

Abstract
We have assessed copy number variation (CNV) in the male-specific part of the human Y chromosome discovered by array comparative genomic hybridization (array-CGH) in 411 apparently healthy UK males, and validated the findings using SNP genotype intensity data available for 149 of them. After manual curation taking account of the complex duplicated structure of Y-chromosomal sequences, we discovered 22 curated CNV events considered validated or likely, mean 0.93 (range 0–4) per individual. 16 of these were novel. Curated CNV events ranged in size from <1 kb to >3 Mb, and in frequency from 1/411 to 107/411. Of the 24 protein-coding genes or gene families tested, nine showed CNV. These included a large duplication encompassing the AMELY and TBL1Y genes that probably has no phenotypic effect, partial deletions of the TSPY cluster and AZFc region that may influence spermatogenesis, and other variants with unknown functional implications, including abundant variation in the number of RBMY genes and/or pseudogenes, and a novel complex duplication of two segments overlapping the AZFa region and including the 3′ end of the UTY gene.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-015-1562-5) contains supplementary material, which is available to authorized users.
Introduction
Copy number variation (CNV) in the human genome contributes to both normal and pathological variation (Freeman et al. 2006). The Y chromosome is the most highly enriched of the human chromosomes for CNV in the general population (Redon et al. 2006), yet studies of Y-CNVs have lagged behind studies of the rest of the genome. For example, the high-resolution hybridization-based survey of Conrad et al. (2010) examined only females, while the sequence-based genomic surveys of the 1000 Genomes Project reported a total of five deletions on the Y, all smaller than 3 kb (Mills et al. 2011; The 1000 Genomes Project Consortium 2012). Similarly, recent surveys of medically relevant CNVs have been limited to specific studies focussed on a small number of known CNVs (Rozen et al. 2012).
This paucity of recent studies contrasts with early work in the field. Early cytogenetic studies revealed that the copy number of the entire Y chromosome in viable individuals could vary from zero (45,X; Turner Syndrome) to four (49,XYYYY) with only moderate phenotypic consequences (Paoloni-Giacobino and Lespinasse 2007; Legro 2012), while abundant variation in length of the Yq heterochromatin and the occasional presence of Nucleolar Organizer Regions (the cytogenetic manifestation of translocations of ribosomal DNA) were detected in surveys of the general population, and transmission observed in families (Jobling 2008). Molecular analyses using pulsed-field gel electrophoresis confirmed the high levels of variation in the heterochromatin in the general population, where detectable differences in the constituent tandemly repeated sequences DYZ1 and DYZ2 were universal, and often found between father and son pairs, and in addition discovered variation in the centromeric alphoid satellite DYZ3 and the tandemly repeated gene TSPY within the DYZ5 array (Oakey and Tyler-Smith 1990; Mathias et al. 1994). Two minisatellites (Jobling et al. 1998; Bao et al. 2000), abundant microsatellites (Kayser et al. 2004) and some retroposon insertions (Hammer 1994; Santos et al. 2000) have been reported. Molecular studies surveying the copy number of Y-specific loci similarly discovered general population duplications and deletions of segments of the chromosome that could be hundreds of kilobases or megabases in size (Jobling et al. 1996; Santos et al. 1998; Saxena et al. 2000; Bosch and Jobling 2003; Fernandes et al. 2004; Repping et al. 2004; Murphy et al. 2007; Balaresque et al. 2008, 2009). Rare pathological CNVs have also been identified, including cytogenetically visible deletions associated with spermatogenetic failure (Tiepolo and Zuffardi 1976) and anomalies of sex determination (Disteche et al. 1986) and three distinct cytogenetically undetectable deletions leading to spermatogenetic failure (Vogt et al. 1996), as well as insertions causing hearing impairment (Wang et al. 2013). In addition, CNVs with milder medically relevant effects have been identified: the gr/gr deletion in the AZFc region of Yq (Repping et al. 2003; Machev et al. 2004) and low TSPY copy number (Giachini et al. 2009), which both slightly increase the risk of spermatogenetic failure, while deletions that remove AMELY have no apparent phenotypic consequences, but confound DNA-based sex tests in forensic analyses (Santos et al. 1998). Thus, early work and later targeted analyses documented a rich variety of CNVs on the Y chromosome.
Subsequently, a genome-wide survey of CNVs using hybridization to BAC arrays revealed both that high levels of CNV associated with the TSPY array, centromere and AZFc region were readily detectable at this level of resolution in HapMap samples with African, European and East Asian ancestry, and also that detectable CNV outside these regions was infrequent in these samples (Redon et al. 2006). A targeted survey of some of the most frequent CNVs known by 2006 (TSPY array, AZFc region and Yq heterochromatin) confirmed the high levels of variation and high mutation rates at these loci in samples chosen to represent diverse branches of the Y-chromosomal phylogeny (Repping et al. 2006). However, as mentioned, our understanding of CNV on the Y chromosome has not benefited from more recent advances in array comparative genomic hybridization (array-CGH) resolution or whole-genome sequencing, and thus lags behind other chromosomes. Further CNV surveys including, or focusing on, the Y chromosome are needed.
We have performed the most comprehensive survey of Y-CNVs in the UK population thus far, discovering Y-CNVs using exome-focused array-CGH and validating them in a subset of samples using SNP-chip genotyping. We report here the rediscovery of several known Y-CNVs, the discovery of many novel ones, and their population-genetic and predicted functional properties.
Materials and methods
Subjects
We studied 411 unrelated apparently healthy UK males drawn from the UK Blood Service controls and the Scottish Family Health Study.
