

Significance
Stable genome maintenance inside viral particles and its controlled delivery to the host are critical for virus infection. We report cryoEM structures of a tailed bacterial virus genome gatekeeper mimicking the states before and after DNA release. The subnanometer resolution allowed precise fitting of individual protein components. We found concerted structural rearrangements in the portal through which DNA traffic occurs. DNA is locked in a capsid by gp16 loops that close the channel by an allosteric mechanism. Gp16 appears to open by a diaphragm-like motion, allowing the genome to exit the capsid through the tail tube to the host cell. We propose a molecular mechanism by which the largest group of viruses on Earth controls its DNA movement.
Keywords: DNA gatekeeper, viral infection, bacteriophage, allosteric mechanism, hybrid methods
Abstract
Many icosahedral viruses use a specialized portal vertex to control genome encapsidation and release from the viral capsid. In tailed bacteriophages, the portal system is connected to a tail structure that provides the pipeline for genome delivery to the host cell. We report the first, to our knowledge, subnanometer structures of the complete portal–phage tail interface that mimic the states before and after DNA release during phage infection. They uncover structural rearrangements associated with intimate protein–DNA interactions. The portal protein gp6 of bacteriophage SPP1 undergoes a concerted reorganization of the structural elements of its central channel during interaction with DNA. A network of protein–protein interactions primes consecutive binding of proteins gp15 and gp16 to extend and close the channel. This critical step that prevents genome leakage from the capsid is achieved by a previously unidentified allosteric mechanism: gp16 binding to two different regions of gp15 drives correct positioning and folding of an inner gp16 loop to interact with equivalent loops of the other gp16 subunits. Together, these loops build a plug that closes the channel. Gp16 then fastens the tail to yield the infectious virion. The gatekeeper system opens for viral genome exit at the beginning of infection but recloses afterward, suggesting a molecular diaphragm-like mechanism to control DNA efflux. The mechanisms described here, controlling the essential steps of phage genome movements during virus assembly and infection, are likely to be conserved among long-tailed phages, the largest group of viruses in the Biosphere.
The dsDNA bacterial viruses (phages or bacteriophages) and herpes viruses keep their genetic information packed at high pressure inside an icosahedral protein capsid. During virus particle assembly the genome is translocated into a prebuilt procapsid through a specialized portal vertex of the capsid (1, 2). Termination of the DNA packaging reaction is coordinated with closure of the portal system to avoid leakage of the viral genome. The outflow of DNA is prevented by conformational changes in the portal protein and binding of head completion proteins building the viral genome gatekeeper (3). In bacteriophages, the resultant complex [connector (4)] provides the connection point for the tail. The head-to-tail interface (HTI), or neck, is composed of the connector and of the tail-completion protein(s) found between the connector and the helical tail tube (Fig. S1A) (3, 5). Phage tails are responsible for host cell recognition and delivery of the viral genome to the host cytoplasm (6). At the beginning of viral infection the phage adsorption apparatus, located at the tail end distal from the capsid, binds to the host receptor, generating a signal that triggers opening of the neck (7). DNA then moves through the tail tube to enter the host cell. That tailed bacteriophages are the most abundant biological entities on Earth indicates the evolutionary advantage of this strategy for infecting bacterial cells. Infection by these viruses plays a central role in microbial ecosystems dynamics and in the horizontal transmission of genetic information within the bacterial world (8).
Bacillus subtilis tailed bacteriophage SPP1 is a paradigm for viruses with a portal system (9). The viral particle is composed of an isometric icosahedral capsid ∼60 nm in diameter, shielding the 45.9-kbp-long viral chromosome (10). The portal protein gp6 (57.3 kDa subunit mass) is incorporated at a single vertex of the procapsid as a circular oligomer with a central channel that serves as a conduit for DNA passage (11). The portal vertex acts as a platform for the assembly of the viral DNA-translocating motor (12). Termination of DNA packaging is coordinated with disassembly of the motor and binding of gp15 subunits (11.6 kDa) to gp6, extending the portal channel that is closed underneath by the gp16 protein (12.5 kDa) (Fig. S1A) (13). The assembled complex represents the 180-Å-high connector that consists of three stacked cyclical homo-oligomers, each composed of 12 subunits of the portal protein gp6, of the adaptor gp15, and of the stopper gp16 (4, 13). Gp16 operates as a docking platform for the SPP1 preassembled tail tapered by the tail-to-head joining protein gp17 (15 kDa) (14, 15). Binding of the flexible 1,600-Å-long helical tail to the connector completes the formation of the HTI (7, 16). The capsid-distal region of the tail features an adsorption apparatus. Binding of this apparatus to the host cell receptor YueB (17, 18) triggers a domino-like cascade of conformational changes within the gp17.1/gp17.1* tail tube (7, 16, 19), signaling for opening of the gp16 stopper to initiate delivery of the SPP1 genome to the host cell.
We report here subnanometer structures of the SPP1 HTI before and after DNA release obtained by cryoEM and single-particle analysis. The EM structures were used for flexible docking of X-ray and NMR atomic models of protein components of the HTI, allowing the uncovering the network of protein–protein and protein–DNA interactions in the complete HTI. The follow-up structure-driven functional analysis unraveled the allosteric mechanism by which the gatekeeper system assembles to lock DNA inside the virion after the genome-packaging reaction. It also provided experimental evidence supporting a model in which reversible diaphragm-like motion is the mechanism that controls viral genome release from the HTI for delivery to the host cell.
