locus for AVS, CoA, HLHS risk locus for AVS, CoA risk locus for HLHS |
[42] |
To identify possible associations of common variants with the etiology of CHD several genome-wide association studies have been initiated. Considering all CHD cases no genome-wide association of a certain risk locus was seen [32]. Therefore, subsequent analyses focused on defined CHD phenotypes.
Tetralogy of Fallot (TOF)
Two recent reports identified genetic variants in the chromosomal region 12q24 as risk factors for TOF [33, 34]. This 1.6 Mb region encompassing 15 genes has previously been reported as a risk factor for several diseases including coronary artery disease [35]. Six different SNPs with strong significance (top SNP rs11065987) were located in this region [33]. The strongest candidate gene is PTPN11, a regulator of Ras/MAP-kinase signaling pathway and associated with Noonan syndrome [36]. Most interestingly, the SNP rs11066320 located in intron 6 of the PTPN11 gene was also reported in the second study which analyzed 22 candidate genes [34]. However, the hypothesis that these SNPs may up-regulate PTPN11 activity thereby increasing the susceptibility for TOF still remains to be validated. More modest significance levels were seen at 13q32 (rs7982677) in intron 7 of GPC5 and the non-synonymous substitution Ile733Val in NRP1 [33]. The latter locus was also confirmed in a Chinese TOF population [37]. Although both genes are involved in cardiac development [38, 39] further studies are necessary to confirm a functional effect of any of these variants.
Grunert and colleagues performed an interesting approach with targeted re-sequencing and whole transcriptome profiling to identify potentially causative genes in TOF [40]. To discriminate causative genes they introduced a gene mutation frequency, calculated by the relation of the number of individuals with deleterious mutations to the total number of individuals with sufficient genotype information. After normalization by the gene-length the method was validated in a retrospective study on a cohort of patients with hypertrophic cardiomyopathy [41]. By this approach they identified deleterious SNPs in 16 TOF genes, six of which have known association with cardiac disease and seven provoke a cardiac phenotype after mutation or knock-out in mice. The vast majority of mutated alleles could be confirmed by mRNASeq in right ventricle tissue of the respective patient. The search for potential interaction of these TOF genes suggested that an array of genes was assembled in a functional network where the alterations may create an imbalance ultimately leading to the phenotype of TOF. They could indeed confirm this imbalance by analyzing the expression profile in the right ventricle of three TOF patients with mutations in the MYOM2 gene. A shared differential expression of four TOF genes, including MYOM2, was observed. Finally they analyzed right ventricle biopsies which confirmed the expected aberrant histological phenotype.
Hypoplastic Left Heart Syndrome (HLHS)
An analysis of 411 microsatellites focused on the identification of genetic susceptibility loci in multiplex CHD families with left ventricular outflow tract (LVOT) malformations [42]. Three novel regions with suggestive linkage were identified on 2p23, 10q21 and 16p12 by a combined analysis of all included malformations. The analysis of subgroups revealed overlapping linkage signals on 16p12 (aortic valve stenosis (AVS), coarctation of the aorta (CoA), HLHS) and on 10q11 (AVS, CoA). Finally a significant peak on 2p15 restricted to the HLHS cohort was identified. While overlapping peaks strengthen the hypothesis of a common genetic etiology of these malformations, further supported by their combined occurrence in families [43] the data also suggest the presence of distinct alterations potentially involved in the development of specific phenotypes.
COPY NUMBER VARIATIONS (TABLE 3)
Table 3.
Copy number variations in CHD patients.
| Platform | Patients | Genetic Alteration | Impact | Reference |
|---|---|---|---|---|
| Affymetrix Genome-Wide Human SNP Array 6.0 |
433 TOF | dup 1q21.1 (GJA5) del 1q32.2 (PLXNA2) |
potential TOF candidate gene potential TOF candidate gene |
[48] |
| Illumina 660W-Quad | 948 TOF | dup 1q21.1 including rare 100-200 bp duplications (GJA5) |
potential TOF candidate gene | [57] |
| Affymetrix Genome-Wide Human SNP Array 6.0 |
59 multiplex families with LS-CHD | 25 candidate regions (e.g. CTHRC1, MFAP4, PLA2G12A) | potential LS-CHD candidate genes | [59] |
| NimbleGen high resolution whole-genome array CGH |
43 HLHS | multiple del/dup CNVs micro dels in 14q23 |
association with HLHS, no cardiac candidate genes | [65] |
del: deletion, dup: duplication, LS CHD: left-sided CHD.
The term copy number variation (CNV) is a form of structural variation which becomes evident when an individual genome is compared to the standard reference genome in the database. Certain sections of the DNA exist in different copies due to deletions, insertions, duplications and complex multi-site variants of the genomic DNA. CNVs ranging from 1 kb to several megabases are found in all humans covering approximately 12% of the human genome [44]. Large CNVs have been detected in all major CHD phenotypes [45, 46] and they typically affect half of the cases [47].
Tetralogy of Fallot (TOF)
The first genome-wide study which analyzed CNVs examined 433 unrelated TOF patients. CNVs were detected at a frequency of 63 CNVs per genome with a median size of approximately 18 kb with no major differences between controls and cases [48]. Focusing on rare large CNVs (<0.1% in control population, > 500 kb) revealed an increased prevalence in TOF patients (OR 1.89), most notable in CNVs which overlapped exons (OR 2.54). Among 47 large CNVs the most compelling in TOF patients were duplications in 1q21.1
Pathway analyses identified several candidate genes including GJA5 thereby confirming previous studies [49, 50]. Most interestingly, the study revealed a novel locus at 1q32.2 in two unrelated TOFs who displayed loss CNVs overlapping the PLXNA2 gene, an important regulator of endothelial cell function during outflow tract septation [51, 52]. Knockout mice deficient for Plxna2 display congenital heart defects including TOF [53]. PLXNA2 is a receptor for semaphorins and two individuals showed a CNV loss in semaphorin genes (SEMA3D, SEMA3E). These genes modify the NOTCH pathway [54] and mutations in SEMA3E can cause Charge syndrome [55]. Pathway analyses allocated important functions of these genes in vasculature development, cell motility, chemotaxis and neuron projection and development.
The role of 1q21.1, first described as causative for CHD a decade ago [56], was analyzed in more than 948 TOF patients and 1488 patients with other forms of CHD [57]. Duplication of the 1q21.1 locus was more common in TOF patients (OR 30.9) while deletion was more common in other forms of CHD (OR 5.5). Rare 100-200 kb duplications encompassing the GJA5 gene were strongly enriched in the TOF population (OR 10.7). An altered expression of this gene might possibly contribute to the TOF phenotype as Gja5-deficient mice show complex heart defects [58]. The impact of these duplications remains unclear since they were present as de novo events and inherited in the patient population.
Hypoplastic Left Heart Syndrome (HLHS)
A large multi-center study, focused on de novo CNVs in patients with left-sided CHD including bicuspid aortic valve, aortic valve stenosis, coarctation of the aorta and HLHS, analyzed 464 individual genomes including 59 multiplex families and eight trios [59]. Almost 7.000 autosomal CNVs were detected in the patient group (average approximately 15/genome) though these numbers did not differ significantly from the controls. However, 73 CNVs remained unique for the patient group with significant enrichment of genes involved in angiogenesis. Three different approaches were used to more specifically identify candidates harboring an important impact during cardiovascular development: i) in silico prediction of gene functionality, ii) screening of SAGE-libraries of embryonic mouse heart libraries for genes with at least three fold higher expression in the outflow tract, and iii) public data base mining. This procedure identified 25 potential candidate genes for left-sided heart disease. These genes include CTHCR1, an activator of the Wnt-pathway and MFAP4, encoding an extracellular matrix protein involved in cell adhesion and interaction, both highly expressed in developing valves and great vessels [60-62]. Finally, PLA2G12A, situated in a locus previously associated with HLHS [63], is an inhibitor of the BMP-pathway [64]. An overlap search with these 25 high priority genes with loci known to be linked to left-sided CHD identified four regions (Xp11.22, 4p16.1, 17p11.2, 1q21) which strengthen the candidacy of the identified genes. In addition, in five different families these unique CNVs segregated with the CHD phenotype.
A small study concentrated on the presence of submicroscopic chromosomal abnormalities in HLHS patients [65]. The frequency of CNVs, predominantly small gains and losses < 60 kb, was increased in the HLHS group (4.60 vs. 2.94 in controls), however, none of them encompassed potential candidate genes for HLHS. Likewise, a unique CNV loss mapping to 14q23.3 which has previously been identified in left heart malformations [63] also does not contain genes associated with cardiac development.
DE NOVO MUTATIONS (TABLE 4)
Table 4.
De novo mutations in CHD patients.
| Platform | Patients | Genetic Alteration | Impact | Reference |
|---|---|---|---|---|
| Agilent customized 400K CGH Array | 55 HLHS + 87 parents |
del 2q33 (BMPR2) dup 16q21 (ZNF423) NOTCH1 (SS, FS, MS) FOXC2 (MS), FOXL1 (MS) |
de novo mutations with cardiac relevance | [69] |
| NimbleGen HD2-2.1 CGH | 219 Trios (HLHS and conotruncal defects | dup 2q21.2 (CFC1) del 10q22.1 (NODAL) de novo dup 16q13.11 (NOMO3) |
potential CHD candidate genes | [70] |
| Illumina 660W SNP Array SNP Array 6.0 |
2256 CHD 283 TOF families |
del 15q11.2 de novo dup 1q21.1 (GJA5) de novo del 4q34 (HAND2) de novo dup 5q14.2 (EDIL3) de novo dup 5q35.3 (CNOT6) |
risc locus for sporadic CHD potential TOF candidate gene potential TOF candidate gene potential TOF candidate gene potential TOF candidate gene |
[50] |
| Affymetrix Human Genome- Wide SNP Array 6.0 | 114 TOF Trios 398 TOF |
de novo dup 1q21.1 (PRKAB2, CHDIL, BCL9, GJA5) |
potential TOF candidate genes | [49] |
| Illumina HiSeq2000 Sequencer | 362 CHD Trios |
de novo mutations (MLL2, WDR5, CHD7, KDM5A, KDM5B, UBE2B, RNF20, USP44, SMAD2) |
potential role of chromatin modeling in CHD cardiac candidate genes |
[71] |
SS: splice-site mutation, FS: frame-shift mutation, MS: missense mutation, del: deletion, dup: duplication.
Several studies have revealed a relatively high number of potentially damaging variations in healthy individuals [66-68] which makes it difficult to identify true disease-related de novo events in affected patients. However, the inclusion of first-degree unaffected relatives can minimize this “natural” genetic variation. De novo occurring genetic alterations in an affected patient might be the last event necessary to develop a CHD in combination with a somehow predisposed genetic background, and such events can be identified by comparative analyses of the genetic material of CHD patients and their unaffected parents (a so-called trio).