Array-CGH
The array-CGH design, experimental procedures, QC, and CNV calling and merging have been described in detail elsewhere (The Deciphering Developmental Disorders Study 2015). Here, we briefly summarize the key features. The platform consisted of 2 × 1 M probe custom Agilent arrays (Amadid Nos. 031220/031221) with the probes targeted to (1) exons of protein-coding genes identified by GENCODE v17, with an average of two probes per exon and only 11 % of exons lacking probes, and (2) a genome-wide backbone with a median probe spacing of 5 kb. For chromosome Y, the platform contained a total of 6152 probes, covering 24 out of the 25 male-specific protein-coding genes/gene families (GENCODE v17). Probes in the X-transposed region and in the pseudoautosomal regions, which are not specific to the Y chromosome, were excluded from this analysis, so we did not call any CNVs from these regions. We were left with 5180 probes, of which 4974 (>96 %) are unique to the Y chromosome using the blastn program in the Blast + suite (http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs), with default parameters. So these probes are Y-specific by this criterion.
The reference sample used in all hybridizations was a pool of 500 males. CNVs were detected by CNsolidate with the default setting of a w-score threshold of 0.5 and the genome-wide false-positive (FPR) and false-negative (FNR) rates for CNsolidate raw calls were estimated by the DDD project (The Deciphering Developmental Disorders Study 2015). First, 73 technical replicates of the HapMap sample NA12878 were examined. True positives were defined as CNVs called in >80 % (i.e. 59) of the technical replicates, and CNVs were defined as the same if they shared greater than a 50 % reciprocal overlap. Overall, 12,634 true positives above the default w-score threshold of 0.5 were defined, 90 % of which (11,372) were classed as common and had been observed during previous studies at a population frequency >1 %. Using the default w-score threshold of 0.5 for CNV calls from CNsolidate resulted in a true-positive rate of 0.82 (FNR is <0.18), and a FPR of 0.052, across all replicates. Second, a custom designed 8 × 60 K Agilent CGH array was used to validate 9008 CNV calls, spanning the w-score range, detected by CNsolidate in 26 samples. Pearson correlation values of the mean log2 ratios across these samples between the discovery and validation arrays were used as the measure of truth. A clear 2-component distribution of correlation values was observed across all CNV calls. A nonparametric EM algorithm was used to determine the mixing proportions for each component and correlation values greater than the mean of the mixing proportions (0.5) were used to define true CNV calls. It showed that the proportion of true (validated) CNV calls was greater than 80 % for both losses and gains at the default 0.5 w-score cut-off for CNsolidate CNV calls.
Subsequently, additional manual curation was performed on the Y-CNVs to take account of the known repeated sequence content of the Y chromosome. Initially, rawCNVs in individual samples were merged into CNV events (CNVEs) when they overlapped, which we refer to as ‘rawCNVEs’. Then, since there is co-occurrence of some rawCNVEs in the same individual in this dataset, we merged some rawCNVEs into curated CNVEs ‘curCNVEs’. The logic was that if, for example, there are two copies of a related region in the reference sequence and most of the population, an individual with a deletion of one of these is expected to show a reduced signal at both locations, while an individual with a duplication will show an increased signal at both locations. The two locations will therefore show correlated signals in the population and do not represent independent events. We therefore examined the rawCNVEs for co-varying effects of this kind, and additional supporting evidence of sharing sequence homology. Several correlated signals were identified, and were noted to affect the CDY, DAZ, PRY, RBMY and TSPY genes; we discuss specific examples in the “Results and discussion” section. In two cases each found in a single individual, two unique rawCNVEs specific to that individual lay close together on the chromosome. The combination of occurrence of the rawCNVEs together in one individual and absence from all other individuals, together with physical proximity, suggested that they were likely to result from a single complex mutational event. These examples (curCNVE8 and curCNVE14) are also discussed further in the “Results and discussion”. Thus, taking all these factors into account, a set of curated CNVEs (curCNVEs) was created and their sizes were refined by manual inspection of probe intensity plots.
SNP-chip genotyping
The SNP genotyping platform and procedures have also been described in detail elsewhere (The Deciphering Developmental Disorders Study 2015). In brief, a customized Illumina Omni-one quad chip was used containing 811,844 mapped and 1734 unmapped markers, with a median intermarker distance of 2378 bp (SangerDDD_OmniExPlusv1_15019773_A). For chromosome Y, the platform contained a total of 1681 probes, covering 12 out of 25 protein-coding genes/gene families (GENCODE v17). Log R ratios (LRR) were used in the current study. Data were available for 149 of the 411 individuals.
Y haplogroup assignment
Y haplogroups were identified from allele calls at standard diagnostic Y-SNPs (Karafet et al. 2008) that were present on the SNP-chip array, as well as other informative markers (Fig. 4, Figure S2; Table S2), and were thus available for 149 individuals. Haplogroups were considered at a maximum phylogenetic resolution of the trinomial level, e.g. R1b. A full Y phylogeny as defined by all markers typed was constructed (Figure S2).
Fig. 4.

Haplogroup distribution of curCNVEs present in more than one individual. Left phylogeny of the Y-chromosomal haplogroups detected in 149 samples; branch lengths are arbitrary. Blue circles haplogroup frequency in the entire 149 individuals, with circle area proportional to frequency. Remaining circles haplogroup frequencies in individual curCNVEs
Results and discussion
CNV calling, validation and curation
Y-chromosomal array-CGH data were analysed from 411 UK males, representative of the general UK population. RawCNVs were called by CNsolidate and combined into rawCNVEs as described in the "Materials and methods" section. In addition, SNP genotype data were available for 149 of the participants, and so when a SNP overlapped with a rawCNVE call, the SNP intensity data could be used to assess whether or not there was independent support for the CNVE call.
