

Atmospheric record in Antarctic blue ice

Waves of blue ice on a cloudy day in the Allan Hills blue ice area, Antarctica. Image courtesy of Mike Waszkiewicz and John A. Higgins.
Ancient atmospheric CO2 and methane (CH4) concentrations, reconstructed from air bubbles in ice cores, link global greenhouse gas concentrations to a 100,000-year glacial cycle dating back 800,000 years. However, deep-sea climate records reveal that this cycle developed only 900,000 years ago, and that glacial cycles prior to this time lasted on average 40,000 years. To better understand both cycles as well as the transition, John Higgins et al. (pp. 6887–6891) reconstructed atmospheric CO2 and CH4 concentrations from roughly 1 million years ago, using a shallow ice core drilled in the Allan Hills blue ice area in Antarctica. Antarctic blue ice forms in areas where mountains force ice to flow upward and strong winds persistently scour material from the surface, allowing the oldest ice at the bottom to rise to the top. Noting that records obtained from blue ice are stratigraphically complex and likely incomplete, the authors show that the amplitude of glacial cycles was lower 1 million years ago, climates were warmer, and peak interglacial CO2 concentrations were higher than any glacial cycle between 400,000 and 800,000 years ago. The findings demonstrate that CO2 and Antarctic temperatures were linked during the warm world of the Mid-Pleistocene, according to the authors. — T.J.
Wheat yield and global warming

Wheat growing in Kansas. Image courtesy of the US Department of Agriculture and David F. Warren.
Climate change is projected to decrease crop yields in many regions. To model the effect of rising global temperatures on US wheat yields, Jesse Tack et al. (pp. 6931–6936) combined results from field trials of wheat varieties in Kansas between 1985 and 2013 with weather data from the site of each field trial. The authors found that extreme heat in spring and freezing temperatures in the fall were the main drivers of wheat yield loss. Although global warming will likely reduce wheat’s exposure to freezing temperature, the authors found that wheat varieties may trade off yield for heat resistance, and that the overall effect of warming on wheat is likely to decrease yield. Recent wheat varieties exhibited less heat resistance than varieties from previous years. Yield loss may be partially offset by increased spring rain, the authors suggest. Rather than calculating temperature exposure from average daily temperatures, the authors calculated exposure from the range of temperatures present each day, and found that the method greatly increased the predictive performance of the wheat yield and temperature effect model. The results suggest that maintaining sustainable agriculture in a warming world will likely require design of wheat crops with both high yield and high heat resistance, according to the authors. — P.G.
Genomic analysis reveals insights on malaria in Africa

Malaria control measures in Senegal.
Around 85% of the roughly 275,000 infants born with sickle cell disease every year hail from Africa, where the genetic affliction has a firm hold due to the well-known resistance it confers against another major disease: malaria. To determine the strength of the link between malaria and the sickle cell trait (SCT), Eric Elguero et al. (pp. 7051–7054) identified hemoglobin variants and malarial parasite species in blood samples from 3,958 people 17–80 years of age from 195 villages in the Republic of Gabon, where SCT prevalence hovers around 28% and up to 2% of children are born with the disease every year. Though no association was found between gender and SCT prevalence, reflecting the autosomal location of the gene encoding hemoglobin and implying equal protection of the sexes against malaria, each 10% rise in the prevalence of Plasmodium falciparum malaria was tied to a 4.3% increase in the number of SCT carriers, and each 10-year spike in age was accompanied by a corresponding surge of 5.5% in SCT prevalence. According to the authors, the strong link between P. falciparum malaria and SCT prevalence, which increases with age, suggests that the two conditions may need to be managed together. In a related article, Rachel Daniels et al. (pp. 7067–7072) demonstrate that an epidemiological model based on genomic analysis of malarial parasite population changes can help uncover changes in transmission rates. Applying a genetic barcode based on 24 SNPs to 1,007 parasite samples collected between 2006 and 2013 in Thiès, Senegal, where a national initiative to control malaria included the use of indoor insecticide spraying, insecticide-treated bed nets, and artemisinin combination treatments, the authors found that malaria transmission dipped from 2006 to 2010, but rebounded between 2012 and 2013. Incidence data corroborated the model’s findings on transmission flux. Traditional methods for estimating transmission rates, such as mosquito sampling, are difficult to implement in regions of low transmission, posing a circular challenge to the monumental goal of malaria eradication. Hence, parasite genomics-based modeling can help monitor transmission and design interventions, according to the authors. — P.N.
Binge drinking and potassium channels in neurons