Hypoplastic Left Heart Syndrome (HLHS)
A first report addressing this question in analyzing HLHS patients used a combined approach of direct sequencing of candidate genes and a genome-wide screening by high resolution array comparative hybridization [69]. In 25 HLHS patients the array CGH analysis identified 33 CNVs. In all cases where the parental DNA could be analyzed, the CNVs were inherited. Two of the identified CNVs (ZNF423, BMPR2) are associated with cardiac development while the remaining CNVs were located in desert regions or genomic regions without known relevance for cardiac development. Direct sequencing analysis identified several splice site changes, frameshift and missense mutations in NOTCH1, FOXL1 and FOXC2 which were, however, predicted to be disease causing only for NOTCH1. In addition, five variations in regulatory regions of FOXC2 and FOXL1 were predicted to be pathogenic. The other two candidate genes (HAND1, NKX2.5) did not show any genetic lesions.
A large trio collection was investigated for the occurrence of de novo and rare CNVs [70]. This study included 148 cases of conotruncal anomalies and 71 cases of HLHS. Despite a rather different pathogenesis the frequency of de novo CNVs was comparable in both groups (approximately 10%) but significantly elevated when compared to unaffected controls (approximately 2%). Within the collection of identified de novo and ultra-rare CNVs the most significant functional category was ‘cardiovascular system development and function’ with 15 candidate genes. By applying different criteria (overlapping de novo events, identical appearance in affected sibs, affecting known CHD candidate genes, occurrence in more than one trio) the detected numbers of CNVs could be reduced to 19 which are considered to be the most relevant. Three of those candidate genes (NODAL, CFC1, NOMO3) are associated with the Nodal-pathway, underlining its potential role in the development of CHD.
A large study of 362 trios including besides others 84 TOF, 65 TGA, 35 DORV and 60 HLHS patients performed a whole-exome sequencing approach to identify de novo mutations in CHD patients [71]. Their results indicate that de novo mutations contribute to approximately 10% of severe CHDs. They hypothesized that genes which contribute to CHD should be expressed in the developing cardiac tissue. Analyzing the top 25% genes of the transcriptome in murine E14.5 hearts (defined as high heart expression, HHE) they identified a significantly increased rate of protein-altering de novo mutations in HHE human ortholog genes in CHD patients (OR 2.53). Focusing on damaging mutations such as premature termination, splice-sites or frame-shifts increased the odds ratios further to 7.50. In contrast, no significant difference in the mutation frequency between CHD and controls was seen in genes with low heart expression (LHE, 75% of the genes). A closer examination of the mutated gene set revealed an enrichment of eight genes involved in the H3K4 network, a mark for both positive and negative gene regulation including key players of embryonic developmental and cardiogenesis [72-74]. Of note, the phenotype of the patients harboring these mutations was quite diverse. Interestingly, a de novo mutation of SMAD2 which also affects the H3K27 methylation pathway was identified twice, a finding unlikely to occur by chance. These results attribute a potential common role to H3K4 methylation as an important regulator of CHD pathogenesis though it could not be assigned to a defined pathogenic phenotype.
Tetralogy of Fallot (TOF)
More than 2.200 CHD patients including 283 TOF families were analyzed for the appearance of CNVs [50]. They report on overall significantly increased (1.8-fold) risk for genic CNVs (defined as CNVs overlapping with RefSeq boundaries), even further pronounced when considering only deletions of > 1Mb (4-fold increase) with no apparent difference between TOF and other CHDs. The number of genes spanned by either deletions or duplications was consistently higher in patients compared to controls. An annotation enrichment analysis on rare deletions showed a statistically significant 2.9-fold enrichment of genes in the Wnt-signaling pathway comprising 13 different genes across all investigated CHD phenotypes. In addition, a 15q11.2 deletion was the strongest indicator for the risk of sporadic non-syndromic CHD. TOF cases showed an almost 2-fold increase of genic duplications which was, however, solely due to a single locus (1q21.1) encoding GJA5, a potential candidate gene for the CHD phenotype [57, 58]. However, this conclusion could not be supported by others [59, 70]. In contrast, a Pro265Ser mutation in GJA5 (Connexin 40) was detected in TOF patients. This mutation prevents gap-junction formation and disrupts the morphology of the heart tube upon injection into zebrafish [75]. In addition, further rare de novo CNVs affecting genes with cardiac importance (HAND2, EDIL3, CNOT6) were identified in a small fraction (approximately 5%) of the TOF population.
A trio analysis of 114 children with TOF and their unaffected parents identified 11 rare de novo CNVs at 10 distinct chromosomal loci [49]. Of interest, the 1q.21.1 locus was affected in multiple cases and four individuals shared a duplicated region encompassing four genes (PRKAB2, CHD1L, BCL9, GJA5) with the highest expression in the right ventricular outflow tract which is malformed in TOF. However, the authors clearly state that none of the studies performed including their own could show a perfect correlation between 1q21 dosage and the CHD phenotype so far.
ANALYSIS OF GENE EXPRESSION (TABLE 5)
Table 5.
Gene expression analyses in CHD patients.
| Platform | Patients | Changes in Expression | Impact | Reference |
|---|---|---|---|---|
| Human Unigene Set-RZPD2 cDNA Array |
55 CHD | 88 dysregulated genes (e.g. SNIP, A2B1, KIAA1437) |
potential relevance for TOF | [76] |
| Applied Microarrays Inc. CodeLink Human Whole Genome Bioarray |
19 TOF | 1062 dysregulated genes (e.g. WNT and NOTCH pathway) |
potential relevance for TOF | [81] |
| Affymetrix Human Exon 1.0 ST v2 | 16 TOF |
NEFH, SST, TF, KRT6A, KRT7,NTRK2, MYL2, PPBP, NRGN, APOC3 |
dysregulated network in TOF | [84] |
| Affymetrix Human Exon 1.0 ST | 6 HLHS | 153 and 96 dysregulated genes vs. normal RV and LV, respectively |
potential relevance for HLHS | [87] |
| Affymetrix GeneChip microRNA 1.0 | 24 TOF | 61 dysregulated miRNAs | dysregulation of 44 cardiac genes in TOF |
[86] |
RV: right ventricle, LV: left ventricle.
Tetralogy of Fallot (TOF)
The analysis of genome-wide expression in tissue of CHD patients started about a decade ago [76]. In this study the expression profiles of the right ventricle obtained from different CHD patients were compared to corresponding normal tissue from donors without cardiac disease. The authors reported a characteristic expression signature of 88 genes in the right ventricle of TOF patients. Interestingly, this TOF-specific signature was different from tissue of right ventricle hypertrophy. Most characteristic was an up-regulation of several ribosomal proteins. In addition, the dysregulation affected several pathways which are involved in cardiac development. Genes such as SNIP and A2BP1 affect central pathways essential for normal cardiac development [77, 78] or play a role in heart embryogenesis [79]. Finally, they have identified a dysregulation of KIAA1437which directly binds K-RAS whose deletion leads to cardiac malformations in mice [80].
A second study included both idiopathic TOF patients and three which harbor the syndromic 22q11.2 deletion [81]. More than 1.000 dysregulated genes were identified in the TOF-derived right ventricles which were associated with protein synthesis, cardiovascular disease, genetic disorder, neurological disease and cell death as the top five networks. The expression of the majority of genes associated with the WNT and NOTCH-signal pathway were significantly reduced, suggesting a mutational convergence leading to general gene suppression of these pathways within the right ventricle. Their results also confirmed an up-regulated expression of VEGF and proteins of the extracellular matrix reported in earlier studies [82]. A significant up-regulation of angiogenic factors including VEGF was also seen by Peters and colleagues who analyzed primary corrected TOF in infants and adult patients who underwent a second corrective surgery for pulmonary regurgitation [83]. The expression of the genes located in the 22q11.2 region was half-reduced in syndromic patients as expected while none of these genes was differentially expressed in any of the idiopathic TOF subjects. Most interestingly, the NOTCH pathway was also suppressed in patients with 22q11.2 deletions [81]. The comparison of idiopathic and syndromic patients strongly suggests that different genetic starting conditions may nevertheless lead to a common pathological outcome.
A different approach to screen for altered gene expression in TOF patients was used by Rodemeyer and colleagues [84]. They started with a panel of 56 genes, a so-called Gene Expression Template (GET), which predicts 24 distinct human tissues under disease-free conditions with very high accuracy [85]. They extracted expression levels of 54 of these genes from previous work [86] and were able to combine the TOF samples in an independent cluster compared to several control tissues. Thirty-three of the analyzed gene showed a significantly altered expression compared to normally developing right ventricles. Nineteen GET genes were significantly altered in TOF compared to normal infant right ventricle. Of these, ten genes were associated with cardiac development, myogenesis and cardiac hypertrophy. Thus, applying the GET signature it was possible to discriminate pathologic TOF tissue from matched normally developing right ventricles.
Hypoplastic Left Heart Syndrome (HLHS)
Recently the first report of a genome-wide expression analysis in tissue derived from HLHS patients was published [87]. They collected right ventricle samples from HLHS patients and right and left ventricles from control neonates. Compared to left and right control ventricles 153 and 96 genes, respectively, were differentially expressed in pathologic tissue with a 66 gene overlap between both comparison groups. The expression profile of HLHS tissues remained constantly different from controls, irrespective of postnatal age. Looking at the top ten candidate genes which showed differential regulation revealed no obvious association with cardiac networks and none of these genes was located near chromosomal regions which have previously been associated with hereditary HLHS [63]. However, the most significantly up-regulated pathway was oxidative phosphorylation suggesting a shift from normal fatty acid towards glucose metabolism, characteristic for myocytes undergoing hypertrophy [88, 89]. Alternative splicing represents an important mechanism to regulate specific gene expression in tissues, including the myocardium [90]. Alternatively spliced mRNAs have been linked to the pathogenesis of certain types of CHD [91, 92] and they have been shown to affect key sarcomere genes in cardiomyopathies and AoS [93]. In HLHS tissue exon array analysis identified almost 1.500 genes which underwent alternative splicing events, significantly influencing the ‘spliceosome’ and ‘arrhythmogenic right ventricle cardiomyopathy’ KEGG pathways [87]. Thus, HLHS-derived tissue is characterized by distinct expression and splicing signatures consistent with the pathophysiological changes in the right ventricle. However, it should be noted at this point that this study describes alterations which appear as a consequence of the altered hemodynamics in HLHS while the genetic defect, preventing the normal development of left heart structures, occurs much earlier.
