

Significance
Traditional methods for estimating malaria transmission based on mosquito sampling are not standardized and are unavailable in many countries in sub-Saharan Africa. Such studies are especially difficult to implement when transmission is low, and low transmission is the goal of malaria elimination. Malaria-control efforts in Senegal have resulted in changes in population genomics evidenced by increased allele sharing among parasite genomes, often including genomic identity between independently sampled parasites. Fitting an epidemiological model to the observed data indicates falling transmission from 2006–2010 with a significant rebound in 2012–2013, an inference confirmed by incidence data. These results demonstrate that genomic approaches may help monitor transmission to assess initial and ongoing effectiveness of interventions to control malaria.
Keywords: malaria, genomics, epidemiology
Abstract
To study the effects of malaria-control interventions on parasite population genomics, we examined a set of 1,007 samples of the malaria parasite Plasmodium falciparum collected in Thiès, Senegal between 2006 and 2013. The parasite samples were genotyped using a molecular barcode of 24 SNPs. About 35% of the samples grouped into subsets with identical barcodes, varying in size by year and sometimes persisting across years. The barcodes also formed networks of related groups. Analysis of 164 completely sequenced parasites revealed extensive sharing of genomic regions. In at least two cases we found first-generation recombinant offspring of parents whose genomes are similar or identical to genomes also present in the sample. An epidemiological model that tracks parasite genotypes can reproduce the observed pattern of barcode subsets. Quantification of likelihoods in the model strongly suggests a reduction of transmission from 2006–2010 with a significant rebound in 2012–2013. The reduced transmission and rebound were confirmed directly by incidence data from Thiès. These findings imply that intensive intervention to control malaria results in rapid and dramatic changes in parasite population genomics. The results also suggest that genomics combined with epidemiological modeling may afford prompt, continuous, and cost-effective tracking of progress toward malaria elimination.
Intensive intervention to reduce the burden of malaria has proven successful in a number of countries in Africa (1). In certain regions of Senegal, implementation of a redesigned National Malaria Control Program (NMCP) in 2006 that included rapid diagnostic tests, artemisinin combination therapies, enhanced insecticide-treated bed nets, and indoor residual spraying resulted in a more than 95% decrease in the number of confirmed cases by 2009 (2). We had been collecting parasite samples in one of these regions annually since 2006. These samples afford a unique opportunity to determine the extent to which intensive intervention is manifested in genetic changes in the parasite population. Genetic changes would be expected to include bottlenecks in the parasite population size, increased random genetic drift, reduced genetic variation, greater self-fertilization during transmission, and increased allele sharing and identity by descent.
A key question for tracking malaria elimination is whether such genomic changes would be large enough to be detected in a cost-effective manner in samples of reasonable size. If changes in parasite population genomics took place rapidly enough after intervention, and if they were large enough to be detected, then parasite genomics could play an important role in malaria elimination. Given sufficiently rapid onset and detectability of changes in parasite genomics, an epidemiological model that incorporates parasite genotypes could in principle be used to estimate the epidemiological parameters that most closely match the genomic observations. Estimates of epidemiological parameters such as transmission intensity would aid in understanding the disease situation on the ground, so that the efficacy of intervention strategies could be evaluated in real time and adjustments made as necessary. This approach could prove especially useful in regions of low transmission where classical epidemiological approaches can be applied only with great difficulty and in regions that are not easily or safely accessed by personnel committed to malaria control.
In this paper, we show that data from a barcode of 24 SNPs in longitudinal samples from Thiès, Senegal over an 8-y period of moderate numbers of samples (100–200 samples/y) reveals rapid and easily detectable signals of changes in parasite population genomics following enhanced intervention. Moreover, an epidemiological model that incorporates parasite genotypes can reproduce the observed barcode patterns. Estimates of epidemiological parameters in the transmission model using likelihoods strongly suggest a reduction of transmission from 2006–2010 with a significant rebound in 2012–2013. The decrease in transmission of malaria in 2006–2010 after enhanced intervention followed by a rebound in 2012–2013 was confirmed directly by incidence data from Thiès. Our findings suggest that genomics combined with epidemiological modeling may afford rapid, continuous, and cost-effective tracking of progress toward malaria elimination.
Results
Genome Relatedness Among Independent Samples.