Results and Discussion
Overall Organization of the SPP1 HTI.
Structural studies of the HTI in complete SPP1 particles before and after DNA release (Fig. S1 B and C) were difficult. First, a portion of the connector is embedded in the capsid, which obscures structural details. Second, after DNA release, we observed increased flexibility between the tail and capsid. Thus, we treated complete viral particles with EDTA. This treatment disrupts the capsids, leaving the connector associated with the tail (Fig. S1 A and D–G). Under these conditions, the last packaged DNA end of the viral chromosome previously was shown to remain attached to the HTI (blue arrow in Fig. S1F) (20, 21). The DNA–connector–tail complexes then were challenged with receptor buffer or with the SPP1 receptor ectodomain YueB780 (22) to obtain HTIFull and HTIEmpty, respectively (Fig. S1). Binding of the SPP1 tail tip (red arrows in Fig. S1 B, D, and F) to YueB780 leads to disassembly of the tip, providing a visual signature of the interaction (yellow arrows in Fig. S1 C, E, and G) (17, 18). This interaction correlates with the release of DNA from the HTI of most connector–tail complexes, as monitored by visualization of DNA after adsorption to mica (Fig. 1G). Therefore, binding of YueB780 to the tail tip triggers a signal transmitted along the complete tail tube that leads to dissociation of the DNA end from the connector region, mimicking the step that initiates release of the genome from phage capsids at the beginning of infection (Fig. S1) (7). Structures of the HTIs in the pre- (HTIFull) and post- (HTIEmpty) DNA release states were determined at ∼7-Å resolution (Fig. 1 and Figs. S2 and S3).
Fig. 1.

Structures of HTIFull and HTIEmpty. (A and B) Fitting of gp6, gp15, gp16, gp17, and gp17.1 models in the cryoEM maps of HTIFull (A) and of HTIEmpty (B). Overlays of pseudoatomic models and EM maps are displayed on the left of A and B. DNA density is rendered in salmon, and the gp18 tape measure density is shown in light green in HTIFull (A). (C) One copy of gp17.1, two copies of gp6, gp16, and gp17, and three copies of gp15 adjacent subunits are highlighted. When more than one copy is highlighted, adjacent subunits are rendered in variations of one similar color in a molecular surface display of the HTIFull atomic model. All other subunits are depicted in gray. Cross-sections show interfaces between rings of subunits.
Detailed pseudoatomic models of the HTIFull and HTIEmpty structures were established by flexible fitting of the available atomic structures of SPP1 proteins (gp6, gp15, gp16, and gp17) into the cryoEM maps (Fig. 1 A and B). An I-TASSER model (23) of the major tail protein gp17.1 (16) was used to define the beginning of the helical tail tube in the reconstructions. A gp17 hexamer is localized between the connector stopper gp16 and the sixfold helical tail (Fig. S3C). However, six subunits of the gp17 hexamer do not account for the complete electron density of this region, suggesting presence of an additional tail protein above the first gp17.1/gp17.1* ring of the tail tube. Each subunit of the gp6, gp15, gp16, gp17, and gp17.1 proteins interacts with at least two subunits of their adjacent rings (Fig. 1C). The alternate distribution of subunits along the structure’s height provides a mechanism for assembly in which oligomerization of one protein creates the interface for stable binding of the following interaction partner. This organization ensures orderly assembly and prevents premature interaction between components of the HTI that are monomeric before assembly (gp15, gp16, and gp17) (13–15) and that do not interact with each other in solution (gp6, gp15, and gp16) (13).
The central channel of the HTI is filled with densities in the pre-DNA release (HTIFull) state. The 20-Å-wide wand of density inside the connector region (shown in salmon in Fig. 1A and Fig. S2C) is consistent with presence of DNA (Fig. S1F) (20) that extends down to the gp16 stopper. Density occupying the channel underneath the stopper exhibits different features and a wider diameter (∼27 Å). This central density, shielded by gp17.1 and on its top by gp17, is attributed to the SPP1 tape measure gp18 (shown in light green in Fig. 1A and Fig. S2C). Such organization differs from that proposed in early studies with phage lambda in which DNA would extend to the tail tube interior (24). Both DNA and gp18 are released from the SPP1 HTI upon challenge with YueB780 (HTIEmpty).
Rearrangements in the Portal Protein Associated with the Presence of DNA in the Central Channel.