Non-coding RNAs
Non-coding RNAs such as microRNAs (miRNAs) have been associated with the development of normal heart and they can modulate cardiovascular disease [94-97]. In TOF samples the expression of 61 miRNAs was significantly changed while none of the miRNAs previously correlated with normal heart development showed an altered gene expression [86]. Noteworthy, the expression of miR-1 and miR-133 which have already been linked to cardiovascular disease by different groups [98-100] remained unchanged in the TOF samples. The top five candidates which showed the largest change in expression between TOF samples and normal tissue were validated by qRT-PCR in all samples used for the array analysis and this result was confirmed in additional 8 independent TOF specimens. Analyzing putative target genes of the 61 miRNAs revealed a clear enrichment in networks associated with cardiac development. Comparing the expression of the 61 miRNAs with expression data obtained from exon arrays performed on the same TOF samples revealed a significant change in expression in 173 genes of these cardiac networks and about 25% of them showed a clear-cut inverse correlation with one or more of the 61 miRNAs. This first analysis of non-coding RNAs in CHD impressively shows the tremendous regulatory changes in the myocardium by such molecules.
DNA METHYLATION (TABLE 6)
Table 6.
Alteration of DNA methylation in CHD patients.
| Platform | Patients | Changes in Methylation | Impact | Reference |
|---|---|---|---|---|
| Illumina Infinium Human Methylation27 Bead Chip |
180 mothers with non- syndromic CHD pregnancy | 425 sites with differential methylation (e.g. EGFR, GATA4, WNT5S) |
risk for CHD pregnancy | [104] |
| Sequenom MassArray EpiTyper | 30 TOF | hypermethylation of NKX2.5 and HAND1 | potential relevance for TOF | [110] |
| Sequenom MassArray EpiTyper | 32 TOF | hypermethylation of NKX2.5 and HAND1,hypomethylation of TBX2 and LINE-1 | potential relevance for TOF increased risk for TOF |
[109] |
| Sequenom MassArray EpiTyper | 42 TOF | 17 genes hypermethylated, 9 genes hypomethylated |
representative methylator phenotype phenotype during TOF development |
[111] |
| MethyLight | 180 mothers with non-syndromic CHD pregnancy | global hypomethylation (LINE-1) | risk for CHD pregnancy | [113] |
DNA methylation is a major mechanism of the cell to reversibly regulate gene expression without altering the nucleotide sequence. It involves covalent addition of a methylgroup to CpG dinucleotides, mostly located in CpG islands of the promoter regions but may also occur in regions of lower CpG density, so-called CpG island shores [101]. Genome-wide methylation profiling of murine embryonic hearts between E11.5 and E14.5 has shown a mostly stable global methylome with a minority of differentially methylated sites. These sites are strongly enriched in genes involved in heart development and affect the expression of cardiac-specific genes such as Has2 required for heart valve development [102].
Importantly, it appears clear that specific methylation patterns can also be inherited [102]. To address potential transgenerational effects of DNA methylation a genome-wide approach analyzed more than 27.000 CpG sites primarily located in the promoter region of more than 14.000 genes in genomic DNA from blood samples drawn after gestationfrom 180 mothers with non-syndromic CHD pregnancies [104]. The cardiac phenotypes included atrial or ventricular defects (47.2%), right- and left-sided obstructed defects (35.4%) and conotruncal defects (16.7%). Compared to the pattern of mothers with unaffected pregnancies 425 sites showed a differential methylation. The vast majority of the sites was located in CpG islands and almost 90% were hypermethylated. The affected genes were strongly enriched in processes affecting fetal development, some of which may influence the development of CHD (e.g. nucleic acid metabolism, signal transduction or anatomical structure development). In addition, a gene set enrichment analysis revealed a functional overlap in pathways involved in embryonic heart development. Several differentially methylated genes have previously been implicated in heart development such as EGFR [105], GATA4 [106, 107] or Wnt5a [108]. However, some limitations of this study should be mentioned as the blood samples were drawn after the pregnancy and there is a lack of data on the methylation status of the infants. Nevertheless, these data strongly suggest an impact of the maternal methylation status for the risk of CHD.
Tetralogy of Fallot (TOF)
Several reports analyzing right ventricular myocardium tissue in collectives of TOF patients have shown an increased methylation status in essential cardiac genes such as NKX2.5 and HAND1 which was also reflected in decreased gene expression [109, 110]. The same group performed an extended study which included the promoter regions of 71 CHD candidate genes which have previously been shown to be differentially methylated [104]. Their results indicated an up-regulated methylation in 17 genes including the above-mentioned NKX2.5, HAND1 and HAS2 (essential for heart valve development, [102]) while nine genes were hypomethylated [111]. Based on their role as key players during heart development, seven CHD candidate genes were further analyzed. Their mRNA expression was clearly down-regulated and the simultaneous hypermethylation of EGFR, EVC2 and NFATC2 was suggested as a representative CpG island methylator phenotype during TOF development [111].
The highly repetitive long interspersed nucleotide element-1 (LINE-1) encompasses up to 25% of the human genome [112]. It is most heavily methylated in its 5’-UTR and can serve as a surrogate marker of global genomic DNA methylation [113, 114]. Applying this marker to mothers who delivered singleton non-syndromic CHD births showed a reduced global methylation status with an almost doubled risk for CHD in the lowest decile [113]. Using the same approach a significantly lower methylation of LINE-1 was detected in TOF patients and those in the lowest quartile had a significantly increased risk for TOF [109]. In good agreement with these data are further results which confirm a decreased expression of DNA methyltransferase in TOF patients [115].
At a first glance it appears contradictory how both hyper- and hypomethylative events may contribute to the development of the CHD phenotype. However, hypomethylation may lead to deleterious changes in gene expression [113], possibly as a consequence of altered binding affinities for transcription factors [116]. In addition, hypomethylation of repetitive elements may generate an overall genomic instability which may be inherited to the progeny [117]. Together with a decreased expression of central cardiac transcription factors [109-111] this may lead to a scenario which increases the risk to develop CHD.
INTERACTIONS BETWEEN THE ENVIRONMENT AND THE GENOME
It has become clear that the environment can have a major influence on human development. Factors such as exposure to environmental air pollution may affect up to 25% of human disorders and may be responsible for a 36% death rate in children up to the age of 14 years [118]. However, except for some particular agents such as radon [119] or asbestos [120] it is difficult to establish a direct connection between an environmental influence on an individual disease. The abnormal formation of heart or the great vessels occurs during the first two months of pregnancy and, therefore, the first trimester of pregnancy is the most crucial period of maternal exposure to environmental pollution in CHD. Parental exposure to pesticides has been associated with the occurrence of different congenital heart disorders [121, 122]. In particular, herbicides and rodenticides have been associated with a transposition of the great arteries [123]. In addition, occupational exposure of the mother to chemicals led to an increased risk of ventricular septal and conotruncal defects [121, 124]. Furthermore, air pollutants such as carbon monoxide may increase the risk of TOF, especially during the first two months of gestation [125]. However, in many cases the validity of these observations is weakened by the low number of reported cases and the lack of an appropriate control group. Other factors which influence the developing embryo come from the mother itself. Smoking especially during the first trimester appears to increase the risk to develop CHD, e.g. ASD, pulmonary valve and artery anomalies [126-128]. A clear risk factor for birth defects including CHD is pregestational maternal diabetes mellitus [129, 130] which may account for more than 2.600 CHD cases in the US per year including TOF (230 cases) and HLHS (75 cases) [131].
In a zebrafish model the effect of exposure to retinoic acid and a dioxin-derivative transcriptional genome-wide profiles were analyzed. Quite rapidly a cluster of cell cycle regulatory genes was dramatically down-regulated together with a massive upregulation of Nr2F5, a transcriptional repressor, and a cessation of heart growth [132]. In humans some information is available concerning the target genes affected by environmental influence. A set of “environmental target genes” (direct targets of teratogens) and “environmental responders” (genes with altered cardiac expression in model organisms with human monogenic CHD mutations) has been defined after compiling datasets from studies of CHD in humans and in animal model organisms [133]. A set of seven environmental target genes was identified. These include several retinoic receptors, aryl hydrocarbon receptors and a folate reductase. The retinoic acid receptors appear to be risk factors for the outflow tract, while TBX5 was identified as a risk target or responder gene for the outflow tract and atrial and ventricular septum [133]. The JAG1 genes which appeared in multiple datasets has previously been associated with TOF [134]. Finally this multi-dataset analysis revealed similar functional networks for CHD risk which include the NOTCH, BMP, TGFβ and PDGFR signaling pathway [133]. Taken together these data suggest that functional interactions between the variation of the genome and the exposure to external environmental factors are rather complex.
SUMMARY
Within the past years the knowledge on the genetics of CHD has tremendously increased though at the same time this information has confirmed the complexity of the etiology. At present our understanding of the genetic reasons strongly suggests a polygenic genetic network and multifaceted interactions. On the genome level these components
include single base-pair variants (SNPs, point and de novo mutations), duplications or deletions of larger sequences (CNVs, InDels) as well as aberrant methylation patterns. On the transcriptome level key players of important developmental and cardiac pathways but also non-coding RNA species are abnormally expressed. An overview of candidate genes with potential relevance for TOF and HLHS is given in (Table 7). The entirety of these alterations and their interaction results in a complex “CHD genotype” ultimately leading to the development of a distinct CHD phenotype (Fig. 2). In addition, hemodynamic changes and environmental influences may complete the complicated picture.
Table 7.
List of genes with potential relevance for TOF and HLHS.