To look for genomic signals associated with intervention, we studied samples of Plasmodium falciparum from Thiès, Senegal during the period 2006–2013.
All samples were genotyped for 24 unlinked SNPs, constituting a molecular barcode, and were assayed for the presence of single genomes (monogenomic infections) or multiple distinct genomes (polygenomic infections) (3–5). Samples with monogenomic infections were used in further analyses. We find that, accompanying intensified intervention after 2006, the allele frequencies of the SNP alleles often change dramatically from year to year. Such fluctuations in allele frequency afford an estimate of the variance effective population size, which is a measure of the uniformity of reproductive success among parasite genomes: The smaller the variance effective size, the greater the variation in reproductive success. Maximum likelihood estimates of the variance effective size indicate a decrease of at least 10-fold after 2006, with estimates of the variance effective size fluctuating around 10–40 thereafter (SI Appendix, Table S1).
Additional evidence for reduced effective population size is the finding that parasites sampled from monogenomic infections in different patients, different households, different places in the catchment area, and different times across the transmission season (August–January) often occur in subsets in which each parasite genome exhibits an identical 24-SNP barcode (SI Appendix, Fig. S1A and Dataset S1). Moreover, the barcode defining some of these subsets is found in parasite samples from different years, in one case in samples separated by 3 y and in another in samples separated by 7 y (SI Appendix, Fig. S2A).
Detailed analysis of the 2006–2013 samples also revealed that many parasite genomes apparently are closely related to others, based on the similarity of their 24-SNP barcodes. A network showing the barcode relatedness among a sample of 65 parasite genomes that also were sequenced in their entirety is shown in Fig. 1A (see also SI Appendix, Fig. S3). Many of the related samples share multiple barcode alleles with the subset. In Fig. 1A, each color except gray corresponds to a barcode repeated three or more times among the samples; gray corresponds to barcodes present in one or two samples. Among the connecting lines, increasing edge thickness indicates a greater degree of relatedness between parasite types, ranging from 95.8–100% relatedness (zero or one SNP difference, indicated by the thickest lines) down to 79–87.4% relatedness (five SNP differences, indicated by the thinnest lines).
Fig. 1.

Relatedness among parasite isolates. (A) Network of barcode relatedness based on genetic distances between barcodes (19), in which edge thickness represents degree of identity. The thickest edges connect samples 95.8–100% related (identical or one SNP difference), and the thinnest edges connect samples that are less than 87.5% related (five SNP differences). Colored dots indicate barcodes present three or more times in the samples; gray indicates those present one or two times. (B) Network of sample relatedness based on full sequence data, in which edge thickness represents the fraction of the genome that is identical by descent; node colors correspond to those in A. The red square and circle indicate clusters containing the parents of parasite sample SenT120.11.
To determine whether the observation of repeated barcodes in monogenomic infections is associated with transmission intensity, we assayed the 24-SNP barcode in parasites from 97 monogenomic infections collected in 2009–2010 from patients in Malawi (Dataset S2). Unlike the samples collected in Senegal, we find no subsets of samples with identical barcodes such as those depicted in Fig. 1A; we attribute this finding to the continuing high transmission rate in Malawi (1).
To exclude the possibility that barcode relatedness is an artifact of genotyping only 24 SNPs, 164 parasite genomes were sequenced completely (Dataset S3), primarily from parasites sampled in 2008–2012 and including all the genomes whose barcodes are depicted in Fig. 1A (6). A network connecting genomes that share significant blocks of sequence is shown in Fig. 1B (see also SI Appendix, Fig. S4), where the colors correspond to the barcodes in Fig. 1A. Not surprisingly, the 24-SNP barcode provides less complete information about allele sharing than does whole-genome sequencing, and some of the weaker relationships detected by barcoding are not confirmed by the genome sequences. Nevertheless, the major groups of related parasites detected by networks among the 24-SNP barcode are clearly related across the entire genome.
Based on an analysis of the 164 fully sequenced parasite genomes from Thiès, an optimal choice of SNPs in a 24-SNP barcode would allow confident detection of parasites that are more than about 70% related across the genome, whereas an optimized expanded barcode of 96 SNPs would allow confident detection of genomic relatedness of 50% or more (SI Appendix, Fig. S5). For both the 24-SNP and the 96-SNP barcodes, the limiting factor in inferring genomewide identity from barcode identity is the wide range of barcode identities when genomewide sequence identity is low.