Representation of GIRK3-containing channels gating binge drinking by modulating the incentive salience of alcohol.
Ion channels called G protein-gated inwardly rectifying potassium (GIRK) channels regulate neuronal excitability. Such channels can be activated by ethanol, but the role of the activation in the behavioral effects of ethanol consumption is unclear. Melissa Herman et al. (pp. 7091–7096) found that the absence of GIRK3, a subunit of GIRK channels, promotes binge drinking in mice by blocking activation of a neural pathway that controls reward seeking. Mice that were genetically engineered to lack GIRK3 consumed more ethanol than their wild-type counterparts. However, the absence of GIRK3 had no effect on ethanol metabolism or the effects of intoxication. Ethanol activates neurons in a brain region called the ventral tegmental area (VTA), triggering a neural pathway called the mesolimbic dopaminergic pathway, which confers incentive properties on reward-associated cues and thereby facilitates reward seeking. In the engineered mice, no such activation was observed. When GIRK3 was reintroduced to the VTA of the engineered mice, ethanol consumption returned to normal levels. Wild-type mice that received similar treatment also showed reduced ethanol consumption. Because previous research has shown that loss of GIRK3 has little effect on behavior other than in response to addictive drugs, the authors suggest that selective targeting of GIRK3-containing channels might help reduce binge drinking with minimal side effects. — B.D.
Linking microbiome samples to human sources
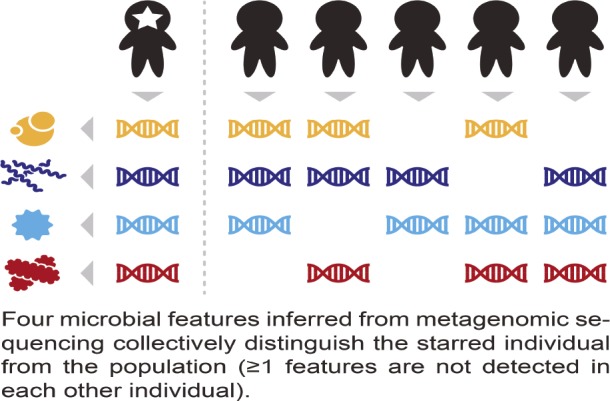
Microbiome fingerprints.
Blood types and genomic variation have long been used to single out individuals in forensics, genealogy, and disaster response, but the microbiome, the complement of microbes found in each person, has so far failed to yield fingerprints, partly due to the lack of robust, stable, and specific codes for individual microbiomes in populations. Eric Franzosa et al. (pp. E2930–E2938) developed an algorithm to create metagenomic codes based on microbial species and genes at various bodily locations and applied the algorithm to samples from 120 people enrolled in the Human Microbiome Project, a government-sponsored consortium to study the human microbiome. The authors pinpointed individual microbiomes among scores of people based on body site-specific microbiome codes alone. When the analysis was repeated on samples collected 30–300 days after each participant’s first visit, around 30% of samples could be uniquely identified using the same codes, with few false positives. Repeat analysis using stool samples correctly identified around 80% of microbiomes up to a year later, suggesting the relative stability of gut microbiome-derived codes. Because a substantial fraction of samples in microbiome repositories can be potentially uniquely identified across databases, some of which may contain sensitive personal information, the anonymity of such samples may not be assumed to be ironclad, according to the authors. — P.N.