| Gene | Chromosome | OMIM | Cellular Function | Related Human Diseases and Known Cardiac Phenotypes | Reference |
|---|---|---|---|---|---|
| Tetrralogy of Fallot | |||||
| BARX1 | 9q22.32 | 603260 | transcription factor | [40] | |
| FOXK1 | 7p22.1 | no entry | transcription factor | [40] | |
| HAND2 | 4q34.1 | 602407 | transcription factor | [50] | |
| CNOT6 | 5q35.3 | 608951 | transcriptional regulation | [50] | |
| A2BP1 | 16p13.3 | 605104 | transcriptional regulation | [76, 81] | |
| TCEB3 | 1p36.1 | 600786 | transcriptional regulation | [40] | |
| PTPN11 | 12q24 | 176876 | modulation of Ras/MAPK signaling | Noonan syndrome, Leopard syndrome | [33, 34] |
| GPC5 | 13q32 | 602446 | modulation of Wnt, Hedgehog, FGF, and BMP signaling | acquired nephrotic syndrome, Hurler syndrome, Hunter syndrome | [33] |
| NRP1 | 10p12 | 602069 | modulation of VEGF signaling | Kallmann syndrome | [33] |
| SEMA3E | 7q21.11 | 608166 | modulation of NOTCH pathway | CHARGE syndrome | [48] |
| BCL9 | 1q21.2 | 602597 | modulation of Wnt signaling | B cell malignancies | [49] |
| TP53BP2 | 1q41 | 602143 | cell communication | [40] | |
| WBSCR16 | 7q11.23 | no entry | cell communication | Williams Beuren syndrome | [40] |
| SNIP | 17q12 | 610786 | cell communication | [76] | |
| BRDG1 | 4q13.2 | 604298 | cell communication | [76] | |
| VEGF | 6p21.1 | 192240 | cell communication | Susceptibility to atherosclerosis and diabetic retinopathy | [81, 82] |
| NPPA | 1p36.21 | 108780 | cell communication | atrial fibrillation, atrial standstill | [81] |
| NPPB | 1p36.22 | 600295 | cell communication | [81] | |
| SST | 3q27.3 | 182450 | cell communication | [84] | |
| PPBP | 4q13,3 | 121010 | cell communication | [84] | |
| NRGN | 11q24.2 | 602350 | cell communication | [84] | |
| MYOM2 | 8p23.3 | 603509 | cell growth and maintenance | [40] | |
| EDIL3 | 5q14 | 606018 | cell growth and maintenance | Ankylosing spondylitis | [50] |
| TEKT2 | 1p34.3 | 608953 | cell growth and/or maintenance | [76] | |
| MYL2 | 12q24.11 | 160781 | cell growth and/or maintenance | familial hypertrophic cardiomyopathy | [84] |
| NEFH | 22q12.2 | 162230 | cell growth and/or maintenance | amyotrophic lateral sclerosis | [84] |
| KRT6A | 12q13.3 | 148041 | cell growth and/or maintenance | Pachyonychia congenita | [84] |
| KRT7 | 12q13.3 | 148059 | cell growth and/or maintenance | [84] | |
| BCCIP | 10q26.1 | 611883 | cell cycle control | [40] | |
| DAG1 | 3p21.31 | 128239 | cell adhesion | Muscular dystrophy | [40] |
| KIAA1437 | 9q34.11 | 608360 | cell adhesion | Agammaglobulinemia | [76] |
| GJA5 | 1q21.1 | 121013 | gap junction | atrial fibrillation | [48, 49], [50, 57] |
| ROCK1 | 18q11.1 | 601702 | Ser/Thr kinase | [40] | |
| Tetrralogy of Fallot | |||||
| STK33 | 11p15.3 | 607670 | Ser/Thr kinase | [76] | |
| NTRK2 | 9q22.1 | 600456 | Tyr kinase | hyperphagia, and developmental delay | [84] |
| PRKAB2 | 1q21.1 | 602741 | AMP-activated protein kinase subunit | [49] | |
| PEX6 | 6p21.1 | 601498 | ATPase activity | Peroxisome biogenesis disorder | [40] |
| FANCL | 2p16.1 | 608111 | ligase activity | Fanconi anemia | [40] |
| FANCM | 14q21.1 | 609644 | DNA repair | Fanconi anemia | [40] |
| CHD1L | 1q21.1 | 613039 | DNA repair | [49] | |
| PLXNA2 | 1q32.2 | 601054 | axon pathfinding | cardiac hypertrophy | [48] |
| SEMA3D | 7q21.11 | 609907 | development of peripheral axons | Hirschsprung disease | [48] |
| TTN | 2q31.2 | 188840 | muscle development | Dilated cardiomyopathy, hereditary myopathy, tibial muscular dystrophy | [40] |
| EDN1 | 6p24.1 | 131240 | vasoconstriction | Auriculocondylar syndrome | [40] |
| HCN2 | 19p13.3 | 602781 | ion channel | Sinus dysrhytmia | [40] |
| FMR1 | Xq27.3 | 309550 | mRNA trafficing | Fragile X mental retardation, syndrome, ataxia syndrome | [40] |
| APOC3 | 11q23.3 | 107720 | transport activity | Apolipoprotein C-III deficiency | [84] |
| TF | 3q22.1 | 190000 | transport activity | Atransferrinemia | [84] |
| Hypoplastic left heart syndrome | |||||
| FOXC2 | 16q24.1 | 602402 | transcription factor | Lymphedema-distichiasis syndrome | [69] |
| FOXL1 | 16q24.1 | 603252 | transcription factorr | [69] | |
| ZNF423 | 16q12 | 604557 | transcription factor | Joubert syndrome, Nephronophthisis | [69] |
| CHD7 | 8q12.2 | 608892 | transcriptional regulation | CHARGE syndrome | [71] |
| MXD1 | 2p13.3 | 600021 | transcriptional regulation | [87] | |
| KDM5B | 1q32.1 | 605393 | transcriptional regulation | [71] | |
| CTHRC1 | 8q22.3 | 610635 | modulation of Wnt pathway | HELLP syndrome | [59] |
| PLA2G12A | 4q25 | 611652 | modulation of BMP pathway | [59] | |
| BMPR2 | 2q32-33 | 600799 | modulation of BMP and TGFb signaling | Pulmonary hypertension | [69] |
| NOTCH1 | 9q34.3 | 190198 | modulation of NOTCH signaling | Aortic valve disease, Adams-Oliver syndrome | [69] |
| NODAL | 10q22.1 | 601265 | modulation of NODAL pathway | Heterotaxy, double-outlet right ventricle, transposition of the great arteries | [70] |
| NOMO3 | 16p13 | 609159 | modulation of NODAL pathway | Heterotaxy | [70] |
| SMAD2 | 18q21.1 | 601366 | modulation of TGFb signaling | Dextrocardia | [71] |
| ANK1 | 8p11.1 | 612641 | cell communication | Spherocytosis | [87] |
| PPFIA2 | 12q21.31 | 603143 | cell communication | [87] | |
| Hypoplastic left heart syndrome | |||||
| PMP22 | 17p12 | 601097 | cell communication | Charcot-Marie-Tooth disease, Dejerine-Sottas disease, Neuropathy, Roussy-Levy syndrome | [87] |
| SPTA1 | 1q32.1 | 182860 | cell growth and/or maintenance | Elliptocytosis, Pyropoikilocytosis, Spherocytosis | [87] |
| CCNJ | 10q23.33 | no entry | cell cycle control | [87] | |
| MFAP4 | 17p11.2 | 600596 | cell adhesion and interaction | Smith Magenis syndrome | [59] |
| MLL2 | 12q13.12 | 602113 | histone methyltransferase | Kabuki syndrome | [71] |
| WDR5 | 9q34 | 609012 | subunit of histone-methyltransferase complex | [71] | |
| KDM5A | 12p13.33 | 180202 | histone demethylation | [71] | |
| RNF20 | 9q31.1 | 607699 | ubiquitination and methylation | [71] | |
| USP44 | 12q22 | 610993 | ubiquitination | [71] | |
| UBE2B | 5q31.1 | 179095 | DNA repair | [71] | |
| GYPB | 4q31.21 | 111740 | immune response | susceptibility to malaria | [87] |
| PLD6 | 17p11.2 | no entry | hydrolase activity | [87] | |
| FUT8 | 14q24.3 | 602589 | fucose transfer | [65] | |
| CFC1 | 2q21.1 | 605194 | patterning of right-left embryonic axis | [70] | |
| GRM1 | 6q24.3 | 604473 | transport activity | autosomal recessive spinocerebellar ataxia | [87] |
| CACNB2 | 10p12.31-33 | 600003 | transport activity | Brugada syndrome | [87] |
Fig. (2).
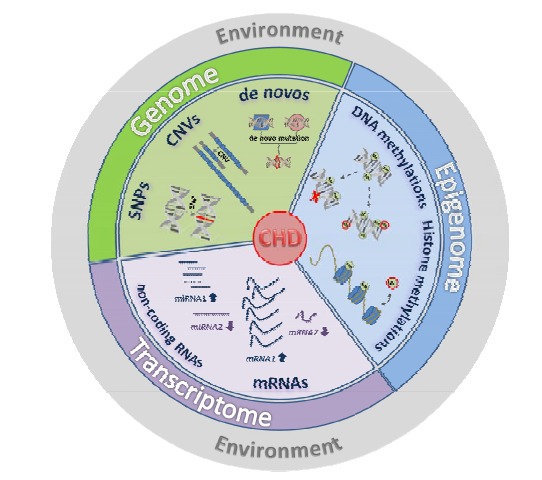
Complex etiology of congenital heart defects. One must assume that a multitude of alterations happen which affect the genomic information directly (SNPs, CNVs, mutations, DNA methylation) or which alter the steady state of the transcriptome (dysregulation of coding and non-coding RNA species). Altogether these multiple factors generate a comprehensive genetic imbalance which ultimately results in the development of a distinct CHD phenotype.
A number of distinct candidate genes have been pinpointed and several genetic risk factors with high significance have been identified, though at present no subsequent downstream experiments exist which substantiate the functional relevance of any of these factors. It has not been possible yet to define a well-defined set of genetic alterations which will reproducibly generate a complex CHD phenotype such as TOF or HLHS in an animal model. Furthermore, alterations in non-coding regions such as promoter or enhancer regions may be involved but such information is only scarcely available [135]. Whole genome sequencing approaches can yield results in that field but appropriate studies have not been conducted yet.
Currently the genetic information on severe CHD phenotypes extracted from genome-wide approaches is almost exclusively restricted to TOF and HLHS. Patients with other CHD subtypes, e.g. double-outlet right ventricle or transposition of the great arteries appear in several of the investigated cohorts as smaller subgroups. However, in all instances specific genetic lesions have not been assigned to those subtypes and their numbers were too low to draw any significant conclusions with regard to a potential relevance of genetic alterations.
Statistically valid data can only be obtained by large-scale multicenter studies. Therefore, several network studies have been launched such as the Pediatric Cardiac Genomics Consortium [136], the Deciphering Developmental Disorders study [137] or the UK10K project [138] to analyze the complex genetics of CHD. High-throughput technologies and next-generation sequencing approaches with high numbers of patients and their parents should shed some light on the still mostly enigmatic components involved in genetic predisposition. It will also be challenging to detect potential loci with recurrent de novo mutations in both syndromic and non-syndromic patients. Finally, it is important to evaluate the functional relevance of these genetic findings in appropriate cellular test systems. The use of induced pluripotent stem cells derived from patients and unaffected first-degree relatives and the differentiation of such cells into the cardiac lineage would represent an ideal model to elucidate apparent functional anomalies in vitro respecting the complete patient-specific background.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by grants from Deutsche Stiftung für Herzforschung, F/37/11 (M. K.), Dr. Rusche Research Grant 2014 from the Deutsche Stiftung für Herzforschung and Deutsche Gesellschaft für Thorax-, Herz- und Gefässchirurgie (M.-A. D.), the German Research Foundation, KR-3770-7/1 and KR-3770-9/1 (M. K.) and the Deutsches Zentrum für Herz- und Kreislaufforschung Säule B Projekt, DZHK B 13-050A and DZHK B 14-013SE (M. K.).
Author contributions: H.L., P. S., M. D., S. D., J. C., M.-A. D. and M. K. Writing of manuscript and design, P. E. and R. L. Conception and design of the manuscript.