Whole-genome sequencing also revealed blocks of genomic sequence shared between independent parasite samples. Fig. 2A shows the size distribution of blocks of genomic sequence shared between pairs of strains. The blocks range in size from 10 kb to >3 Mb (the parasite genome size is 23 Mb), and the distribution of shared block length is approximately exponential. These blocks of shared sequence also seem to be associated with transmission intensity, because an analysis of 23 of the parasite genomes from Malawi that were sequenced in their entirely (Dataset S3) reveals no evidence of blocks of shared sequence such as those depicted in Fig. 2A.
Fig. 2.

Size distribution of shared sequence blocks. (A) Size distribution of regions of identity by descent (blocks of shared sequence) between nonidentical pairs of isolates among the sequenced isolates. (B) Parental origin of genomic segments for sample SENT120.11. Colored segments show identity by descent (as assigned by a hidden Markov model) to the two parental types (blue and orange), to both types (black), or to neither (gray).
One of the sequenced genomes (SenT120.11, indicated in Fig. 1B) is obviously the offspring of a cross between parents related to the two adjacent groups, and indeed genomes virtually identical to the parents are included in the two groups (SenT036.10 in the oval and SenT136.11 in the square). Fig. 2B shows the clear segregation of large blocks of genome from the parents. Some chromosomes did not undergo recombination in this particular meiosis: For example, chromosome 3 derives entirely from the SenT036.10 parent and chromosome 9 entirely from the SenT136.11 parent. Comparison of apicoplast and mitochondrial DNA shows that the SenT036.10 parent provided the female gametocyte for this cross. SI Appendix, Fig. S6 shows a similar pattern of large, shared blocks in the genome of SenT069.11, as is consistent with this strain being the offspring of a cross between parents essentially identical to SenT131.10 (the female gametocyte) and SenT058.10 (the male gametocyte). In this case five chromosomes derive from one or the other parent with no evidence of recombination.
Implications for Malaria Epidemiology.
The results in Figs. 1 and 2, as well as those in SI Appendix, Figs. S1A, S2A, S3, and S4 and Table S1, exhibit the genomic signatures expected to accompany a drastic reduction in parasite population size, including increased random genetic drift, allele sharing, inbreeding, and identity by descent. Suppose, however, that one did not know that Senegal had instituted aggressive malaria-control measures during the sampling period. Would the genomic signs alone imply that there had been some dramatic decrease in incidence? Or suppose that such genomic signatures began to change in a population under control measures. Would that change imply that the control was losing effectiveness?
Based on a simplification of the malaria-transmission dynamics in an existing agent-based model (7), parasite genome dynamics were added to construct a combined genetic-epidemiological model to address these questions. The genetic-epidemiological model is stochastic and includes parasite lineage extinction, survival with clonal reproduction, outcrossing, and immigration. In the model, (i) individual human hosts experience the same risk of infection; (ii) vector-to-host cotransmission of multiple parasites from polygenomic infections is permitted; (iii) genetic recombination between unrelated parasites in multiply infected individuals is equivalent to recombination between two genomes chosen randomly from the existing population; and (iv) the 24-SNP barcode loci are unlinked so that each pair of SNP alleles segregates independently.
The free parameters used in the calibration to the collected barcode data include the lineage extinction rate, the immigration rate, the size of the human population, the reproductive rate at the beginning of the multiyear period (), the value reached after a linear transition between 2006 and 2010 (), and the value from 2012 onwards (). Note that R0 is the maximum value of a seasonally varying reproduction rate (SI Appendix, SI Supporting Information). This piecewise function of R0 allows us to assess whether there were significant increases or reductions in transmission between these fixed intervals.
An incremental mixture importance sampling (IMIS) algorithm was implemented to fit the parameters of the epidemiological model to the observed barcode patterns in Thiès efficiently and accurately (8, 9). First, an initial set of simulation parameters was sampled from a uniform prior in the six free dimensions of the parameter space. Based on the likelihood values calculated at each sample point in parameter space, an iterative process was conducted until the weighted mixture of samples was sufficient to represent the posterior probability distribution to a specified accuracy (SI Appendix, SI Supporting Information and Table S2).