We took a three-stage approach to evaluating the starting rawCNVE calls. First, we used the comparison of array-CGH probe intensity with SNP intensity, together with manual examination of the two datasets by two independent individuals, to identify a set of validated rawCNVEs, where the array-CGH calls were supported by the SNP data. The two most ‘borderline’ examples, which illustrate the procedure, were curCNVE7 and curCNVE12 (Fig. 1). curCNVE7 was considered validated because it had strong support from two SNPs, which showed the highest SNP intensity values in the 1 Mb window illustrated. For curCNVE12, where the array-CGH signal was very strong, a single SNP showing the highest SNP intensity value in the 400 kb window illustrated was considered sufficient to validate it. Second, we applied the array-CGH probe intensity criteria established in the first stage to the remaining rawCNVE calls, which did not have SNP intensity data, again with manual examination, to identify a set of likely rawCNVEs. Third, we used literature data to determine whether or not the validated and likely rawCNVEs were supported by previous work, and also whether known common CNVs were being called.
Fig. 1.

Validated examples of CNVs based on both CNV probe and SNP intensity data. Top panel male-specific euchromatic region of the Y chromosome derived from the UCSC genome browser showing gaps in the reference sequence (black bars), chromosomal regions (yellow Y-specific unique, red X–Y transposed, blue Y-specific repeated, purple heterochromatic), segmental duplications and the CNVEs illustrated in the rest of the figure. Remaining panels paired probe intensity (blue) and SNP intensity (red) plots for seven curCNVEs. Each rawCNVE is indicated by buff shading, chromosomal coordinates in Mb are shown at the bottom and overlapping protein-coding genes within the plot regions are included below the SNP intensities
Manual curation of the rawCNVE calls in the 149 individuals with both array-CGH and SNP data identified a set of calls with strong evidence for variation in copy number, and examples of these validated rawCNVEs are shown in Fig. 1. All are >7 kb in size, as expected from the requirement to contain both multiple array and SNP probes. Similar curation using the array probe intensity data alone in these and the remaining 262 individuals identified additional CNVs in regions that did not overlap with SNPs, or in the individuals without SNP data. Examples of these likely rawCNVEs are shown in Fig. 2. They included some smaller CNVEs, such as rawCNVE5.2 where three probes lay within 165 bp (Fig. 2d, f). During curation, we combined some of the rawCNVEs into curCNVEs. In deciding whether or not rawCNVEs should be combined, we considered correlations between probe intensity signals in different individuals to determine whether multiple individual rawCNVEs co-vary in the population: for example, when one rawCNVE shows a log2 ratio increase in a particular individual, do other rawCNVEs show this pattern as well; and similarly when one shows a decrease? If they did this consistently, they were combined in the same curCNVE. Additional supporting information taken into account was whether or not all co-varying CNVs were known to share sequence homology (as for the shared RBMY elements of curCNVE16, Fig. 2) or if the co-varying CNVs lay close together on the chromosome, so that a single mutational event could plausibly have affected them all. For example, rawCNVEs 5.1, 5.2 and 5.3 all showed decreased signal indicating a decrease in copy number in one individual (Fig. 2d), and increased signal and copy number in a different individual (Fig. 2f). A similar pattern was seen for rawCNVEs 16.1, 16.2, 16.3 and 16.4 (Fig. 2e, g). These rawCNVEs are therefore each likely to represent a single event where probes cross-hybridize, as the (moderately) repeated regions show the copy number change, while the unique regions do not. We therefore conclude that in cases like these, a single copy number change in a TSPY-related sequence (curCNVE5) or an RBMY gene (curCNVE16) could generate the observed signal because of cross-hybridization.
Fig. 2.

Likely examples of CNVs based on CNV probe intensity data. a Male-specific euchromatic region of the Y chromosome derived from the UCSC genome browser showing gaps in the reference sequence (black bars), chromosomal regions (yellow Y-specific unique, red X–Y transposed, blue Y-specific repeated, purple heterochromatic), segmental duplications and the CNVEs illustrated in the rest of the figure (green duplications, orange deletions, purple both). b curCNVE1. c curCNVEs 10 and 11. d and f curCNVE5 in two different individuals showing the coordinated decrease or increase of rawCNVEs 5.1, 5.2 and 5.3. e and g similar plots for curCNVE16 and its corresponding rawCNVEs. Each rawCNVE is indicated by buff shading, chromosomal coordinates in Mb are shown at the bottom and overlapping protein-coding genes are plotted at the bottom of b and c, f and g
In addition, some rawCNVEs were detected in single individuals, lay close together in the genome and showed similar changes: rawCNVE8.1 and rawCNVE8.2 (Fig. 1), and rawCNVE14.1 and 14.2 (Fig. 1). The two distinct rawCNVEs in both cases behaved in the same way in the population (duplication one individual, no change in the rest of the individuals investigated); also, in both cases, the two regions lay within 1 Mb on the chromosome. In cases like the four discussed, we considered the rawCNVEs as likely to reflect the same mutational event, and in a second round of curation grouped them together as curCNVEs: 5, 16, 8 and 14, respectively. When nearby rawCNVEs in the same single individual showed contrasting signals, such as the deletion at rawCNVE10 and duplication at rawCNVE11 (Fig. 2c), we did not group them, and retained them as curCNVE10 and curCNVE11, respectively.