REFERENCES
- 1.Hoffmann JI, Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002;39(12):1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 2.Lindinger A, Schwedler G, Hense HW. Prevalence of congenital heart defects in newborns in Germany: Results of the first registration year of the PAN study (July 2006 to June 2007) Klin. Padiatr. 2010;222(5):321–326. doi: 10.1055/s-0030-1254155. [DOI] [PubMed] [Google Scholar]
- 3.Morris CD, Outcalt J, Menashe VD. Hypoplastic left heart syndrome: natural history in a geographically defined population. Pediatrics. 1990;85(6):977–983. [PubMed] [Google Scholar]
- 4.McGuirk SP, Griselli M, Stumper O F, Rumball E M, Miller P, Dhillon R, de Giovanni JV, Wright JG, Barron DJ, Brawn W J. Staged surgical management of hypoplastic left heart syndrome: a single institution 12 year experience. Heart. 2006;92(3):364–370. doi: 10.1136/hrt.2005.068684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azakie A, McCrindle BW, Benson LN, Van Arsdell GS, Russell J L, Coles J G, Nykanen D, Freedom R M, Williams W G. Total cavopulmonary connections in children with a previous Norwood procedure. Ann. Thorac. Surg. 2001;71(5):1541–1546. doi: 10.1016/s0003-4975(01)02465-1. [DOI] [PubMed] [Google Scholar]
- 6.Mahle W T, Spray T L, Wernovsky G, Gaynor JW, Clark B J., 3rd Survival after reconstructive surgery for hypoplastic left heart syndrome. A 15-year experience from a single institution. Circulation. 2000;102(19) Suppl 3:III136–141. doi: 10.1161/01.cir.102.suppl_3.iii-136. [DOI] [PubMed] [Google Scholar]
- 7.Cuypers JA, Menting ME, Konings EE, Opic P, Utens E M, Helbing WA, Witsenburg M, van den Bosch AE, Ouhlous M, van Domburg RT, Rizopoulos D, Meijboom F J, Boersma E, Bogers AJ, Roos-Hesselink JW. The unnatural history of Tetralogy of Fallot: prospective follow-up of 40 years after surgical correction. Circulation. 2014;130(22):1944–1953. doi: 10.1161/CIRCULATIONAHA.114.009454. [DOI] [PubMed] [Google Scholar]
- 8.Nora JJ. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases. Circulation. 1968;38(3):604–617. doi: 10.1161/01.cir.38.3.604. [DOI] [PubMed] [Google Scholar]
- 9.Whittemore R, Hobbins JC, Englr MA. Pregnancy and its outcome in women with and without surgical treatment of congenital heart disease. Am. J. Cardiol. 1982;50(3):641–651. doi: 10.1016/0002-9149(82)90334-4. [DOI] [PubMed] [Google Scholar]
- 10.Nabulsi M M, Tanim H, Sabbagh M, Obeid M Y, Yunis K A, Bitar F F. Parental consanguinitiy and congenital heart malformations in a developing country. Am. J. Med. Genet. A. 2003;116A(4):342–347. doi: 10.1002/ajmg.a.10020. [DOI] [PubMed] [Google Scholar]
- 11.Yunis K, Mumtaz G, Bitar F, Chamseddine F, Kassar M, Rashkidi J, Makhoul G, Tamim H. Consanguineous marriage and congenital heart defects: a case-control study in the neonatal period. Am. J. Med. Genet. A. 2006;140(14):1524–1530. doi: 10.1002/ajmg.a.31309. [DOI] [PubMed] [Google Scholar]
- 12.Schott J J, Benson D W, Basson C T, Pease W, Silberbach G M, Moak J P, Maron B J, Seidman C E, Seidman J G. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281(5373):08–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 13.Garg V, Kathiriya I S, Barnes R, Schluterman M K, King I N, Butler C A, Rothrock C R, Eapen R S, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen J C, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 14.Hinton R B , Jr, Martin L J, Tabangin M E, Mazwi M L, Cripe L H, Benson D W. Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 2007;50(16):1590–1595. doi: 10.1016/j.jacc.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein D. Evaluation of the cardiovascular system Nelson Textbook of Pediatrics. In: Behrman R E, Kliegman R M, Jenson H B, editors; 17th ed. Philadelphia Saunders; 2004. pp. 1481–1488. [Google Scholar]
- 16.Pierpont ME, Basson CT, Benson DW, Jr, Gelb BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D, Webb C L. American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):3015–3038. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 17.Hartman R J, Rasmussen S A, Botto L D, Riehle-Colarusso T, Martin C L, Cragan J D, Shin M, Correa A. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr.Cardiol. 2011;32(8):1147–1157. doi: 10.1007/s00246-011-0034-5. [DOI] [PubMed] [Google Scholar]
- 18.Goldmuntz E, Clark B J, Mitchell L E, Jawad A F, Cuneo B F, Reed L, McDonald-McGinn D, Chien P, Feuer J, Zackai E H, Emanuel B S, Driscoll D A. Frequency of 22q11 deletions in patients with conotruncal defects. J. Am. Coll. Cardiol. 1998;32(3):492–498. doi: 10.1016/s0735-1097(98)00259-9. [DOI] [PubMed] [Google Scholar]
- 19.Lalani S R, Belmont J W. Genetic basis of congenital cardiovascular malformations. Eur. J. Med. Genet. 2014;57(4):402–413. doi: 10.1016/j.ejmg.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori AD, Bruneau B G. TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr. Opin. Cardiol. 2004;19(3):211–215. doi: 10.1097/00001573-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Lahm H, Deutsch M A, Dreßen M, Doppler S, Werner A, Hörer J, Cleuziou J, Schreiber C, Böhm J, Laugwitz K L, Lange R, Krane M. Mutational analysis of the human MESP1 gene in patients with congenital heart disease reveals a highly variable sequence in exon 1. ur. J. Med. Genet. 2013;56(11):591–598. doi: 10.1016/j.ejmg.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 22.McElhinney DB, Geiger E, Blinder J, Benson D W, Goldmuntz E. NKX2 mutations in patients with congenital heart disease. J. Am. Coll. Cardiol. 2003,;42(9):1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Reamon-Buettner S, Borlak J. NKX2-5: an update on this hypermutable homeodomain protein and its role in congenital heart disease (CHD) Hum. Mutat. 2010;31(11):1185–1194. doi: 10.1002/humu.21345. [DOI] [PubMed] [Google Scholar]
- 24.Stallmeyer B, Fenge H, Nowak-Göttl U, Schulze-Bahr E. Mutational spectrum in the cardiac transcription factor gene NKX2 (CSX) associated with congenital heart disease. Clin. Genet. 2010;78(6):533–540. doi: 10.1111/j.1399-0004.2010.01422.x. [DOI] [PubMed] [Google Scholar]
- 25.Tomita-Mitchell A, Maslen CL, Morris CD, Garg V, Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J. Med. Genet. 2007;44(12):779–783. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao H, Yan B, Liu Y, Yang Y, Chen YH. A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J. Hum. Genet. 2010;55(10):662–667. doi: 10.1038/jhg.2010.84. [DOI] [PubMed] [Google Scholar]
- 27.Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, Mackay J P, Waddell L B, Cole A D, Hayward C, Keogh A, Macdonald P, Griffiths L, Fatkin D, Sholler G F, Zorn A M, Fenely M P, Winlaw D A, Harvey R P. Mutations in the cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of spetation and valvulogensis and cardiomyopathy. Am. J. Hum. Genet. 2007;81(3):280–291. doi: 10.1086/519530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granados-Riveron J T, Pope M, Bu’Lock F A, Thornborough C, Eason J, Setchfield K, Ketley A, Kirk E P, Fatkin D, Feneley M P, Harvey R P, Brook J D. Combined mutation screening of NKX2-5, GATA4, and TBX5 in congenital heart disease: multiple heterozygosity and novel mutations. Congenit. Heart Dis. 2012;7(3):151–159. doi: 10.1111/j.1747-0803.2011.00573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren H, Francis W, Boys A, Chueh A C, Wong N, La P, Wong L H, Ryan J, Slater H R, Choo K H. BAC-based PCR fragment microarray: high-resolution detection of chromosomal deletion and duplication breakpoints. Hum. Mut. 2005;25(5):476–285. doi: 10.1002/humu.20164. [DOI] [PubMed] [Google Scholar]
- 30.Urban A E, Korbel J O, Selzer R, Richmond T, Hacker A, Popescu G V, Cubells J F, Green R, Emanuel B S, Gerstein M B, Weissman S M, Snyder M. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc. Natl. Acad. Sci. USA. 2006;103(12):4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards A A, Garg V. Genetics of congenital heart disease. Curr. Cardiol. Rev. 2010;6(3):91–97. doi: 10.2174/157340310791162703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cordell H J, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, Cosgrove C, Blue G, Granados-Riveron J, Setchfield K, Thornborough C, Breckpot J, Soemedi R, Martin R, Rahman T J, Hall D, van Engelen K, Moorman A F, Zwinderman A H, Barnett P, Koopmann T T, Adriaens M E, Varro A, George A L , Jr, dos Remedios C, Bishopric N H, Bezzina C R, O'Sullivan J, Gewillig M, Bu'Lock F A, Winlaw D, Bhattacharya S, Devriendt K, Brook J D, Mulder B J, Mital S, Postma A V, Lathrop G M, Farrall M, Goodship J A, Keavney B D. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 2013;45(7):822–824. doi: 10.1038/ng.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cordell H J, Töpf A, Mamasoula C, Postma A V, Bentham J, Zelenika D, Heath S, Blue G, Cosgrove C, Granados Riveron J, Darlay R, Soemedi R, Wilson I J, Ayers K L, Rahman T J, Hall D, Mulder B J, Zwinderman A H, van Engelen K, Brook J D, Setchfield K, Bu'Lock F A, Thornborough C, O'Sullivan J, Stuart A G, Parsons J, Bhattacharya S, Winlaw D, Mital S, Gewillig M, Breckpot J, Devriendt K, Moorman A F, Rauch A, Lathrop GM, Keavney B D, Goodship J A. Genome-wide association study identifies loci on 12q24 and 13q32 associated with tetralogy of Fallot. Hum. Mol. Genet. 2013;22(7):1473–1481. doi: 10.1093/hmg/dds552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodship J A, Hall D, Topf A, Mamasoula C, Griffin H, Rahman T J, Glen E, Tan H, Palomino Doza J, Relton C L, Bentham J, Bhattacharya S, Cosgrove C, Brook D, Granados-Riveron J, Bu'Lock F A, O'Sullivan J, Stuart A G, Parsons J, Cordell H J, Keavney B. A common variant in the PTPN11 gene contributes to the risk of tetralogy of Fallot. Circ. Cardiovasc. Genet. 