For each point in parameter space, 20 simulations were carried out starting from different random seeds, and random parasite barcodes were sampled annually during the peak season and analyzed as described for the actual data from Thiès. A likelihood metric was constructed from deviations between measurements and model simulations using the following seven summary statistics: (i) the fraction of polygenomic infections; (ii) the number of sampled barcodes that are unique within a measurement year; (iii) the number of barcodes sampled twice in a year; (iv) the number of barcodes sampled more than twice in a year; (v) the number of barcodes persisting over 2, 3, 4, 5, and >5 y (allowing for missing years within the interval); (vi) the number of new barcodes that persist for more than 2 y; and (vii) the number of persisting barcodes that disappear after at least 2 y. Details are given in SI Appendix, SI Supporting Information.
For each of these features, an individual measure of deviation was calculated as the sum of squared differences normalized to the estimated variance. Variances in the simulated data were estimated from multiple stochastic realizations, and uncertainties in the actual data were calculated from binomial statistics with the assumption that different years constituted independent measurements.
Results for a single high-likelihood simulation sampled from the model fitting are illustrated in Fig. 3. The bar graphs in Fig. 3A show the yearly proportion of parasite barcodes that are unique in the sample or are present in subsets of the actual data, compared with the simulated data shown in Fig. 3B. More details on the number and sizes of subsets for the simulated data are presented in SI Appendix, Fig. S1B, and persistence across years is shown in SI Appendix, Fig. S2B. The dynamics of simulated parasite genomes for the same simulation are shown in Fig. 4A in response to a seasonally varying reproductive rate that falls from 2006–2010 and rebounds in 2012–2013.
Fig. 3.

Model output calibrated to observed barcode data. (A) Unique and repeated subsets of barcodes in observed data. (B) Unique and repeated subsets of barcodes in data output from the fitted model.
Fig. 4.

Changing transmission dynamics in the epidemiological model fitted to barcode data. (A) Seasonal changes in relative number of affected individuals and unique barcodes across years. is the initial maximum rate of parasite increase, which is assumed to decrease linearly to a minimum rate () and then to rebound to a rate . The curves shown are for the maximum likelihood estimates of the three rates of parasite increase. (B) Log-likelihoods for values of versus . (C) Log-likelihoods for values of versus .
A key finding from the epidemiological model is that the observed barcode data are sufficient in themselves to imply a decreasing and then rebounding reproductive rate (R0) across years. Fig. 4B shows a projection of the result of iterative six-dimensional sampling in parameter space, sampling most densely in the region of maximum likelihood. As indicated by the diagonal line, essentially all the posterior probability distribution requires a significant drop in transmission intensity over the years 2006–2010.
The simulations also imply a significant rebound in reproductive rate in 2012–2013 (Fig. 4C). A separate set of 2,400 simulations based on Latin hypercube sampling of the three R0 dimensions, carried out at the most likely values of population size, generation time, and import rate, verified the significance of the rebound in 2012–2013 (P = 0.0039) (SI Appendix, Fig. S7). Estimating the annual maximum reproduction rate R0 by iterative resampling confirms that the minimum R0 is in 2010–2011 with the rebound detectable only in 2012–2013 (methods are described in SI Appendix, SI Supporting Information and results in SI Appendix, Fig. S7).
Validation from Incidence Data.
Do the inferences from genomic epidemiology coincide with observed rates of incidence of malaria in Thiès in 2006–2013? To address this question, we analyzed data compiled under the auspices of the Senegal National Malaria Control Program (2). The 2006–2013 data from Thiès alone and from all of Senegal excluding Thiès were normalized to the malaria incidence per person observed in 2006 (when incidence was 0.114 per person in Thiès and 0.132 per person in Senegal excluding Thiès) and then were fitted to a nonlinear model consisting of an exponential decrease in incidence per person plus a rebound term. The results for Thiès are shown in Fig. 5A. Although there is a slight rebound starting in about 2009–2010, this rebound is not statistically significant until 2012 and then is only marginally so (P = 0.04, t test). By 2013 the rebound is highly statistically significant (P = 0.007, t test), consistent with the genomic signals and inferences from epidemiological modeling in Fig. 4. The rebound in Thiès is not observed nationwide and hence probably results from a change in local conditions. The relative incidence for all of Senegal excluding Thiès is shown in Fig. 5B; in this case there is no significant rebound (P = 0.129, t test).