DNA is confined in the portal protein gp6 mainly by close contacts with the portal tunnel loops (Figs. 1A and 2A). DNA exit is associated with rearrangements in the portal channel (Fig. 2 B–D). Helix α6, which traverses the entire gp6 wing domain, undergoes reorientation of segment α6′, rotating ∼40° from its position in HTIFull to an orientation quasi-parallel to the central axis in HTIEmpty. Segment α6′′ shifts slightly clockwise (Fig. 2B, Upper Inset). Helix α6′ is connected to helix α5 via the tunnel loop. Density-bridging tunnel loops to DNA indicate a direct interaction (Figs. 1A and 2A, Left). An additional gp6–DNA contact might occur between the ring formed by K331 residues of α5 that localize within a close distance to DNA in HTIFull (Fig. 2A and Fig. S4A). These interactions are consistent with the requirement for helix α5 motion, possibly coordinated with the movement of tunnel loops, during viral DNA packaging (11, 12, 25). They also can prevent DNA reflux from the capsid during DNA translocation and before the portal system is closed. Tunnel loops and rings of lysines lining the portal channel are found in gp6-related proteins and in other portal structures, suggesting that they play the same DNA-anchoring role in the central channel (Fig. S4 B–D) (26–30). These elements are too far from the central channel in the portal of phage P22 for interaction with DNA (Fig. S4E) (31). In the group of P22-like phages, DNA may be held by a long (∼200 Å) α-helical barrel protruding from the portal complex to the capsid interior (31), in a way analogous to the protein–DNA interactions proposed for the structurally related tube of H-protein in bacteriophage ΦX174 (32). Helix α5 in SPP1 HTIFull is translated ∼6 Å counterclockwise to reach the HTIEmpty state (Fig. 2 B and C), and the helix α3 region proximal to the wing shifts ∼6 Å inwards, acquiring a more upright position relative to the molecule central axis (Fig. 2 B and D). Release of DNA thus is accompanied by structural reorganization of the portal channel where a change in position of tunnel loops is concerted with reorientation of α3, α5, and segment α6′ from α6 (Fig. 2). Pumping of DNA through the portal channel during genome packaging conceivably leads to coordinated movements of these structural elements.
Fig. 2.

Structure of the portal protein gp6 in the presence and absence of DNA. (A) Flexible fitting of one gp6 subunit and corresponding densities in HTIFull (Left) and HTIEmpty (Right). Lysine side chains exposed to the portal channel in helix α5 (K342 and K331) and tunnel loop residues (T353 and I354) are displayed as orange spheres. E294 involved in the interaction with gp15 is shown in cyan. (Insets: α5 viewed from the channel interior with side chains.) (B, Left) Overlay of the gp6 HTIFull (orange) and HTIEmpty (blue) models. (Right) Top views corresponding to the dotted rectangles. Y367 and G384 are hinge points in helix α6. (C and D) Details of the overlays along the planes a and b in B are shown viewed from inside the central channel in C and D, respectively.
Mechanism of HTI Assembly.
Fitting of the gp15 monomer NMR structure in the HTIFull and HTIEmpty EM maps reveals that helices α0 and α1 form the outer surface in the head-to-tail complex and helices α2 and α3 rotate to acquire a position parallel to the central channel (Fig. 1 A and B and Fig. S5A). This rearrangement of gp15 during virus assembly is less dramatic than previously proposed (13). GP15 contacts gp6 via its loop α1–α2 (brown arrow in Fig. 1C, section 1) that is sandwiched between loops of the clip and helix a4 of gp6-adjacent subunits (Fig. 3A). Mutation gp6E294G in one of these loops (shown as cyan spheres in Fig. 2 and as sticks in Fig. 3A) specifically abolishes the gp6–gp15 interaction (33). Gp15 interactions were investigated further by structure-driven mutagenesis and analysis of the composition of purified capsids that were assembled in presence of mutant gp15 forms (Fig. 3B). These experiments showed that the other region of gp15 necessary for binding to the portal is the C terminus where elimination of its basic residues (changing RKMAR102 to MAG) prevents assembly of the gp15 ring at the portal vertex (Ct in Figs. 1C, section 1, and 3 A and B). The gp15 analog in bacteriophage P22 is the adaptor protein gp4 that has a similar overall fold but features a longer C-terminal helix extended by 28 residues (Fig. S5B) (31). In contrast to gp15, the loop of gp4 from P22 that is topologically equivalent to loop α1–α2 of SPP1 gp15 does not interact with the portal. Contacts between loop α1–α2 and the portal clip likely compensate for the absence of the long C-terminal extension in the adaptor proteins of phages SPP1 (gp15) and HK97 [gp6 (34)] (Fig. S5 A and D), suggesting that a common fold developed two structural solutions to achieve robust binding to the portal protein.
Fig. 3.

Gp15 interaction with gp6 and gp16. (A) Interface of two subunits of gp6 (ribbons in orange and surface representation in yellow) with one gp15 subunit (magenta) in HTIFull. The gp15 subunit is viewed from outside the complex and is rotated slightly clockwise. Side chains of gp6 and gp15 residues which when mutated prevent the gp6–gp15 interaction are displayed as sticks and are labeled in red. (B) Composition of purified capsids assembled in bacteria producing different mutant versions of gp15 as labeled above each gel lane and infected with the gp15-defective phage SPP1sus128. Capsids produced during infection with the gp16-defective mutant SPP1sus117 are a control (far left lane) showing that gp15 incorporation at the portal vertex precedes gp16 binding (4, 21). Gp13, the major capsid protein, gp15, and gp16 were detected by Western blot. Note that infection of the strain producing wild-type gp15 by SPP1sus128 leads to complementation, yielding complete virions that do not copurify with capsids. In such cases tailless capsids carrying gp15 and gp16 are present in reduced amounts (lane gp15wt). (C) Interface of gp15 subunit (magenta) with two subunits of gp16 (ribbons in green and surface representation in light green) of HTIFull. Gp15 residues which when mutated disrupt assembly are displayed as in A. Gp16 residues mutated to cysteine (Figs. 5 and 6) are shown as green spheres. (D) DNA protected from DNase inside viral particles assembled in the presence of different gp15 mutant proteins, as labeled above each gel lane. The assay was carried out in crude lysates of cells infected with wild-type SPP1 and SPP1sus128 as marked below the gel. Purified SPP1 DNA (far left lane) was used as control.