2012;5(3):287–292. doi: 10.1161/CIRCGENETICS.111.962035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soranzo N, Spector T D, Mangino M, Kühnel B, Rendon A, Teumer A, Willenborg C, Wright B, Chen L, Li M, Salo P, Voight B F, Burns P, Laskowski R A, Xue Y, Menzel S, Altshuler D, Bradley J R, Bumpstead S, Burnett M S, Devaney J, Döring A, Elosua R, Epstein S E, Erber W, Falchi M, Garner S F, Ghori M J, Goodall A H, Gwilliam R, Hakonarson H H, Hall A S, Hammond N, Hengstenberg C, Illig T, König I R, Knouff C W, McPherson R, Melander O, Mooser V, Nauck M, Nieminen M S, O'Donnell C J, Peltonen L, Potter S C, Prokisch H, Rader D J, Rice C M, Roberts R, Salomaa V, Sambrook J, Schreiber S, Schunkert H, Schwartz S M, Serbanovic-Canic J, Sinisalo J, Siscovick D S, Stark K, Surakka I, Stephens J, Thompson J R, Völker U, Völzke H, Watkins N A, Wells G A, Wichmann H E, Van Heel D A, Tyler-Smith C, Thein S L, Kathiresan S, Perola M, Reilly M P, Stewart A F, Erdmann J, Samani N J, Meisinger C, Greinacher A, Deloukas P, Ouwehand W H, Gieger C. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 2009;41(11):1182–1190. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tartaglia M, Mehler E L, Goldberg R, Zampino G, Brunner H G, Kremer H, van der Burgt I, Crosby A H, Ion A, Jeffery S, Kalidas K, Patton M A, Kucherlapati R S, Gelb B D. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001;29(4):465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Lin Y, Si L, Jin G, Dai J, Wang C, Chen J, Da M, Hu Y, Yi C, Hu Z, Shen H, Mo X, Chen Y, Wang X. Genetic variants at 10p11 confer risk of Tetralogy of Fallot in Chinese of Nanjing. PLoS One. 2014;9(3):e89636. doi: 10.1371/journal.pone.0089636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9(5):224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu C, Rodriguez E R, Reimert D V, Shu T, Fritzsch B, Richards L J, Kolodkin A L, Ginty D D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell. 2003;5(1):45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grunert M, Dorn C, Schueler M, Dunkel I, Schlesinger J, Mebus S, Alexi-Meskishvili V, Perrot A, Wassilew K, Timmermann B, Hetzer R, Berger F, Sperling S R. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum. Mol. Genet. 2014;23(12):3115–3128. doi: 10.1093/hmg/ddu021. [DOI] [PubMed] [Google Scholar]
- 41.Maron B J, Maron M S. Hypertrophic cardiomyopathy. Lancet. 2013;381(9862):242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 42.McBride K L, Zender G A, Fitzgerald-Butt S M, Koehler D, Menesses-Diaz A, Fernbach S, Lee K, Towbin J A, Leal S, Belmont J W. Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome) Eur. J. Hum. Genet. 2009;17(6):811–819. doi: 10.1038/ejhg.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loffredo C A, Chokkalingam A, Sill A M, Boughman J A, Clark E B, Scheel J, Brenner J I. Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coarctation of the aorta, and d-transposition of the great arteries. Am. J. Med. Genet. A. 2004;124A(3):225–230. doi: 10.1002/ajmg.a.20366. [DOI] [PubMed] [Google Scholar]
- 44.Redon R, Ishikawa S, Fitch K R, Feuk L, Perry G H, Andrews T D, Fiegler H, Shapero M H, Carson A R, Chen W, Cho E K, Dallaire S, Freeman J L, González J R, Gratacòs M, Huang J, Kalaitzopoulos D, Komura D, MacDonald J R, Marshall C R, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville M J, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad D F, Estivill X, Tyler-Smith C, Carter N P, Aburatani H, Lee C, Jones K W, Scherer S W, Hurles M E. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, Fryns J P, Gewillig M, Vermeesch J R, Devriendt K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 2007;28(22):2778–2784. doi: 10.1093/eurheartj/ehl560. [DOI] [PubMed] [Google Scholar]
- 46.Richards A A, Santos L J, Nichols H A, Crider B P, Elder F F, Hauser N S, Zinn A R, Garg V. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatr. Res. 2008;64(4):358–363. doi: 10.1203/PDR.0b013e31818095d0. [DOI] [PubMed] [Google Scholar]
- 47.Loffredo C A. Epidemiology of cardiovascular malformations: prevalence and risk factors. Am. J. Med. Genet. 2000;97(4):319–325. doi: 10.1002/1096-8628(200024)97:4<319::aid-ajmg1283>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 48.Silversides C K, Lionel A C, Costain G, Merico D, Migita O, Liu B, Yuen T, Rickaby J, Thiruvahindrapuram B, Marshall C R, Scherer S W, Bassett A S. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8(8):e1002843. doi: 10.1371/journal.pgen.1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenway S C, Pereira A C, Lin J C, DePalma S R, Israel S J, Mesquita S M, Ergul E, Conta J H, Korn J M, McCarroll S A, Gorham J. M Gabriel, S Altshuler DM, Quintanilla-Dieck Mde L Artunduaga MA, Eavey RD Plenge RM, Shadick N A, Weinblatt M E, De Jager P L, Hafler D A, Breitbart R E, Seidman J G, Seidman C E. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41(8):931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soemedi R, Wilson I J, Bentham J, Darlay R, Töpf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue G M, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman T J, Hall D, Rauch A, Devriendt K, Gewillig M, O' Sullivan J, Winlaw D S, Bu'Lock F, Brook J D, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell H J, Goodship J A, Keavney B D. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012;91(3):489–501. doi: 10.1016/j.ajhg.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perala N. More than nervous: The emerging role of plexins. Differentiation. 2012;83:77–91. doi: 10.1016/j.diff.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Feiner L, Webber A L, Brown C B, Lu M M, Jia L, Feinstein P, Mombaerts P, Epstein J A, Raper J A. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128(16):3061–3070. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- 53.Brown C B, Feiner L, Lu M M, Li J, Ma X, Webber A L, Jia L, Raper J A, Epstein J A. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128(16):3071–3080. doi: 10.1242/dev.128.16.3071. [DOI] [PubMed] [Google Scholar]
- 54.Kim J, Oh W-J, Gaiaono N, Yoshida Y, Gu C. Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanisms. Genes Dev. 2011;25(13):1399–1411. doi: 10.1101/gad.2042011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lalani S R, Safiullah A M, Fernbach S D, Harutyunyan K G, Thaller C, Peterson L E, McPherson J D, Gibbs R A, White L D, Hefner M, Davenport S L, Graham J M, Bacino C A, Glass N L, Towbin J A, Craigen W J, Neish S R, Lin A E, Belmont J W. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 2006;78(3):303–314. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christiansen J, Dyck J D, Elyas B G, Lilley M, Bamforth J S, Hicks M, Sprysak K A, Tomaszewski R, Haase S M, Vicen-Wyhony L M, Somerville M J. Chromosome 1q211 contiguous gene deletion is associated with congenital heart disease. Circ. Res. 2004;94(11):1429–1435. doi: 10.1161/01.RES.0000130528.72330.5c. [DOI] [PubMed] [Google Scholar]
- 57.Soemedi R, Töpf A, Wilson I J, Darlay R, Rahman T J, Glen E, Hall D, Huang N, Bentham J, Bhattacharya S, Cosgrove C, Brook J D, Granados-Riveron J, Setchfield K Bu Lock, F Thornborough C, Devriendt K, Breckpot J Hofbeck, M Lathrop, M Rauch, A Blue G M, Winlaw D S, Hurles M, Santibanez-Koref M, Cordell H J, Goodship J A, Keavney B D. Phenotype-specific effect of chromosome 1q21 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum. Mol. Genet. 2012;21(7):1513–1520. doi: 10.1093/hmg/ddr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu H, Smith F C, Taffet S M, Delmar M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003;93(3):201–206. doi: 10.1161/01.RES.0000084852.65396.70. [DOI] [PubMed] [Google Scholar]
- 59.Hitz M-P, Lemieux-Perreualt L-P, Marshall C, Feroz-Zada Y, Davies R, Yang S W, Lionel A C, D’Amours G, Lemyre E, Cullum R, Bigras J-L, Thibeualt M, Chetaille P, Montpetit A, Khairy P, Overduin B, Klaassen S, Hoodless P, Nemer M, Stewart A F R, Boerkoel C, Scherer S W, Richter A, Dubé M-P, Andelfinger G. Rare copy number variants contribute to congenital left-sided heart disease. PLOS Genetics. 2012;8(9):e1002903. doi: 10.1371/journal.pgen.1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wulf-Johansson H, Lock Johansson S, Schlosser A, Trommelholt Holm A, Rasmussen L M, Mickley H, Diederichsen A C, Munkholm H, Poulsen T S, Tornøe I, Nielsen V, Marcussen N, Vestbo J, Sækmose S G, Holmskov U, Sorensen G L. Localization of microfibrillar-associated protein 4 (MFAP4) in human tissues: clinical evaluation of serum MFAP4 and its association with various cardiovascular conditions. PLoS One. 2013;8(12):e82243. doi: 10.1371/journal.pone.0082243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamamoto S, Nishimura O, Misaki K, Nishita M, Minami Y, Yonemura S, Tarui H, Sasaki H. Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev. Cell. 2008;15(1):23–36. doi: 10.1016/j.devcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 62.Toyoshima T, Nishi N, Kusama H, Kobayashi R, Itano T. 36-kDa microfibrill-associated glycoprotein (MAGP-36) is an elastin-binding protein increased in chick aortae during development and growth. Exp. Cell Res. 2005;307(1):224–230. doi: 10.1016/j.yexcr.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 63.Hinton R B, Martin L J, Tabangin M E, Mazwi M L, Cripe L H, Benson D W. Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 2009;53(12):1065–1071. doi: 10.1016/j.jacc.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munoz-Sanjuan I, Brivanlou AH. Induction of ectopic olfactory structures and bone morphogenic protein inhibition by Rossy, a group XII secreted phospholipase A2. Mol. Cell. Biol. 2005;25(9):3608–3619. doi: 10.1128/MCB.25.9.3608-3619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Payne A R, Chang S W, Koenig S N, Zinn AR, Garg V. Submicroscopic chromosomal copy number variations identified in children with hypoplastic left heart syndrome. Pediatr. Cardiol. 2012;33(5):757–763. doi: 10.1007/s00246-012-0208-9. [DOI] [PubMed] [Google Scholar]