Fig. 5.
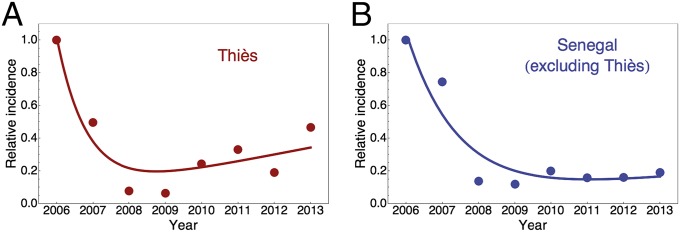
Malaria incidence per person, 2006−2013, normalized to that observed in 2006 and fitted to a model with an exponential decrease plus a rebound. (A) Data from Thiès, in which the rebound is statistically significant. (B) Data from all of Senegal excluding Thiès, which shows no significant rebound. Incidence data are from the Senegal National Malaria Control Program (2).
Discussion
Several aspects of our results and analysis warrant emphasis. One is that the expected genomic signatures of reduced transmission are detected surprisingly rapidly following intervention. This timeline closely follows implementation of control efforts by the NMCP in Senegal. After significant restructuring in 2005, the NMCP developed a control strategy for 2006–2010 that involved supplying rapid diagnostic tests to all health centers (2007), nationwide access to artemisinin combination therapies (2007 and 2008), and distribution of insecticide-treated bednets (2007–2009) (2).
Although such genetic signatures have been observed in parasite populations associated with sustained low transmission in South America (10, 11) and Southeast Asia (12), the situation in Thiès represents the first time (to our knowledge) that they have been observed in African parasite populations. In Thiès, the genetic-epidemiological simulations, fitted solely to population-genomic signatures, demonstrate a dramatic reduction in transmission intensity in 2006–2010 followed by a recent rebound. As an added benefit, the genetic-epidemiological model yields estimates not only of the transmission intensity year-on-year but also the uncertainty of each of these estimates (SI Appendix, Fig. S8).
In contrast to the observations in Thiès, no genomic signatures of parasite relatedness were noted in a set of samples analyzed from a region in Malawi in which no significant reduction in transmission has occurred. The reasons for the observed rebound in Thiès are not yet clear; however, vector resistance to insecticide-treated nets, failure of the insecticide in older nets, or change in the relative importance of vector mosquito species may contribute. In addition, with effective control strategies, the at-risk population may shift in response to reduced parasite exposure, with adolescents and adults losing their acquired partial immunity (13).
Our results do establish a foundational link between observations of parasite population genomics and epidemiological models that incorporate genetic mechanisms. Combining genomic observations with epidemiological modeling provides a powerful and complementary tool for elucidating population-level details of transmission in low-prevalence settings from a small sample of parasite genomes. In particular, modeling parasite genetics allows one to take a collection of many different types of measurement—effective population size, multilocus linkage disequilibrium, heterozygosity of mixed infections, complexity of infection—and from these measurements form a quantitative assessment of the transmission dynamics most consistent with the full information available.
Although our simplified parasite barcode model has allowed a robust and intuitive interpretation of how changes in population genomics track with changes in transmission intensity through time, the addition of different layers of complexity in future studies has the potential to clarify other features of these data. For example, the few very large clusters of repeated sequences in 2008 and 2011 are a challenge to reproduce within the simple model structure. Nevertheless, different types of heterogeneity and complexity can be added to explore their qualitative effects on genetic signatures: multiple weakly linked subpopulations experiencing different levels of exposure, interactions between acquired immunity in a population, and strain-specific genetics, as well as selection pressure from antimalarial drug use. More complex models will require more detailed data to constrain the parameterization, but as sample sizes grow and the resolution of patient metadata increases, our ability to resolve features of malaria transmission vulnerable to intervention strategies will improve.
It also is noteworthy that genomewide relatedness can be detected based on only the number of shared alleles in a 24-SNP barcode. However, full-sequence data provide additional insights. Analysis of 164 complete genome sequences indicates substantial allele sharing across independent isolates. Approximately half of all genomes share significant sequence identity with at least one other genome, with the extent of sequence identity ranging from 10–90% or more (Fig. 2A). Genomes sharing a significant number of SNPs with common barcodes in large subsets or that persist across multiple years account for a substantial proportion of the genomewide identity by descent.