381 raw Y-specific CNVE calls from 185 individuals were accepted at 34 rawCNVE loci, an average of 0.93 per individual (range 0–4). No rawCNVE was accepted in 226 individuals: 36 had no raw calls at all on the Y, 10 had raw calls only outside the MSY region, and 180 individuals with raw calls in MSY region did not pass the manual check. Overall, the raw calls were consolidated into 22 curCNVE loci. A single curCNVE could include both duplications and deletions of a particular region. The full call set is shown in Table S1 and examples of each are illustrated in Supplementary Fig. 1.
General characteristics of validated and likely CNVEs
The sizes of rawCNVEs ranged from <1 kb to >3 Mb [Table 1; Fig. 3a; the mean size was 309 kb (median 72 kb)]. These large sizes reflect the low probe and SNP densities, and the need for a signal at multiple probes/SNPs to make confident calls; since curCNVEs can be discontinuous, their summed sizes are not easily interpreted and we do not consider them. Frequencies ranged from 1/411 (0.24 %) to 107/411 (26.0 %) (Fig. 3b). More than half (12/22, ~55 %) were observed in just one individual, but six were called in more than 5 % (Fig. 3b). Among the 381 curCNVE calls across all samples, deletions (240) outnumbered duplications (141), a statistic dominated by the 76 deletions at curCNVE16 and 68 deletions at curCNVE5 (Table 1). Six of the curated CNVEs have been reported previously and the remaining 16 are novel.
Table 1.
Summary of 34 rawCNVEs and 22 curCNVEs called in this study
| curCNVE | rawCNVE | rawCNVE | curCNVE | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Size (bp) | SNP support | Known | Duplications | Deletions | Total frequency | Protein-coding gene contenta | ||
| curCNVE1 | rawCNVE1 | 2,891,036 | 2,903,671 | 12,635 | 1 | 0.0024 | ||||
| curCNVE2 | rawCNVE2 | 6,138,072 | 9,161,980 | 3,023,908 | + | Murphy et al. (2007) | 1 | 0.0024 | AMELY, TBL1Y | |
| curCNVE3 | rawCNVE3 | 7,659,321 | 8,497,398 | 838,077 | 1 | 0.0024 | ||||
| curCNVE4 | rawCNVE4 | 9,170,730 | 9,175,364 | 4634 | 53 | 11 | 0.1557 | TSPY4 | ||
| curCNVE5 | rawCNVE5.1 | 9,196,977 | 9,198,235 | 1258 | 8 | 68 | 0.1849 | TSPY8 | ||
| curCNVE5 | rawCNVE5.2 | 9,311,600 | 9,311,765 | 165 | ||||||
| curCNVE5 | rawCNVE5.3 | 9,383,079 | 9,384,475 | 1396 | ||||||
| curCNVE6 | rawCNVE6 | 9,196,977 | 9,382,943 | 185,966 | Oakey and Tyler-Smith (1990) | 2 | 29 | 0.0754 | TSPY cluster | |
| curCNVE7 | rawCNVE7 | 9,785,127 | 9,792,677 | 7550 | + | 1 | 0.0024 | |||
| curCNVE8 | rawCNVE8.1 | 14,588,389 | 14,745,226 | 156,837 | + | 1 | 0.0024 | |||
| curCNVE8 | rawCNVE8.2 | 15,034,145 | 15,475,430 | 441,285 | + | UTY | ||||
| curCNVE9 | rawCNVE9 | 15,144,435 | 15,146,222 | 1787 | 1 | 0.0024 | ||||
| curCNVE10 | rawCNVE10 | 15,869,445 | 16,096,260 | 226,815 | 1 | 0.0024 | ||||
| curCNVE11 | rawCNVE11 | 16,170,165 | 16,233,113 | 62,948 | 1 | 0.0024 | ||||
| curCNVE12 | rawCNVE12 | 18,733,053 | 18,762,614 | 29,561 | + | 1 | 0.0024 | |||
| curCNVE13 | rawCNVE13 | 21,032,549 | 21,074,621 | 42,072 | + | 1 | 0.0024 | |||
| curCNVE14 | rawCNVE14.1 | 23,441,081 | 23,649,415 | 208,334 | + | 1 | 0.0024 | |||
| curCNVE14 | rawCNVE14.2 | 23,756,420 | 24,005,801 | 249,381 | + | |||||
| curCNVE15 | rawCNVE15 | 24,218,723 | 24,218,783 | 60 | 18 | 1 | 0.0462 | PRY2 | ||
| curCNVE16 | rawCNVE16.1 | 23,660,808 | 23,709,077 | 48,269 | 31 | 76 | 0.2603 | RBMY1B, RBMY1A1 | ||
| curCNVE16 | rawCNVE16.2 | 24,005,497 | 24,062,091 | 56,594 | RBMY1D, RBMY1E | |||||
| curCNVE16 | rawCNVE16.3 | 24,316,281 | 24,327,019 | 10,738 | RBMY1F | |||||
| curCNVE16 | rawCNVE16.4 | 24,551,695 | 24,562,435 | 10,740 | RBMY1J | |||||
| curCNVE17 | rawCNVE17.1 | 24,551,695 | 24,658,825 | 107,130 | 4 | 19 | 0.0560 | RBMY1J, PRY | ||
| curCNVE17 | rawCNVE17.2 | 24,551,695 | 24,795,554 | 243,859 | RBMY1J, PRY | |||||