- 66.Tennessen J A, Bigham A W, O'Connor T D, Fu W, Kenny E E, Gravel S, McGee S, Do R, Liu X, Jun G, Kang H M, Jordan D, Leal S M, Gabriel S, Rieder M J, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante C D, Bamshad M J, Akey J M, Broad G O. Seattle GO NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337(6090):64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marth G T, Yu F, Indap A R, Garimella K, Gravel S, Leong W F, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, Chen Y, Challis D, Clarke L, Ball E V, Cibulskis K, Cooper D N, Fulton B, Hartl C, Koboldt D, Muzny D, Smith R, Sougnez C, Stewart C, Ward A, Yu J, Xue Y, Altshuler D, Bustamante C D, Clark A G, Daly M, DePristo M, Flicek P, Gabriel S, Mardis E, Palotie A, Gibbs R. 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12(9):R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T, Grarup N, Guo Y, Hellman I, Jin X, Li Q, Liu J, Liu X, Sparsø T, Tang M, Wu H, Wu R, Yu C, Zheng H, Astrup A, Bolund L, Holmkvist J, Jørgensen T, Kristiansen K, Schmitz O, Schwartz T W, Zhang X, Li R, Yang H, Wang J, Hansen T, Pedersen O, Nielsen R, Wang J. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010;42(11):969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 69.Iascone M, Ciccone R, Galletti L, Marchetti D, Seddio F, Lincesso A R, Pezzoli L, Vetro A, Barachetti D, Boni L, Federici D, Soto A M, Comas J V, Ferrazzi P, Zuffardi O. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin. Genet. 2012;81(6):542–554. doi: 10.1111/j.1399-0004.2011.01674.x. [DOI] [PubMed] [Google Scholar]
- 70.Warburton D, Ronemus M, Kline J, Robanputra V, Williams I, Anyane-Yeboa K, Chung W, Yu L, Wang N, Awad D, Yu C, Leotta A, Kendall J, Yamrom B, Lee Y, Wigler M, Levy D. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypolplastic left heart disease. Hum. Genet. 2014;133(1):11–27. doi: 10.1007/s00439-013-1353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson R D, Breitbart R E, Brown K K, Carriero N J, Cheung Y H, Deanfield J, DePalma S, Fakhro K A, Glessner J, Hakonarson H, Italia M J, Kaltman J R, Kaski J, Kim R, Kline J K, Lee T, Leipzig J, Lopez A, Mane S M, Mitchell L E, Newburger J W, Parfenov M, Pe'er I, Porter G, Roberts A E, Sachidanandam R, Sanders S J, Seiden H S, State M W, Subramanian S, Tikhonova I R, Wang W, Warburton D, White P S, Williams I A, Zhao H, Seidman J G, Brueckner M, Chung W K, Gelb B D, Goldmuntz E, Seidman C E, Lifton R P. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pinskaya M, Morillon A. Histone H3 lysine 4 di-methylation: a novel landmark for transcriptional fidelityκ. Epigenetics. 2009;4(5):302–306. doi: 10.4161/epi.4.5.9369. [DOI] [PubMed] [Google Scholar]
- 73.Bernstein B E, Mikkelsen T S, Xie X, Kamal M, Huebert D J, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber S L, Lander E S. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(3):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 74.Paige SL, Thomas S, Stoick-Cooper C L, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, Moon RT, Stamatoyannopoulos J, Murry C E. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151(1):221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guida V, Ferese R, Rocchetti M, Bonetti M, Sarkozy A, Cecchetti S, Gelmetti V, Lepri F, Copetti M, Lamorte G, Cristina Digilio M, Marino B, Zaza A, den Hertog J, Dallapiccola B, De Luca A. A variant in the carboxyl-terminus of connexin 40 alters GAP junctions and increases risk for tetralogy of Fallot. Eur. J. Hum. Genet. 2013;21(1):69–75. doi: 10.1038/ejhg.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaynak B, von Heydebreck A, Mebus S, Seelow D, Hennig S, Vogel J, Sperling H-P, Pregla R, Alexi-Meskishvilli V, Hetzer R, Lange P E, Vingron M, Lehrach H, Sperling S. Genome-wide array analysis of normal and malformed human hearts. Circulation. 2003;107(19):2467–2474. doi: 10.1161/01.CIR.0000066694.21510.E2. [DOI] [PubMed] [Google Scholar]
- 77.Klaus A, Saga Y, Taketo M M, Tzahor E, Birchmeier W. Distinct roles of Wnt/beta-catenin and Bmp signaling during early cardiogenesis. Proc. Natl.Acad. Sci. USA. 2007;104(47):18531–18536. doi: 10.1073/pnas.0703113104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ladd A N, Yatskievych T A, Antin P B. Regulation of an avian cardiac morphogenesis by activin/TGFbeta and bone morphogenic proteins. Dev. Biol. 1998;204(3):407–410. doi: 10.1006/dbio.1998.9094. [DOI] [PubMed] [Google Scholar]
- 79.Kiehl T R, Shibata H, Vo T, Huynh D P, Pulst S M. Identification and expression of a mouse ortholog of A2BP1. Mamm. Genome. 2001;12(8):595–601. doi: 10.1007/s00335-001-2056-4. [DOI] [PubMed] [Google Scholar]
- 80.Koera K, Nakamura K, Nakao K, Miyoshi J, Toyoshima K, Hatta T, Otani H, Aiba A, Katsuki M. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15(10):1151–1159. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 81.Bittel D C, Butler M G, Kibiryeva N, Marshall J A, Chen J, Lofland G K, O’Brien J E ., Jr Gene expression in cardiac tissues from infants with idiopathic conotruncal defects. BMC Med. Genomics. 2011;4:1. doi: 10.1186/1755-8794-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma H S, Peters T H F, Moorhouse M J, van der Spek P J, Bogers A J J C. DNA microarray analysis for human congenital heart disease. Cell Biochem.Biophy. 2006;44(1):1–10. doi: 10.1385/CBB:44:1:001. [DOI] [PubMed] [Google Scholar]
- 83.Peters T H F, Sharma V, Yilmaz E, Mooi W J, Bogers A J J C, Sharma H S. DNA microarray and quantitative analysis reveal enhanced myocardial VEGF expression with stunted angiogenesis in human tetralogy of Fallot. Cell. Biochem. Biophys. 2013;67(3):305–316. doi: 10.1007/s12013-013-9710-9. [DOI] [PubMed] [Google Scholar]
- 84.Rodemoyer A, Kibiryeva N, Bair A, Marshall J, O’Brien Jr J E, Bittel D C. A tissue-specific gene expression template portrays heart development and pathology. Hum. Genomics. 2014;8:6. doi: 10.1186/1479-7364-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hwang P-I, Wu H-B, Wang C-D, Lin B-L, Chen C-T, Yuan S, Wu G, Li K-C. Tissue-specific gene expression templates for accurate molecular characterization of the normal physiological states of multiple human tissues with implication in development and cancer. BMC Genomics. 2011;12:439. doi: 10.1186/1471-2164-12-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O'Brien J E , Jr, Kibiryeva N, Zhou X G, Marshall J A, Lofland G K, Artman M, Chen J, Bittel D C. Noncoding RNA expression in myocardium from infants with tetralogy of Fallot. irc. Cardiovasc. Genet. 2012;5(3):279–286. doi: 10.1161/CIRCGENETICS.111.961474. [DOI] [PubMed] [Google Scholar]
- 87.Ricci M, Xu Y, Hammond H L, Wiloughby D A, Nathanson L, Rodriguez M M, Vatta M, Lipshultz SE, Lincoln J. Myocardial alternative RNA splicing and gene expression profiling in early stage hypoplastic left heart syndrome. PLOS One. 2012;7(1):e29784. doi: 10.1371/journal.pone.0029784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosca M G, Vazquez E J, Kerner J, Parland W, Chandler M P, Stanley W, Sabbah H N, Hoppel C L. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008;80(1):30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Doenst T, Pytel G, Schrepper A, Amorim P, Färber G, Shingu Y, Mohr F W, Schwarzer M. Decreased rates of and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010;86(3):461–470. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- 90.Margulies K B, Bednarik D P, Dries D L. Genomics, transcriptional profiling, and heart failure. J. Am. Coll. Cardiol. 2009;53(19):1752–1759. doi: 10.1016/j.jacc.2008.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pavan M, Ruiz V F, Silva F A, Sobreira T J, Cravo R M, Vasconcelos M, Marques LP, Mesquita S M, Krieger J E, Lopes A A, Oliveira P S, Pereira A C, Xavier-Neto J. ALDH1A2 (RALDH2) genetic variation in human congenital heart disease. BMC Med. Genet. 2009;10:113. doi: 10.1186/1471-2350-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Georges R, Nemer G, Morin M, Lefebvre C, Nemer M. Distinct expression and function of alternatively spliced Tbx5 isoforms in cell growth an,d differentiation. Mol. Cell Biol. 2008;28(12):4052–4067. doi: 10.1128/MCB.02100-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kong S W, Hu Y W, Ho J W K, Ikeda S, Polster S, ohn R, Hall J L, Bisping E, Pieske B, dos Remedios C G, Pu W T. Heart failure-associated changes in RNA splicing of sarcomere genes. Circ. Cardiovasc. Genet. 2010;3(3):138–146. doi: 10.1161/CIRCGENETICS.109.904698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Quiat D, Olson E N. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J. Clin. Invest. 2013;123(1):11–18. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jentzsch C, Leierseder S, Loyer X, Flohrschütz I, Sassi Y, Hartmann D, Thum T, Laggerbauer B, Engelhardt S. A phenotypic screen to identify hypertrophy-modulating microRNAs in primary cardiomyocytes. J. Mol. Cell. Cardiol. 2012;52(1):13–20. doi: 10.1016/j.yjmcc.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 96.Liu N, Olson E N. MicroRNA regulatory networks in cardiovascular development. Dev. Cell. 2010;18(4):510–525. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake L W, Doevendans P A, Mummery C L, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116(3):258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 98.Ferreira L R, Frade A F, Santos R H, Teixeira P C, Baron M A, Navarro I C, Benvenuti L A, Fiorelli A I, Bocchi E A, Stolf N A, Chevillard C, Kalil J, Cunha-Neto E. MicroRNAs miR-1, miR-133a, miR-133b, miR-208a and miR-208b are dysregulated in Chronic Chagas disease Cardiomyopathy. Int. J. Cardiol. 2014;175(3):409–417. doi: 10.1016/j.ijcard.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 99.Rau F, Freyermuth F, Fugier C, Villemin J P, Fischer M C, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, Wahbi K, Day J W, Fujimura H, Takahashi M P, Auboeuf D, Dreumont N, Furling D, Charlet-Berguerand N. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. at.Struct. Mol. Biol. 2011;18(7):840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- 100.Bostjancic E, Zidar N, Stajer D, Glavac D. MicroRNAs miR-1, miR133a, miR-133b and miR-208 are dsregulated in human myocardial infarction. Cardiology. 2010;115(3):163–169. doi: 10.1159/000268088. [DOI] [PubMed] [Google Scholar]