The gp15 region distal from gp6 interacts with gp16. The gp15 tilted helix α0 binds to the outer bending region of the L-shaped gp16 protein, and the gp15 loop α2–α3 contacts the internal region of neighboring gp16 subunits (section 2 in Figs. 1C and 3C). This arrangement is consistent with biochemical results (Fig. 3B). Mutating the residue in loop α2–α3 that mediates this interaction, gp15R73E prevents binding of gp16 to viral capsids (Fig. 3B). Interestingly, capsids carrying the double mutant gp15R5ER8E in α0 contain gp16 (Fig. 3B) but do not retain the packaged SPP1 chromosome as shown by DNase protection (Fig. 3D) and cryoEM observation (Fig. S6). Thus the additional gp15–gp16 interaction at the connector periphery is necessary to accomplish closure of the portal channel by the stopper, effectively bringing residues gp16K48 into close proximity to the end of the DNA density (Fig. S7A). We hypothesize that gp16 binding to α0 of gp15 is necessary for the correct positioning of the gp16 central β-sheet core of each subunit in the gp16 dodecamer. This structural constellation directs folding of the gp16 β2′–β3 loops to assemble the intermolecular stopper structure that locks the portal central channel (Fig. 4) (13). This 20-Å-long distance effect reveals an allosteric mechanism for closure of the phage DNA gatekeeper system (Fig. 4A).
Fig. 4.

Gp16 stopper gating mechanism. (A) A diagram of gp16 structural rearrangements during morphogenesis of the viral particle and DNA release. (B) Top views of gp16 fitted in the EM maps of the connector (Left), HTIFull (Center Left), and HTIEmpty (Right). The diaphragm model of gp16 stopper opening for DNA release is depicted as a cartoon between HTIFull and HTIEmpty.
Gp16 plays dual roles as a DNA gatekeeper for the capsid portal system and as a docking interface for the tail during virus assembly. Binding of the tail tapered by gp17 (14, 15) induces changes in the position of loops β1/β2 of gp16 that protrude downward at the molecule periphery (Fig. S7B). The β-sheet β1/β2 and the long loop connecting its two strands adopt more vertical positions than in the connector of tailless particles (13). They stretch down by ∼8 Å to embrace gp17 and shift away from their 12-fold symmetrical positions to sixfold symmetry (section 3 in Figs. 1C and 4B, Center Left). This shift may be caused by the interaction with helices α1 and α2 of the gp17 subunit, with each helix contacting a different gp16 subunit (Fig. S7B). The central channel of the HTI remains closed by the gp16 stopper whose structure is maintained to delimit a narrow pore ∼17 Å in diameter (Fig. 4B).
Gp16 Regulates DNA Release from the Capsid.
The gp16 stopper is closed in both the HTIFull and HTIEmpty states. Although the HTIEmpty state is triggered by interaction of the receptor ectodomain YueB780 with the tail tip, it cannot be concluded that the stopper opens and closes during this process because the DNA might have exited through the connector rather than moving down the tail. We also note that disruption of the phage capsid during preparation of the HTI samples leads to dissipation of the pressure that drives DNA ejection through the tail tube in intact virions (35). To determine if the stopper opens for DNA ejection and recloses afterward, we performed disulfide cross-linking experiments using infectious phage particles. The stopper was proposed to be a propeller-like β-sheet composed of 12 parallel strands, each contributed by a single gp16 subunit (13). The parallel β-strands can be cross-linked by intersubunit disulfide bridges between cysteines, substituting the same residue of the stopper in adjacent gp16 subunits (lanes 1 in Fig. 5) (13). The very precise distance and geometry constraints between sulfhydryl groups required to form disulfide bridges renders them exquisite molecular rulers to probe a specific protein conformation (refs. 25 and 36 and references therein). Stopper intersubunit disulfide bonds in mutant SPP1 virions were reduced (“red.” in Fig. 5, lanes 2–9) in control experiments (Fig. 5, lanes 2–5) and in experiments in which SPP1 DNA subsequently was ejected through the stopper upon incubation of phages with YueB780 (“DNA rel.” in Fig. 5, lanes 6–9). Oxidation after genome release from viral particles led to efficient reformation of disulfide bridges between adjacent gp16 subunits in phages that had ejected their DNA (“oxid.” in Fig. 5, lane 8). After DNA passage, the stopper cysteine residues thus repositioned back to a precise 3D arrangement. This conformation allowed effective intersubunit disulfide cross-linking, indicating that the stopper reacquired its closed state. The mechanism of gp16 opening for DNA movement through the tail tube thus requires dissociation and reassembly of the stopper propeller β-sheet. Reformation of disulfide bridges is less efficient in intact phages (control not incubated with YueB780) whose stopper is only partially cross-linked (mutants gp16Q51C and gp16Q43CQ51C; Fig. 5, lane 4). This reduced efficiency is probably caused by DNA interacting with the stopper which disturbs the favorable positioning of cysteines for reformation of disulfide bridges.
Fig. 5.

Reclosure of the gp16 stopper after DNA ejection. Wild-type SPP1 and phages with gp16 cysteine mutations were reduced (red.) with DTT and challenged or not (controls) with YueB780 to trigger DNA ejection (DNA rel.) followed, or not, by oxidation (oxid.) with DTNB. A final step of reduction with DTT (red.) was carried out, or not, to control that intersubunit cross-links were caused by disulfide bridges that are sensitive to reduction. The complete experimental procedure is detailed in Materials and Methods. Gp16 forms with n cross-linked subunits are labeled on the left of the Western blots.