| curCNVE18 | rawCNVE18 | 25,130,433 | 27,895,495 | 2,765,062 | Fernandes et al. (2004) | 8 | 17 | 0.0608 | BPY2, DAZ1, DAZ2, CDY1B, BPY2B, DAZ3, DAZ4, BPY2C, CDY1 | |
| curCNVE19 | rawCNVE19 | 24,658,743 | 25,428,575 | 769,832 | Repping et al. (2003) | 2 | 9 | 0.0268 | BPY2, DAZ1, DAZ2 | |
| curCNVE20 | rawCNVE20.1 | 25,284,428 | 25,428,580 | 144,152 | Saxena et al. (2000) | 7 | 6 | 0.0316 | DAZ1, DAZ2 | |
| curCNVE20 | rawCNVE20.2 | 26,950,819 | 27,177,168 | 226,349 | DAZ3, DAZ4 | |||||
| curCNVE21 | rawCNVE21.1 | 25,829,578 | 26,194,226 | 364,648 | Machev et al. (2004) | 1 | 1 | 0.0049 | CDY1B | |
| curCNVE21 | rawCNVE21.2 | 27,768,203 | 27,768,295 | 92 | CDY1 | |||||
| curCNVE22 | rawCNVE22.1 | 28,472,070 | 28,654,473 | 182,403 | + | 1 | 0.0024 | |||
| curCNVE22 | rawCNVE22.2 | 28,688,829 | 28,704,081 | 15,252 | + | |||||
| curCNVE22 | rawCNVE22.3 | 28,723,589 | 28,804,541 | 80,952 | + | |||||
Genome coordinates are based on GRCh37/hg19. Gene names are from GENCODE v20
aGenes showing CNV. For genes that are members of families, the copy that is actually duplicated or deleted is unknown because of shadowing effects
Fig. 3.
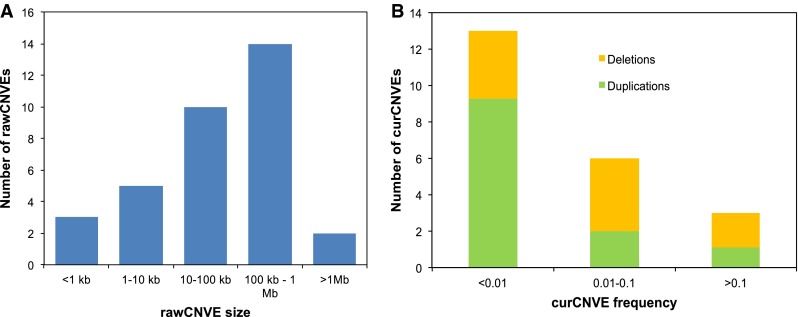
Size and frequency distribution of validated and likely CNVs in 238 individuals. a rawCNVE size distribution. b curCNVE frequency distribution; each frequency bar is coloured according to the proportion of duplications (green) and deletions (orange) among the total 381 curCNVE calls
Distribution of curCNVEs among Y haplogroups
The absence of recombination in the male-specific portion of the Y chromosome results in a simple phylogenetic tree that can be defined by Y-SNPs (Jobling and Tyler-Smith 2003; Wei et al. 2013). With the SNPs available in this study, we could assign all 149 samples with SNP genotype data to trinomial level haplogroups, and the haplogroup distribution is as expected in the UK population (Fig. 4; Table S1; Figure S2; Table S2) (Capelli et al. 2003). curCNVEs can then be placed on the known tree, and a minimal number of mutational events leading to each curCNVE can be deduced. curCNVEs confined to a single haplogroup, or cluster of phylogenetically related haplogroups, can be most parsimoniously explained by a single mutational event, while curCNVEs dispersed among unrelated haplogroups require multiple mutational events.
Applying this reasoning to the 10 curCNVEs present in more than one individual, involving 62 of the 149 samples (Fig. 4), shows that all require multiple mutations to explain their phylogenetic distribution, a conclusion reinforced by the observation that both duplications and deletions were called at all of these 10 loci (Table 1), although haplogroup assignments were not available in all cases (Fig. 4).
Biological impact of curCNVEs
The Y chromosome codes for 25 male-specific proteins, and 24 of these are covered by probes on the CGH array. The 22 curCNVEs together overlap with genes that code for nine of these proteins (AMELY, TBL1Y, TSPY, UTY, CDY, RBMY, PRY, BPY2 and DAZ), with the caveat that due to shadowing effects (where there are multiple copies of these genes or pseudogenes in the reference sequence), we cannot always be sure whether the copy number variation affects the functional gene(s) or non-functional pseudogenes(s).
Since duplication and deletion of the AMELY/TBL1Y (Santos et al. 1998; Murphy et al. 2007), TSPY (Oakey and Tyler-Smith 1990; Mathias et al. 1994; Repping et al. 2006) and the Yq region containing the CDY, BPY2 and DAZ genes (Jobling et al. 1996; Repping et al. 2003, 2004, 2006; Fernandes et al. 2004) have been extensively documented in the literature, and can have subtle biological consequences (Repping et al. 2003; Machev et al. 2004; Giachini et al. 2009), we focus here on the remaining UTY, RBMY and PRY genes.