- 101.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Ciu H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg A P. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41(3):178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chamberlain A A, Lin M, Lister R L, Maslov A A, Wang Y, Suzuki M, Wu B, Greally J M, Zheng D, Zhou B. DNA methylation is developmentally regulated for genes essential for cardiogenesis. J. Am. Heart Assoc. 2014;3(3):e000976. doi: 10.1161/JAHA.114.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morgan D K, Whitelaw E. The case for transgenerational epigenetic inheritance in humans. Mamm. Genome. 2008;19(6):394–397. doi: 10.1007/s00335-008-9124-y. [DOI] [PubMed] [Google Scholar]
- 104.Chowdhury S, Erickson S W, Macleod S L, Cleves M A, Hu P, Karim M A, Hobbs C A. Maternal genome-wide DNA methylation patterns and congenital heart defects. PLoS One. 2011;6(1):e16506. doi: 10.1371/journal.pone.0016506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Makki N, Thiel K W, Miller F J ., Jr The epidermal growth factor receptor and its ligands in cardiovascular disease. nt. J. Mol. Sci. 2013;14(10):20597–20613. doi: 10.3390/ijms141020597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xin M, Olson E N, Bassel-Dulby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell. Biol. 2013;14(8):529–541. doi: 10.1038/nrm3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pikkarainen S, Tokola H, Kerkelä R, Ruskoaho H. GATA transcription factors in the developing an dadult heart. Cardiovasc. Res. 2004;63(3):196–207. doi: 10.1016/j.cardiores.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 108.Cohen E D, Miller M F, Wang Z, Moon R T, Morrisey E E. Wnt5a and Wnt11 are essential for second heart field progenitor development. Development. 2012;139(11):1931–1940. doi: 10.1242/dev.069377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sheng W, Wang H, Ma X, Qian Y, Zhang P, Wu Y, Zheng F, Chen L, Huang G, Ma D. LINE-1 methylation status and its association with tetralogy of fallot in infants. BMC Med. Genomics. 2012;5:20. doi: 10.1186/1755-8794-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sheng W, Qian Y, Wang H, Ma X, Zhang P, Diao L, An Q, Chen L, Ma D, Huang G. DNA methylation status of NKX2-5, GATA4 and HAND1 in patients with tetralogy of fallot. BMC Med. Genomics. 2013;6:46. doi: 10.1186/1755-8794-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sheng W, Qian Y, Zhang P, Wu Y, Wang H, Ma X, Chen L, Ma D, Huang G. Association of promoter methylation statuses of congenital heart defect genes with Tetralogy of Fallot. J. Translat. Med. 2014;12:31. doi: 10.1186/1479-5876-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kazazian Jr H H. Mobile elements: drivers of genome evolution. Science. 2004;303(5664):1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 113.Chowdhury S, Cleves M A, MacLeod S L, James S J, Zhao W, Hobbs C A. Maternal DNA hypomethylation and congenital hearts defects. Birth Defects Res. (Part A) 2011;91(3):69–76. doi: 10.1002/bdra.20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang T, Wang I, Wang F, Guan J, Le J, Wu L H, Zou J Z, Zhao H Z, Pei L J, Zheng X Y. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am. J. Clin. Nutr. 2010;91(5):1359–1367. doi: 10.3945/ajcn.2009.28858. [DOI] [PubMed] [Google Scholar]
- 115.Sheng W, Qian Y, Wang H, Ma X, Zhang P, Chen L, Ma D, Huang G. Association between mRNA levels of DNMT1, DNMT3A, DNMT3B, MBD2 and LINE-1 methylation status in infants with tetralogy of Fallot. Int. J. Mol. Med. 2013;32(3):694–702. doi: 10.3892/ijmm.2013.1427. [DOI] [PubMed] [Google Scholar]
- 116.Cao Y X, Jean JC, Williams M C. Cytosine methylation of an Sp1 site contributes to organ-specific and cell-specific regulation of expression of the lung epithelial gene t1alpha. Biochem J. 2000;350Pt3:883–890. [PMC free article] [PubMed] [Google Scholar]
- 117.Filikowski JN, Ilnytskyy Y, Tamminga J, Koturbash I, Golubov A, Bagnyukova T, Pogribny I P, Kovalchuk O. Hypomethylation and genome instability in the germline of exposed parents and their progeny is associated with altered miRNA expression. Carcinoge. 2010; 31(6):1110–1115. doi: 10.1093/carcin/bgp300. [DOI] [PubMed] [Google Scholar]
- 118.Rizwan m. WHO Preventing disease through healthy environments: toward an estimate of the environmental burden of disease. WHO Press Geneva. 2006:1–106. [Google Scholar]
- 119.Sethi TK, El-Gramry MN, Kloecker GH. Radon and lung cancer. Clin. Adv. Hematol. Oncol. 2012;10(3):157–164. [PubMed] [Google Scholar]
- 120.Lin R, Takahashi K, Karjalainen A, Hoshuyama T, Wilson D, Kameda T, Chan CC, Wen CP, Furuya S, Higashi T, Chien LC, Ohtaki M. Ecological associations between asbestos-related diseases and historical asbestos consumption: an international analysis. Lancet. 2012;369(9564):844–849. doi: 10.1016/S0140-6736(07)60412-7. [DOI] [PubMed] [Google Scholar]
- 121.Shaw GM, Nelson V, Iovannisci DM, Finnell RH, Lammer EJ. Maternal occupational chemical exposures and biotransformation genotypes as risk factors for selected congenital anomalies. Am. J. Epidemiol. 2003;157(6):475–484. doi: 10.1093/aje/kwg013. [DOI] [PubMed] [Google Scholar]
- 122.Wilson PD, Loffredo CA, Correa-Villasenor A, Ferencz C. Attributible fraction for cardiac malformation. m. J. Epidemiol. 1998;148(5):414–423. doi: 10.1093/oxfordjournals.aje.a009666. [DOI] [PubMed] [Google Scholar]
- 123.Loffredo CA, Silbergeld EK, Ferencz C, Zhang J. Association of transposition of the great arteries in infants with maternal exposures to herbicides and rodenticides. Am. J. Epidemiol. 2001;153(6):529–536. doi: 10.1093/aje/153.6.529. [DOI] [PubMed] [Google Scholar]
- 124.Tikkanen J, Heinonen OP. Risk factors for ventricular septal defects in Finland. Public Health. 1999;105(3):99–112. doi: 10.1016/s0033-3506(05)80283-5. [DOI] [PubMed] [Google Scholar]
- 125.Gilboa M, Mendola P, Olshan AF, Langlois PH, Savitz DA, Loomis D, Herring AH, Fixler DE. Relation between ambient air quality and selected birth defects, seven county study, Texas 1997 – 2000. 2005. Am. J. Epidemiol. 2005;162(3):238–252. doi: 10.1093/aje/kwi189. [DOI] [PubMed] [Google Scholar]
- 126.Sullivan PM, Dervan LA, Reiger S, Buddhe S, Schwartz SM. Risk of congenital heart defects in the offspring of smoking mothers: a population-based study. J. Pediatr. 2015;pii:S0022-3476(14):01117–-2. doi: 10.1016/j.jpeds.2014.11.042. [DOI] [PubMed] [Google Scholar]
- 127.Alverson CJ, Strickland MJ, Gilboa SM, Correa A. Maternal smoking and congenital heart defects in the Baltimore-Washington infant study. Pediatrics. 2011;127(3):e647–e653. doi: 10.1542/peds.2010-1399. [DOI] [PubMed] [Google Scholar]
- 128.Malik S, Cleves MA, Honein MA, Romitti PA, Botto LD, Yang S, Hobbs CA. National Birth Defects Prevention Study. Pediatrics. 2008;121(4):e810–e816. doi: 10.1542/peds.2007-1519. [DOI] [PubMed] [Google Scholar]
- 129.Lisowski LA, Verheijen PM, Copel JA, Kleinman CS, Wassink S, Visser GH, Meijboom EJ. Congenital heart disease in pregnancies complicated by maternal diabetes mellitus An international clinical collaboration, literature review, and meta-analysis. Herz. 2010;35(1):19–26. doi: 10.1007/s00059-010-3244-3. [DOI] [PubMed] [Google Scholar]
- 130.Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Waller DK, Reece EA. Diabetes mellitus and birth defects. Am. J. Obstet. Gynecol. 2008;199(3):e1–e9. doi: 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Simeone RM, Devine OJ, Marcinkevage JA, Gilboa SM, Razzaghi H, Bardenheier BH, Sharma AJ, Honein MA. Diabetes and congenital heart defects: a systematic review, meta-analysis, and modeling project. Am. J. Prev. Med. 2015;48(3):195–204. doi: 10.1016/j.amepre.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen J, Carney SA, Peterson RE, Heideman W. Comparative genomics identifies genes mediating cardiotoxicity in the embryonic zebrafish heart. Physiol. Genomics. 2008;33(3):148–58. doi: 10.1152/physiolgenomics.00214.2007. [DOI] [PubMed] [Google Scholar]
- 133.Lage K, Greenway SC, Rosenfeld JA, Wakimoto H, Gorham JM, Segrè AV, Roberts AE, Smoot LB, Pu WT, Pereira AC, Mesquita SM, Tommerup N, Brunak S, Ballif BC, Shaffer LG, Donahoe PK, Daly MJ, Seidman JG, Seidman CE, Larsen LA. Genetic and environmental risk factors in congenital heart disease functionally converge in protein networks driving heart development. Proc. Natl. Acad. Sci. USA. 2012;109(35):14035–14040. doi: 10.1073/pnas.1210730109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bauer RC, Laney AO, Smith R, Gerfen J, Morrissette JJ, Woyciechowski S, Garbarini J, Loomes KM, Krantz ID, Urban Z, Gelb BD, Goldmuntz E, Spinner NB. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat. 2010;31(5):594–601. doi: 10.1002/humu.21231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Smemo S, Campos L C, Moskowitz I P, Krieger J E, Pereira A C, Nobrega A M. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 2012;21(14):3255–3263. doi: 10.1093/hmg/dds165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.rizwan m. Pediatric Cardiac Genomics Consortium. The congenital heart disease genetic network study.Rationale, design, and early results. Circ. Res. 2013;112(4):698–706. doi: 10.1161/CIRCRESAHA.111.300297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Firth H V, Wright C F. DDD Study The Deciphering Developmental Disorders (DDD) study. Dev. Med. Child Neurol. 2011;53(8):702–703. doi: 10.1111/j.1469-8749.2011.04032.x. [DOI] [PubMed] [Google Scholar]
- 138.Muddyman D, Smee C, Griffin H, Kaye J. UK10K project Implementing a successful data-management framework: the UK10K managed access model. Genome Med. 2013;5(11):100. doi: 10.1186/gm504. [DOI] [PMC free article] [PubMed] [Google Scholar]