To define an optimum number of SNPs to detect the emergence of closely related parasites in a population, we performed an in silico analysis. The minimum proportion of genome sharing that can be detected is limited by the probability that two independent barcodes might share multiple SNP alleles by chance alone. SI Appendix, Fig. S5 shows the ability of a 24-SNP barcode to detect genomewide sequence concordance when the SNPs are chosen to be maximally informative, which in this case means having minor allele frequencies in Thiès as close as possible to 0.5. Genomewide sequence concordance of ≥70% can be detected quite confidently. Genomewide concordance of ≥50% can be achieved with a maximally informative barcode of consisting of 96 SNPs (SI Appendix, Fig. S5).
Major advantages of barcode genotyping are that it is rapid, inexpensive, sensitive, reliable, and fully deployable in the field. Although whole-genome sequencing costs continue to decrease, library construction, sample preparation, and analysis costs remain significant. SNP genotyping technologies such as the molecular barcode are deployed currently in field settings in Senegal, Malawi, Zambia, and Mozambique using existing real-time and dedicated high-resolution systems (4). These instruments offer high throughput, straightforward analysis, and relatively low costs (less than $10 per sample for barcoding, including reagents and consumables).
In principle, the advantageous features of SNP barcoding would allow continuous monitoring of progress in malaria elimination independent of entomological or prevalence surveys. With steadily increasing throughput of genomic technologies and decreasing cost, population genomics also potentially could be used to track parasite genotypes through time and geographically across routes of human or vector migration. These methods would allow the source or sources of parasite resurgence to be identified and controlled so that elimination could be maintained.
A major implication of this work is that intensification of existing prevention and treatment interventions can impact the parasite population dramatically, resulting in the survival of a smaller, less diverse parasite population. It is important to recognize that this reduction in diversity could result in the emergence of parasites with altered biological properties, including the selection of parasites with an enhanced propensity for transmission. These results give us new insights into the transmission network in the Thiès region of Senegal. The observation that more than half of the parasites analyzed at the full-sequence level share some portion of their genome indicates a limited transmission network. More generally, in areas of low transmission, it might be possible to identify the likely sources of imported infections by comparing the parasite genomes in the imported infections with those from possible sources inferred from patients’ travel history derived from analysis of data from cellular telephone global-positioning systems or from more traditional questionnaires. Limited local transmission implies that parasites from imported infections and those from the source often may share significant portions of their genomes.
Measuring transmission is necessary to enable adaptable control and elimination campaigns that respond to changes in malaria epidemiology and as, paradoxically, intervention reduces transmission to low levels, standard methods of measurement become less feasible. At about the time that improved situational awareness of transmission rates is required to understand how much transmission has decreased, how much further it needs to be reduced, and how effective current measures have been, our ability to measure transmission accurately decreases. The mosquito entomological inoculation rate (EIR) often is assayed by rate of human biting or incidence of sporozoites. The methods are not standardized across studies, and data on transmission intensities are available for only about half the countries of sub-Saharan Africa (14). An additional complication is that entomological parameters become very difficult to estimate as transmission drops to lower levels and finding sporozoite-positive mosquitoes through standard sampling becomes rare. In Thiès, for example, the EIR is thought to be at the low end of the range 1−5 (15). With continuously low transmission intensity, parasite rates and incidence can become sparse, heterogeneous, and clustered in time or space. At the other end of the spectrum, when transmission intensity is high, human parasite infection rates and incidence begin to saturate. In addition, the incidence of detected clinical cases depends highly on acquired population immunity in addition to transmission, and thus the relationship between clinical incidence and population immunity will change as local transmission rates depart from historical equilibriums. For these reasons, among others, when transmission is low and traditional measures of transmission become unreliable, prevalence surveys either can be expensive and labor intensive or else may be performed only on a biased subsample of the population. Our results suggest that new approaches combining genomics with epidemiological modeling have the potential to provide accurate and timely transmission estimates without costly surveys or changing incidence relationships. Our results demonstrate that genomic approaches can serve to monitor transmission to gauge the initial and ongoing effectiveness of interventions to control malaria. Improved measurements of transmission will enable adaptive campaign measures that respond to changing conditions and therefore improve outcomes.