We have found that some preparations of virions carrying gp16Q43CQ51C contain two subpopulations of particles. One subpopulation contains particles that fully eject DNA upon challenge with YueB780 under oxidizing conditions. Presence of these phages is identifiable by a reduction in the total amount of DNA protected from DNase attack inside viral particles (lanes 7 and 9 of the gel in Fig. 6A). The other subpopulation did not eject (full-length chromosome band protected from DNase) or ejected DNA only partially (smear of shorter protected DNA molecules highlighted by white brackets in lanes 7 and 9 of Fig. 6A). The presence of partially DNA-filled SPP1 particles was confirmed by EM observation (Fig. 6B). Reduction of disulfide bridges with DTT led to complete DNA ejection in the great majority of phage particles (lanes 8 and 10). Cross-links in the gp16 stopper thus impaired (no ejection) or imposed a physical constraint for DNA movement to exit from the viral particle. Viruses trapped during genome release are attributed to slow DNA passage through partially cross-linked gp16 whose presence is detected in Western blots of the corresponding phage particle preparations (e.g., lane 1 of the third gel from top in Fig. 5).
Fig. 6.
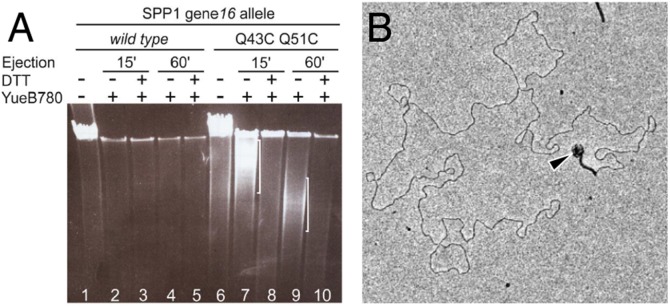
DNA ejection in gp16 stopper mutant virions. (A) DNA ejection from SPP1 gp16Q43CQ51C monitored by DNase protection after incubation with YueB780. DNA was separated by pulse-field gel electrophoresis. White brackets identify protected DNA in phage particles that have partially ejected their genome. SPP1 with wild-type gp16 was used as a control. (B) EM of a SPP1 gp16Q43CQ51C virion challenged with YueB780. The phage particle capsid partially filled with DNA is indicated by an arrowhead.
Conclusion
Our results demonstrate that assembly of the HTI and DNA release from the SPP1 virion is accomplished by subtle structural rearrangements in the HTI proteins. A crucial player is gp16 that forms the stopper by an allosteric mechanism to retain the viral genome and that opens for DNA ejection during infection (Fig. 4). Hindrance with DNA flow by the gp16 stopper can reduce the rate of DNA exit from the virion (the gp16Q43CQ51C mutant; Fig. 6). After DNA exit the gp16 stopper recloses (Fig. 5), suggesting motion by its central propeller intersubunit parallel β-sheet resembling that of a camera diaphragm of (Fig. 4B). A diaphragm-like opening movement also was proposed for gp15 of phage T4 upon contraction of the T4 tail sheath (37). T4 gp15 is structurally related to SPP1 gp17 (5, 14). We propose that the closed conformation of SPP1 gp16 is the system-stable state whose opening is imposed by signaling for genome release. Reclosure of the stopper after DNA ejection might be a mechanism to prevent the loss of cellular components, including ions, when a continuous hydrophilic channel is established between the phage interior and the bacterial cytoplasm. The coordinated action of the different HTI components thus is essential to ensure the timing of stopper opening for free flow of DNA. This sophisticated part of mechanics combines features of a flexible joint that can act as a camera aperture with robust architecture that can withstand the strong forces produced by the internal pressure of packed DNA at the beginning of the DNA ejection process [47 ± 6 atm for wild-type SPP1 (35)]. The ability of the HTI to serve as gatekeeper for the viral genome is a key requirement for building viruses with a capsid container carrying tightly packed DNA combined with a long tail tube device for delivery of genetic information to the host cell, the most successful virion design for infecting bacteria.
Materials and Methods
Microbiology, Molecular Biology, and Genetic Methods.
A detailed description of microbiology methods, cloning procedures, mutagenesis, transfer of mutations to the SPP1 phage genome, and purification of viral particles is provided in SI Materials and Methods. Plasmids and oligonucleotides are listed in Tables S1 and S2, respectively.
Assay for Stable DNA Packaging in SPP1 Particles in Vivo.
Detection of DNA packaged in vivo was carried out by a DNase protection assay in infected cell lysates as described in SI Materials and Methods.
DNA Ejection Assays in Vitro.
Ejection of DNA triggered by incubation with receptor YueB780 (22) was monitored by DNase protection as described in SI Materials and Methods.
Disulfide Bridges Assay.