Deletion of a region of the Y chromosome between 14,434,311 and 15,228,218 carrying the USP9Y and DDX3Y genes (the AZFa region) leads to azoospermia (Tyler-Smith and Krausz 2009), but duplication of the same region is present in the general population and compatible with male fertility (Bosch and Jobling 2003). Partial AZFa deletions have consequences that range from azoospermia to normozoospermia, but partial duplications have not previously been reported. curCNVE8, found in a single individual, consists of duplications of two discontinuous regions overlapping with AZFa although these do not include USP9Y or DDX3Y. curCNVE8 does, however, extend beyond the distal boundary of AZFa and duplicate the 3′ end of the UTY gene (Fig. 5). UTY is a histone demethylase (Walport et al. 2014) but this partially duplicated copy seems unlikely to be expressed and should be considered a variant of unknown significance.
Fig. 5.

Novel partial duplication of UTY. curCNVE8 showing the relationship with the AZFa deletion and the protein-coding genes in the region
RBMY and PRY both form multicopy gene families on the Y chromosome containing pseudogenes as well as six and two active genes, respectively. curCNVEs 14–17 include members of these families, and curCNVE16 containing RBMY genes (Fig. 2e, g) is the most common CNV detected (Table 1). However, because the CNV analysis does not distinguish between genes and pseudogenes, and there is an RBMY pseudogene located at 9,148,467–9,162,451 (which contains only a single probe and thus does not permit reliable CNV measurement), the biological implication of the variation detected remains uncertain.
The DDD project has carried out false-positive and false-negative rate assessments using both technical replicates and custom designed array validation (see “Materials and methods”), which showed that the FNR is <20 % and FPR is about 5 %. However, these measurements have only limited application to our study, as we did not use the automatic calls in their raw state as our final call set. Instead, we manually examined all the rawCNV calls one by one, a procedure which we regard as gold standard. We also took into account the complication of repeated Y chromosome structures, and the evidence for the 22 curCNVEs we accepted was compelling. So the false-positive rate among our curCNVEs is likely to be even lower (Figs. 1, 2), perhaps zero. In contrast, the false-negative rate is unknown but likely to be high. This is an inevitable consequence of the limited probe coverage. Mills et al. (2011) showed that genome-wide numbers of CNVs increase as size decreases, at least down to the 100 bp resolution limit of their analysis. Since we have essentially no power to discover CNVs of 100 bp, we must be missing a lot of small ones from our call set. We also potentially miss CNVs in sequences present on some Y chromosomes but absent from haplogroup R1b, since the array-CGH probes were designed based on the reference sequence, which is mostly derived from an R1b chromosome. Even with the current ‘next generation’ sequencing technologies, which still rely on mapping reads to the reference sequence, we would not detect such regions even if we sequenced the whole Y chromosome. However, with third generation sequencing technologies with much longer reads combined with de novo assembly, future work may discover such new sequences, not only on the Y chromosome, but also in the whole genome. Our approach of discovering CNVs by array-CGH limits the precision with which the endpoints can be determined and alternative methods, such as sequenced-based ones (Mills et al. 2011) need to be used for this.
Despite a number of limitations, CNVs affecting protein-coding genes have been effectively discovered in this study, and indeed the well-known common CNVs involving TSPY and the gr/gr and b1/b3 regions expected to be present were all detected. Because of the limited phenotypes associated with complete loss or duplication of the entire Y chromosome, the chance of actionable incidental findings from Y studies is low, but some of the variants discovered have potential implications for spermatogenesis.
Conclusions
We have analysed the distribution of Y-chromosomal CNVs in apparently healthy UK males. Although there are limitations to our dataset, including low sensitivity to small events and a resulting bias towards detecting large CNVs, we show that Y-CNVs can readily be detected. We confirm the abundance of this form of variation on the Y chromosome, where over 6 Mb of sequence is copy number variable and affects over one-third of the male-specific Y proteins. Novel CNVs, both common and rare, continue to be discovered and some of these may have implications for phenotypes, especially spermatogenesis.
Electronic supplementary material
Acknowledgments
We thank the families for their participation, the DDD Study for providing the data, the UK National Blood Service and the Generation Scotland: Scottish Family Health Study for access to DNA from controls. Generation Scotland has received core funding from the Chief Scientist Office of the Scottish Government Health Directorates CZD/16/6 and the Scottish Funding Council HR03006. The Deciphering Developmental Disorders study presents independent research commissioned by the Health Innovation Challenge Fund (Grant Number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (Grant Number WT098051), which also supported the further study of the Y-CNVs.