Materials and Methods
Sample Collection.
All human samples were collected from individuals after recruitment and written consent of either the subject or a parent/guardian. This protocol was reviewed and approved by the ethical committees of the Senegal Ministry of Health (Senegal) and the Harvard School of Public Health (16330, 2008) for Senegalese subjects and University of Malawi College of Medicine (Blantyre) and The Brigham and Women's Hospital (2006-P-002031).
Samples were collected passively from patients reporting to the clinic for suspected malaria between approximately September and December each year. Patients over the age of 12 y with acute fevers within the past 24 h of visiting the clinic and with no reported history of antimalarial use were considered; they were diagnosed with malaria based on microscopic examination of thick slide smears and rapid diagnostic tests.
Sequencing and Analysis.
Extracted genomic DNA from patient samples from Malawi and Senegal were sequenced using Illumina Hi-Seq (Illumina, Inc., San Diego, CA) machines. Reads were aligned using the Burrows-Wheeler Aligner version 0.5.9-r16 (16) against the 3D7 reference assembly (PlasmoDB v7.1; 17). A consensus sequence was called for each strain using the GATK Unified Genotyper (18) (see SI Appendix, SI Supporting Information for parameter values and quality-score thresholds).
A total of 190 fully sequenced samples from Senegal were available for study; all were identified as monogenomic infections by barcode. Of these, 176 were collected in a clinic at Thiès. One sample was removed because it had a very low call rate (3%). To screen out possible cryptic polygenomic infections and cross-sample contamination, samples also were eliminated if they had an unusually low rate of calls (<0.3%) with the minor allele (the mean rate for the rest of the sample was 0.8%); this screen removed 11 samples. The remaining 164 samples were analyzed.
All SNPs with a call rate of at least 80% were used. Triallelic SNPs (1% of the total) were treated as biallelic, with the most common allele treated as the major allele. Details of the hidden Markov model to identify specific regions of genomes that were identical by descent are reported in SI Appendix, SI Supporting Information.
Epidemiological Modeling.
The general features of the epidemiological model and the parameters estimated to fit the observed barcode data are summarized in Results. Details of the model as to state, time dependence, initialization, sampling, and fitting to observed data are described in SI Appendix, SI Supporting Information.
Malaria Incidence Analysis.
Malaria incidence per person 2006−2013 was normalized to the values observed in 2006 and analyzed for Thiès alone or for all Senegal excluding Thiès. The relative incidence data were fitted to a nonlinear model with an exponential decrease plus a rebound of the form y = a × Exp(−bx) + cx, where y = relative incidence. Curve fitting and statistical analysis were performed using the NonlinearModelFit package in Mathematica. Details of the parameter estimates, SEs, and statistical tests are summarized in Table S3.
Optimized Barcodes.
To address the extent to which genomewide identity by descent can be estimated through the use of molecular barcodes, we set out to identify and characterize optimal barcodes based on the fully sequenced samples from Thiès, Senegal. We excluded highly discordant strains and screened the others for all polymorphic sites to include SNPs with a minor allele frequency >0.2, and from these screens we compiled 24-SNP and 96-SNP optimal barcodes of sites ranked by highest minor allele frequency and in which ≥80% of samples had no ambiguous or missing calls. We then calculated the barcode similarity for each pair of strains among all sequenced strains, counting ambiguous or missing calls as mismatches. To avoid biasing the percent similarity because of matching major alleles, we restricted the percent-similarity calculations to include only sites where the minor allele was present. The similarity index therefore was calculated as the number of sites where the minor allele matched divided by the total number of sites where the minor allele was present. The barcode similarity indices then were compared with genomewide sequence concordance.
Supplementary Material
Acknowledgments
E.A.W., J.L.P., and P.A.E. thank Bill and Melinda Gates for their active support of the Institute for Disease Modeling and their sponsorship through the Global Good Fund. We also thank Mike Famulare, Daniel Klein, Jaline Gerardin, Kevin McCarthy, Eli Moss, and Danny Park for their contributions. Funding for this work at the Harvard School of Public Health and the Broad Institute was provided by a grant from the Bill and Melinda Gates Foundation and by National Institutes of Health (NIH) Grants AI099105 (to D.F.W. and D.L.H.) and AI106734 (to D.L.H.). Sample collection was supported in part by Grant 5D43TW001503 from the Fogarty International Center, Grant U19AI089696 from the International Centers of Excellence for Malaria Research, and NIH Grants K23AI072033 (to D.A.M.) and AI034969 (to T.E.T.). H.-H.C. is supported by NIH Grant U54GM088558.