The presence of disulfide bridges in phage particles with mutated gp16 that contains cysteine residues was assayed as described (13). To assess reformation of disulfide bridges in the stopper after DNA ejection (Fig. 5), phages carrying the engineered gp16 forms were first incubated with 4 mM DL-DTT (Euromedex) for 15 min at 37 °C for reduction of the bridges. The total NaCl concentration then was raised to 300 mM, and phages were incubated with YueB780 (12 µg/mL), or with receptor buffer (22) as control, for 1 h at 37 °C in the presence of 3 µg/µL DNase (Sigma) for DNA ejection. DNase was tested to be active enough under these reducing conditions to digest free DNA fully. The reactions were dialyzed overnight in microdialysis filters (Pierce) against 300 mM NaCl, 100 mM Tris-Cl, 10 mM MgCl2, pH 7.5, and then were incubated with or without 100 µM 5,5′-dithio-bis(2-nitrobenzoic acid (DTNB) (Sigma) for 30 min at 20 °C. After incubation with or without DTT (4 mM, 15 min, 37 °C), the different samples were treated with 10 mM N-ethylmaleimide (Sigma) (1 h, 37 °C) to alkylate free sulfhydryl groups. Gp16 was detected by Western blot after separation of phage proteins by SDS/PAGE in the absence of reducing agents (25). The experiment was repeated at least three times for each SPP1 mutant phage.
Samples Preparation of Samples for EM.
SPP1 wild-type bacteriophages (38) (∼1011 infectious particles in a volume of 30–60 µL) were incubated with 50 mM EDTA at 55 °C for 30 min for particle disruption (Fig. S1A). MgCl2 (100 mM) was added, and free DNA was digested with 37.5 U Benzonase. An additional 37.5 U of Benzonase (Merck) were added after 1 h, and the incubation was continued in a 37 °C room overnight. The method’s efficiency in yielding disrupted capsids and tails with connectors was monitored by EM of negatively stained samples to observe viral structures and of samples, untreated with Benzonase, that were adsorbed to mica to visualize DNA (20). The sample was split in two and the salt concentration was raised to 300 mM NaCl. One sample was incubated with YueB780 (109 µg/mL), and the other was mixed with receptor buffer (22). After incubation for 2 h at 37 °C the samples were diluted threefold with 50 mM NaCl, 50 mM Tris-Cl, 5 mM MgCl2 (pH 7.5) and applied immediately to a grid for freezing in liquid nitrogen in an FEI Vitrobot system. Controls with SPP1 particles processed in parallel but not treated with EDTA maintained their infectivity, structural integrity, and ability to eject DNA when challenged with YueB780. Key parameters for good preparations for cryoEM were the full digestion of DNA and reduction of the sample concentration by dilution. Additional purification steps by differential centrifugation, sedimentation, ion exchange, or size-exclusion chromatography failed to improve the quality and homogeneity of the preparation.
EM and Image Analysis.
EM and data collection were carried out on a Tecnai FEG Polara Microscope (FEI) operated at 300 kV acceleration voltage. Data were recorded on Kodak SO-163 film at a magnification of 39,000. The defocus range for the data was 0.9–3.6 μm. Micrographs were scanned using a Heidelberg Primescan drum scanner at a resulting pixel size of 1.15 Å at the specimen level. The contrast transfer function (CTF) was estimated using the CTFIT program [EMAN (39)], and correction was done by phase flipping using SPIDER (40). Before CTF correction, boxes were normalized, and density variation in each box was limited to 5σ. The initial dataset of ∼8,000 particles was selected manually with BOXER (EMAN). After a preliminary 3D model became available (SI Materials and Methods), further particle selection was done semiautomatically using SPIDER (40) based on cross-correlation to the projections of the 3D model. Altogether, ∼22,000 particles of SPP1 HTI regions were selected from the EDTA-treated SPP1 sample, and ∼24,000 particles were selected from the EDTA/YueB780-treated sample. Particle alignment steps, multivariate statistical analysis (MSA), angular reconstitution, 3D reconstruction, and structure refinement were performed using the IMAGIC-5 software package (41). The image processing steps are described in SI Materials and Methods. The final reconstructions included ∼14,000 and ∼18,000 particle images for HTIFull and HTIEmpty complexes, respectively. Resolution of reconstructions has been asses using the Fourier Shell Correlation at the threshold 0.5.
Fitting of Atomic Models into EM 3D Maps.
Initial rigid body fit of 3D models of gp6 [Protein Data Bank (PDB) ID code 2JES (11)], gp15 [PDB ID code 2KBZ (13)], gp16 [PDB ID code 2KCA (13)], and gp17 [PDB ID code 2LFP (14)] into the EM density maps was done using local automated fitting in Chimera (42). After symmetrization of the fitted atomic models, clashes between adjacent subunits were removed using VEDA (UROX) software (43). Residues 170–238 were not defined in the portal gp6 crystallographic structure, so their initial atomic model was obtained using I-TASSER (23); then their position was refined during the fitting. To obtain the initial model of the gp17.1 tail protein whose atomic structure is not available, we used the I-TASSER server to predict its atomic organization, which is closely related to gpV of lambda (44) and Hcp1 of the type VI secretion system (45). Several models for gp17.1 were generated by I-TASSER. To choose the best one, we considered the correlation coefficient in the corresponding density in the cryo-EM maps and also the position of the N and C termini in the model. The model with the highest correlation was selected for further refinement of docking. To improve the fit and to localize protein–protein contacts, flexible fitting was done using Modeler FlexEM software (see Table S3) (46). For details see SI Materials and Methods.
Structure illustrations were prepared with PyMOL (47) and Chimera (42).