Footnotes
W. Wei and T. Fitzgerald have equally contributed to this study.
References
- Balaresque P, Bowden GR, Parkin EJ, Omran GA, Heyer E, Quintana-Murci L, Roewer L, Stoneking M, Nasidze I, Carvalho-Silva DR, Tyler-Smith C, de Knijff P, Jobling MA. Dynamic nature of the proximal AZFc region of the human Y chromosome: multiple independent deletion and duplication events revealed by microsatellite analysis. Hum Mutat. 2008;29:1171–1180. doi: 10.1002/humu.20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaresque P, Parkin EJ, Roewer L, Carvalho-Silva DR, Mitchell RJ, van Oorschot RA, Henke J, Stoneking M, Nasidze I, Wetton J, de Knijff P, Tyler-Smith C, Jobling MA. Genomic complexity of the Y-STR DYS19: inversions, deletions and founder lineages carrying duplications. Int J Legal Med. 2009;123:15–23. doi: 10.1007/s00414-008-0253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Zhu S, Pandya A, Zerjal T, Xu J, Shu Q, Du R, Yang H, Tyler-Smith C. MSY2: a slowly evolving minisatellite on the human Y chromosome which provides a useful polymorphic marker in Chinese populations. Gene. 2000;244:29–33. doi: 10.1016/S0378-1119(00)00021-4. [DOI] [PubMed] [Google Scholar]
- Bosch E, Jobling MA. Duplications of the AZFa region of the human Y chromosome are mediated by homologous recombination between HERVs and are compatible with male fertility. Hum Mol Genet. 2003;12:341–347. doi: 10.1093/hmg/ddg031. [DOI] [PubMed] [Google Scholar]
- Capelli C, Redhead N, Abernethy JK, Gratrix F, Wilson JF, Moen T, Hervig T, Richards M, Stumpf MP, Underhill PA, Bradshaw P, Shaha A, Thomas MG, Bradman N, Goldstein DB. A Y chromosome census of the British Isles. Curr Biol. 2003;13:979–984. doi: 10.1016/S0960-9822(03)00373-7. [DOI] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche CM, Casanova M, Saal H, Friedman C, Sybert V, Graham J, Thuline H, Page DC, Fellous M. Small deletions of the short arm of the Y chromosome in 46, XY females. Proc Natl Acad Sci USA. 1986;83:7841–7844. doi: 10.1073/pnas.83.20.7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes S, Paracchini S, Meyer LH, Floridia G, Tyler-Smith C, Vogt PH. A large AZFc deletion removes DAZ3/DAZ4 and nearby genes from men in Y haplogroup N. Am J Hum Genet. 2004;74:180–187. doi: 10.1086/381132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, Carter NP, Scherer SW, Lee C. Copy number variation: new insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- Giachini C, Nuti F, Turner DJ, Laface I, Xue Y, Daguin F, Forti G, Tyler-Smith C, Krausz C. TSPY1 copy number variation influences spermatogenesis and shows differences among Y lineages. J Clin Endocrinol Metab. 2009;94:4016–4022. doi: 10.1210/jc.2009-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer MF. A recent insertion of an Alu element on the Y chromosome is a useful marker for human population studies. Mol Biol Evol. 1994;11:749–761. doi: 10.1093/oxfordjournals.molbev.a040155. [DOI] [PubMed] [Google Scholar]
- Jobling MA. Copy number variation on the human Y chromosome. Cytogenet Genome Res. 2008;123:253–262. doi: 10.1159/000184715. [DOI] [PubMed] [Google Scholar]
- Jobling MA, Tyler-Smith C. The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet. 2003;4:598–612. doi: 10.1038/nrg1124. [DOI] [PubMed] [Google Scholar]
- Jobling MA, Samara V, Pandya A, Fretwell N, Bernasconi B, Mitchell RJ, Gerelsaikhan T, Dashnyam B, Sajantila A, Salo PJ, Nakahori Y, Disteche CM, Thangaraj K, Singh L, Crawford MH, Tyler-Smith C. Recurrent duplication and deletion polymorphisms on the long arm of the Y chromosome in normal males. Hum Mol Genet. 1996;5:1767–1775. doi: 10.1093/hmg/5.11.1767. [DOI] [PubMed] [Google Scholar]
- Jobling MA, Bouzekri N, Taylor PG. Hypervariable digital DNA codes for human paternal lineages: MVR-PCR at the Y-specific minisatellite, MSY1 (DYF155S1) Hum Mol Genet. 1998;7:643–653. doi: 10.1093/hmg/7.4.643. [DOI] [PubMed] [Google Scholar]
- Karafet TM, Mendez FL, Meilerman MB, Underhill PA, Zegura SL, Hammer MF. New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree. Genome Res. 2008;18:830–838. doi: 10.1101/gr.7172008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser M, Kittler R, Erler A, Hedman M, Lee AC, Mohyuddin A, Mehdi SQ, Rosser Z, Stoneking M, Jobling MA, Sajantila A, Tyler-Smith C. A comprehensive survey of human Y-chromosomal microsatellites. Am J Hum Genet. 2004;74:1183–1197. doi: 10.1086/421531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legro RS. Turner syndrome: new insights into an old disorder. Fertil Steril. 2012;98:773–774. doi: 10.1016/j.fertnstert.2012.07.1138. [DOI] [PubMed] [Google Scholar]