Footnotes
The authors declare no conflict of interest.
Data deposition: Molecular barcodes are available in Datasets S1 and S2. Dataset S3 contains the whole-genome sequence accession numbers described in the paper.
See Commentary on page 6782.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1505691112/-/DCSupplemental.
References
- 1.Noor AM, et al. The changing risk of Plasmodium falciparum malaria infection in Africa: 2000-10: A spatial and temporal analysis of transmission intensity. Lancet. 2014;383(9930):1739–1747. doi: 10.1016/S0140-6736(13)62566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mouzin E, Thior PM, Diouf MB, Sambou B. Focus on Senegal Roll Back Malaria: Progress and Impact Series. Vol 4 World Health Organization; Geneva: 2010. [Google Scholar]
- 3.Daniels R, et al. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J. 2008;7:223. doi: 10.1186/1475-2875-7-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daniels R, et al. Rapid, field-deployable method for genotyping and discovery of single-nucleotide polymorphisms associated with drug resistance in Plasmodium falciparum. Antimicrob Agents Chemother. 2012;56(6):2976–2986. doi: 10.1128/AAC.05737-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daniels R, et al. Genetic surveillance detects both clonal and epidemic transmission of malaria following enhanced intervention in Senegal. PLoS ONE. 2013;8(4):e60780. doi: 10.1371/journal.pone.0060780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang HH, et al. Malaria life cycle intensifies both natural selection and random genetic drift. Proc Natl Acad Sci USA. 2013;110(50):20129–20134. doi: 10.1073/pnas.1319857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eckhoff PA. Malaria parasite diversity and transmission intensity affect development of parasitological immunity in a mathematical model. Malar J. 2012;11:419. doi: 10.1186/1475-2875-11-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steele SJ, Raftery AE, Emond MJ. Computing normalizing constants for finite mixture models via incremental mixture importance sampling (IMIS) J Comput Graph Stat. 2006;15(3):712–734. [Google Scholar]
- 9.Raftery AE, Bao L. Estimating and projecting trends in HIV/AIDS generalized epidemics using Incremental Mixture Importance Sampling. Biometrics. 2010;66(4):1162–1173. doi: 10.1111/j.1541-0420.2010.01399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branch OH, et al. Plasmodium falciparum genetic diversity maintained and amplified over 5 years of a low transmission endemic in the Peruvian Amazon. Mol Biol Evol. 2011;28(7):1973–1986. doi: 10.1093/molbev/msq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obaldia N, et al. Clonal Outbreak of Plasmodium falciparum Infection in Eastern Panama. J Infect Dis. 2015;211(7):1087–1096. doi: 10.1093/infdis/jiu575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nkhoma SC, et al. Population genetic correlates of declining transmission in a human pathogen. Mol Ecol. 2013;22(2):273–285. doi: 10.1111/mec.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trape JF, et al. Malaria morbidity and pyrethroid resistance after the introduction of insecticide-treated bednets and artemisinin-based combination therapies: A longitudinal study. Lancet Infect Dis. 2011;11(12):925–932. doi: 10.1016/S1473-3099(11)70194-3. [DOI] [PubMed] [Google Scholar]
- 14.Kelly-Hope LA, McKenzie FE. The multiplicity of malaria transmission: A review of entomological inoculation rate measurements and methods across sub-Saharan Africa. Malar J. 2009;8:19. doi: 10.1186/1475-2875-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ndiaye D, et al. Polymorphism in dhfr/dhps genes, parasite density and ex vivo response to pyrimethamine in Plasmodium falciparum malaria parasites in Thies, Senegal. Int. J. Parasitol. Drugs Drug. Resist. 2013;3:135–142. doi: 10.1016/j.ijpddr.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aurrecoechea CJ, et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 2009;37:D539–D543. doi: 10.1093/nar/gkn814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004;101(30):11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.