Supplementary Material
Acknowledgments
We thank Dr. M.-C. Vaney for fruitful discussions and important advice in structure analysis; Dr. T. Mielke and J. Bürger for support in sample preparation and cryoEM; and an anonymous referee for the suggestion that stopper closure acts to prevent release of cytoplasm components from the infected bacterium. Research was supported by Biotechnology and Biological Sciences Research Council Grant BB/F012705/1 (to E.V.O.) and by French National Research Agency Grant “DNA Gating” (to P.T. and S.Z.-J.). Institutional funding was provided by CNRS (P.T. and S.Z.-J.), the Max Planck Institute for Molecular Genetics (R.L.), and Alternative Energies and Atomic Energy Commission (S.Z.-J.). The Wellcome trust provided the EM equipment for E.V.O.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.E.J. is a guest editor invited by the Editorial Board.
Data deposition: The EM maps of the HTI complexes have been deposited in the Electron Microscopy Data Bank, www.ebi.ac.uk/pdbe/ (accession codes EMD2993 and EMD2994) and in the Protein Data Bank, www.pdb.org (ID codes 5A20 and 5A21).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1504039112/-/DCSupplemental.
References
- 1.Bazinet C, King J. The DNA translocating vertex of dsDNA bacteriophage. Annu Rev Microbiol. 1985;39:109–129. doi: 10.1146/annurev.mi.39.100185.000545. [DOI] [PubMed] [Google Scholar]
- 2.Rao VB, Feiss M. The bacteriophage DNA packaging motor. Annu Rev Genet. 2008;42:647–681. doi: 10.1146/annurev.genet.42.110807.091545. [DOI] [PubMed] [Google Scholar]
- 3.Tavares P, Zinn-Justin S, Orlova EV. Genome gating in tailed bacteriophage capsids. Adv Exp Med Biol. 2012;726:585–600. doi: 10.1007/978-1-4614-0980-9_25. [DOI] [PubMed] [Google Scholar]
- 4.Orlova EV, et al. Structure of a viral DNA gatekeeper at 10 A resolution by cryo-electron microscopy. EMBO J. 2003;22(6):1255–1262. doi: 10.1093/emboj/cdg123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fokine A, et al. The molecular architecture of the bacteriophage T4 neck. J Mol Biol. 2013;425(10):1731–1744. doi: 10.1016/j.jmb.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veesler D, Cambillau C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol Mol Biol Rev. 2011;75(3):423–433. doi: 10.1128/MMBR.00014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plisson C, et al. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 2007;26(15):3720–3728. doi: 10.1038/sj.emboj.7601786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hambly E, Suttle CA. The viriosphere, diversity, and genetic exchange within phage communities. Curr Opin Microbiol. 2005;8(4):444–450. doi: 10.1016/j.mib.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Oliveira L, Tavares P, Alonso JC. Headful DNA packaging: Bacteriophage SPP1 as a model system. Virus Res. 2013;173(2):247–259. doi: 10.1016/j.virusres.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 10.White HE, et al. Capsid structure and its stability at the late stages of bacteriophage SPP1 assembly. J Virol. 2012;86(12):6768–6777. doi: 10.1128/JVI.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebedev AA, et al. Structural framework for DNA translocation via the viral portal protein. EMBO J. 2007;26(7):1984–1994. doi: 10.1038/sj.emboj.7601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliveira L, Cuervo A, Tavares P. Direct interaction of the bacteriophage SPP1 packaging ATPase with the portal protein. J Biol Chem. 2010;285(10):7366–7373. doi: 10.1074/jbc.M109.061010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lhuillier S, et al. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc Natl Acad Sci USA. 2009;106(21):8507–8512. doi: 10.1073/pnas.0812407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chagot B, et al. Solution structure of gp17 from the Siphoviridae bacteriophage SPP1: Insights into its role in virion assembly. Proteins. 2012;80(1):319–326. doi: 10.1002/prot.23191. [DOI] [PubMed] [Google Scholar]
- 15.Auzat I, Petitpas I, Lurz R, Weise F, Tavares P. A touch of glue to complete bacteriophage assembly: The tail-to-head joining protein (THJP) family. Mol Microbiol. 2014;91(6):1164–1178. doi: 10.1111/mmi.12526. [DOI] [PubMed] [Google Scholar]
- 16.Auzat I, Dröge A, Weise F, Lurz R, Tavares P. Origin and function of the two major tail proteins of bacteriophage SPP1. Mol Microbiol. 2008;70(3):557–569. doi: 10.1111/j.1365-2958.2008.06435.x. [DOI] [PubMed] [Google Scholar]
- 17.Goulet A, et al. The opening of the SPP1 bacteriophage tail, a prevalent mechanism in Gram-positive-infecting siphophages. J Biol Chem. 2011;286(28):25397–25405. doi: 10.1074/jbc.M111.243360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vinga I, et al. Role of bacteriophage SPP1 tail spike protein gp21 on host cell receptor binding and trigger of phage DNA ejection. Mol Microbiol. 2012;83(2):289–303. doi: 10.1111/j.1365-2958.2011.07931.x. [DOI] [PubMed] [Google Scholar]
- 19.Langlois C, et al. Bacteriophage SPP1 tail tube protein self-assembles into β-structure-rich tubes. J Biol Chem. 2015;290(6):3836–3849. doi: 10.1074/jbc.M114.613166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tavares P, Lurz R, Stiege A, Rückert B, Trautner TA. Sequential headful packaging and fate of the cleaved DNA ends in bacteriophage SPP1. J Mol Biol. 1996;264(5):954–967. doi: 10.1006/jmbi.1996.0689. [DOI] [PubMed] [Google Scholar]