- Machev N, Saut N, Longepied G, Terriou P, Navarro A, Levy N, Guichaoua M, Metzler-Guillemain C, Collignon P, Frances AM, Belougne J, Clemente E, Chiaroni J, Chevillard C, Durand C, Ducourneau A, Pech N, McElreavey K, Mattei MG, Mitchell MJ. Sequence family variant loss from the AZFc interval of the human Y chromosome, but not gene copy loss, is strongly associated with male infertility. J Med Genet. 2004;41:814–825. doi: 10.1136/jmg.2004.022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias N, Bayes M, Tyler-Smith C. Highly informative compound haplotypes for the human Y chromosome. Hum Mol Genet. 1994;3:115–123. doi: 10.1093/hmg/3.1.115. [DOI] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, Chinwalla A, Conrad DF, Fu Y, Grubert F, Hajirasouliha I, Hormozdiari F, Iakoucheva LM, Iqbal Z, Kang S, Kidd JM, Konkel MK, Korn J, Khurana E, Kural D, Lam HY, Leng J, Li R, Li Y, Lin CY, Luo R, Mu XJ, Nemesh J, Peckham HE, Rausch T, Scally A, Shi X, Stromberg MP, Stutz AM, Urban AE, Walker JA, Wu J, Zhang Y, Zhang ZD, Batzer MA, Ding L, Marth GT, McVean G, Sebat J, Snyder M, Wang J, Eichler EE, Gerstein MB, Hurles ME, Lee C, McCarroll SA, Korbel JO. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KM, Cohen JS, Goodrich A, Long PP, Griffin CA. Constitutional duplication of a region of chromosome Yp encoding AMELY, PRKY, and TBL1Y: implications for sex chromosome analysis and bone marrow engraftment analysis. J Mol Diagn. 2007;9:408–413. doi: 10.2353/jmoldx.2007.060198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakey R, Tyler-Smith C. Y chromosome DNA haplotyping suggests that most European and Asian men are descended from one of two males. Genomics. 1990;7:325–330. doi: 10.1016/0888-7543(90)90165-Q. [DOI] [PubMed] [Google Scholar]
- Paoloni-Giacobino A, Lespinasse J. Chromosome Y polysomy: a non-mosaic 49,XYYYY case. Clin Dysmorphol. 2007;16:65–66. doi: 10.1097/01.mcd.0000228423.04908.0c. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repping S, Skaletsky H, Brown L, van Daalen SK, Korver CM, Pyntikova T, Kuroda-Kawaguchi T, de Vries JW, Oates RD, Silber S, van der Veen F, Page DC, Rozen S. Polymorphism for a 1.6-Mb deletion of the human Y chromosome persists through balance between recurrent mutation and haploid selection. Nat Genet. 2003;35:247–251. doi: 10.1038/ng1250. [DOI] [PubMed] [Google Scholar]
- Repping S, van Daalen SK, Korver CM, Brown LG, Marszalek JD, Gianotten J, Oates RD, Silber S, van der Veen F, Page DC, Rozen S. A family of human Y chromosomes has dispersed throughout northern Eurasia despite a 1.8-Mb deletion in the azoospermia factor c region. Genomics. 2004;83:1046–1052. doi: 10.1016/j.ygeno.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Repping S, van Daalen SK, Brown LG, Korver CM, Lange J, Marszalek JD, Pyntikova T, van der Veen F, Skaletsky H, Page DC, Rozen S. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat Genet. 2006;38:463–467. doi: 10.1038/ng1754. [DOI] [PubMed] [Google Scholar]
- Rozen SG, Marszalek JD, Irenze K, Skaletsky H, Brown LG, Oates RD, Silber SJ, Ardlie K, Page DC. AZFc deletions and spermatogenic failure: a population-based survey of 20,000 Y chromosomes. Am J Hum Genet. 2012;91:890–896. doi: 10.1016/j.ajhg.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos FR, Pandya A, Tyler-Smith C. Reliability of DNA-based sex tests. Nat Genet. 1998;18:103. doi: 10.1038/ng0298-103. [DOI] [PubMed] [Google Scholar]
- Santos FR, Pandya A, Kayser M, Mitchell RJ, Liu A, Singh L, Destro-Bisol G, Novelletto A, Qamar R, Mehdi SQ, Adhikari R, de Knijff P, Tyler-Smith C. A polymorphic L1 retroposon insertion in the centromere of the human Y chromosome. Hum Mol Genet. 2000;9:421–430. doi: 10.1093/hmg/9.3.421. [DOI] [PubMed] [Google Scholar]
- Saxena R, de Vries JW, Repping S, Alagappan RK, Skaletsky H, Brown LG, Ma P, Chen E, Hoovers JM, Page DC. Four DAZ genes in two clusters found in the AZFc region of the human Y chromosome. Genomics. 2000;67:256–267. doi: 10.1006/geno.2000.6260. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiepolo L, Zuffardi O. Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human Y chromosome long arm. Hum Genet. 1976;34:119–124. doi: 10.1007/BF00278879. [DOI] [PubMed] [Google Scholar]
- Tyler-Smith C, Krausz C. The will-o’-the-wisp of genetics—hunting for the azoospermia factor gene. N Engl J Med. 2009;360:925–927. doi: 10.1056/NEJMe0900301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt PH, Edelmann A, Kirsch S, Henegariu O, Hirschmann P, Kiesewetter F, Kohn FM, Schill WB, Farah S, Ramos C, Hartmann M, Hartschuh W, Meschede D, Behre HM, Castel A, Nieschlag E, Weidner W, Grone HJ, Jung A, Engel W, Haidl G. Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet. 1996;5:933–943. doi: 10.1093/hmg/5.7.933. [DOI] [PubMed] [Google Scholar]
- Walport LJ, Hopkinson RJ, Vollmar M, Madden SK, Gileadi C, Oppermann U, Schofield CJ, Johansson C. Human UTY(KDM6C) is a male-specific N-methyl lysyl demethylase. J Biol Chem. 2014;289:18302–18313. doi: 10.1074/jbc.M114.555052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xue Y, Zhang Y, Long Q, Yang F, Turner DJ, Fitzgerald T, Ng BL, Zhao Y, Chen Y, Liu Q, Yang W, Han D, Quail MA, Swerdlow H, Burton J, Fahey C, Ning Z, Hurles ME, Carter NP, Yang H, Tyler-Smith C. Genetic basis of Y-linked hearing impairment. Am J Hum Genet. 2013;92:301–306. doi: 10.1016/j.ajhg.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ayub Q, Chen Y, McCarthy S, Hou Y, Carbone I, Xue Y, Tyler-Smith C. A calibrated human Y-chromosomal phylogeny based on resequencing. Genome Res. 2013;23:388–395. doi: 10.1101/gr.143198.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.