- 21.Lurz R, et al. Structural organisation of the head-to-tail interface of a bacterial virus. J Mol Biol. 2001;310(5):1027–1037. doi: 10.1006/jmbi.2001.4800. [DOI] [PubMed] [Google Scholar]
- 22.São-José C, et al. The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J Biol Chem. 2006;281(17):11464–11470. doi: 10.1074/jbc.M513625200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40–49. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas JO. Chemical linkage of the tail to the right-hand end of bacteriophage lambda DNA. J Mol Biol. 1974;87(1):1–9. doi: 10.1016/0022-2836(74)90555-5. [DOI] [PubMed] [Google Scholar]
- 25.Cuervo A, Vaney MC, Antson AA, Tavares P, Oliveira L. Structural rearrangements between portal protein subunits are essential for viral DNA translocation. J Biol Chem. 2007;282(26):18907–18913. doi: 10.1074/jbc.M701808200. [DOI] [PubMed] [Google Scholar]
- 26.Simpson AA, et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature. 2000;408(6813):745–750. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guasch A, et al. Detailed architecture of a DNA translocating machine: The high-resolution structure of the bacteriophage phi29 connector particle. J Mol Biol. 2002;315(4):663–676. doi: 10.1006/jmbi.2001.5278. [DOI] [PubMed] [Google Scholar]
- 28.Grimes S, Ma S, Gao J, Atz R, Jardine PJ. Role of φ29 connector channel loops in late-stage DNA packaging. J Mol Biol. 2011;410(1):50–59. doi: 10.1016/j.jmb.2011.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fang H, Jing P, Haque F, Guo P. Role of channel lysines and the “push through a one-way valve” mechanism of the viral DNA packaging motor. Biophys J. 2012;102(1):127–135. doi: 10.1016/j.bpj.2011.11.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Padilla-Sanchez V, et al. Structure-function analysis of the DNA translocating portal of the bacteriophage T4 packaging machine. J Mol Biol. 2014;426(5):1019–1038. doi: 10.1016/j.jmb.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olia AS, Prevelige PE, Jr, Johnson JE, Cingolani G. Three-dimensional structure of a viral genome-delivery portal vertex. Nat Struct Mol Biol. 2011;18(5):597–603. doi: 10.1038/nsmb.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L, et al. Icosahedral bacteriophage ΦX174 forms a tail for DNA transport during infection. Nature. 2014;505(7483):432–435. doi: 10.1038/nature12816. [DOI] [PubMed] [Google Scholar]
- 33.Isidro A, Henriques AO, Tavares P. The portal protein plays essential roles at different steps of the SPP1 DNA packaging process. Virology. 2004;322(2):253–263. doi: 10.1016/j.virol.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 34.Cardarelli L, et al. The crystal structure of bacteriophage HK97 gp6: Defining a large family of head-tail connector proteins. J Mol Biol. 2010;395(4):754–768. doi: 10.1016/j.jmb.2009.10.067. [DOI] [PubMed] [Google Scholar]
- 35.São-José C, de Frutos M, Raspaud E, Santos MA, Tavares P. Pressure built by DNA packing inside virions: Enough to drive DNA ejection in vitro, largely insufficient for delivery into the bacterial cytoplasm. J Mol Biol. 2007;374(2):346–355. doi: 10.1016/j.jmb.2007.09.045. [DOI] [PubMed] [Google Scholar]
- 36.Petersen MTN, Jonson PH, Petersen SB. Amino acid neighbours and detailed conformational analysis of cysteines in proteins. Protein Eng. 1999;12(7):535–548. doi: 10.1093/protein/12.7.535. [DOI] [PubMed] [Google Scholar]
- 37.Kostyuchenko VA, et al. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat Struct Mol Biol. 2005;12(9):810–813. doi: 10.1038/nsmb975. [DOI] [PubMed] [Google Scholar]
- 38.Riva S, Polsinelli M, Falaschi A. A new phage of Bacillus subtilis with infectious DNA having separable strands. J Mol Biol. 1968;35(2):347–356. doi: 10.1016/s0022-2836(68)80029-4. [DOI] [PubMed] [Google Scholar]
- 39.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128(1):82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 40.Frank J, et al. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116(1):190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 41.van Heel M, et al. Single-particle electron cryo-microscopy: Towards atomic resolution. Q Rev Biophys. 2000;33(4):307–369. doi: 10.1017/s0033583500003644. [DOI] [PubMed] [Google Scholar]
- 42.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Siebert X, Navaza J. UROX 2.0: An interactive tool for fitting atomic models into electron-microscopy reconstructions. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 7):651–658. doi: 10.1107/S0907444909008671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pell LG, Kanelis V, Donaldson LW, Howell PL, Davidson AR. The phage lambda major tail protein structure reveals a common evolution for long-tailed phages and the type VI bacterial secretion system. Proc Natl Acad Sci USA. 2009;106(11):4160–4165. doi: 10.1073/pnas.0900044106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mougous JD, et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science. 2006;312(5779):1526–1530. doi: 10.1126/science.1128393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topf M, et al. Protein structure fitting and refinement guided by cryo-EM density. Structure. 2008;16(2):295–307. doi: 10.1016/j.str.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.