

Abstract
It has been nearly 40 y since it was suggested that genomic methylation patterns could be transmitted via maintenance methylation during S phase and might play a role in the dynamic regulation of gene expression during development [Holliday R, Pugh JE (1975) Science 187(4173):226–232; Riggs AD (1975) Cytogenet Cell Genet 14(1):9–25]. This revolutionary proposal was justified by “... our almost complete ignorance of the mechanism for the unfolding of the genetic program during development” that prevailed at the time. Many correlations between transcriptional activation and demethylation have since been reported, but causation has not been demonstrated and to date there is no reasonable proof of the existence of a complex biochemical system that activates and represses genes via reversible DNA methylation. Such a system would supplement or replace the conserved web of transcription factors that regulate cellular differentiation in organisms that have unmethylated genomes (such as Caenorhaditis elegans and the Dipteran insects) and those that methylate their genomes. DNA methylation does have essential roles in irreversible promoter silencing, as in the monoallelic expression of imprinted genes, in the silencing of transposons, and in X chromosome inactivation in female mammals. Rather than reinforcing or replacing regulatory pathways that are conserved between organisms that have either methylated or unmethylated genomes, DNA methylation endows genomes with the ability to subject specific sequences to irreversible transcriptional silencing even in the presence of all of the factors required for their expression, an ability that is generally unavailable to organisms that have unmethylated genomes.
Keywords: DNA methylation, differentiation, development
Structure of Genomic Methylation Patterns
The addition of a fifth base (5-methylcytosine or m5C) increases the maximum potential information content of DNA from 2 bits per base pair to 2.32 bits; the addition of naturally occurring oxidized forms of m5C (5-hydroxymethycytosine, 5-formylcytosine, and 5-carboxylcytosine) increases the information content still further, although m5C is much more abundant than the oxidized derivatives. The assembled and annotated fraction of the human genome contains ∼29 million CpG dinucleotides, each of which can exist in the methylated or unmethylated state. The number of possible methylation patterns per haploid genome is therefore 2 to the power of 29 million, or ∼108,700,000. DNA methylation increases the information content of the genome, promoter methylation represses transcription in experimental systems (1, 2), and regulated changes in genomic methylation patterns could in principle form the basis of a system that regulates gene expression. However, it is far from certain that such a regulatory system exists, and a set of uniform criteria that could be applied to identify genes that are regulated by dynamic DNA methylation has yet to be formulated and applied.
The CpG dinucleotide occurs at only ∼20% of the expected frequency and varies in density to a much greater degree than any other dinucleotide. Introns, 3′ untranslated regions, and intergenic sequences are severely depleted in CpG, whereas coding exons have a somewhat higher density with an unexplained spike in CpG density near splice donor and acceptor sites (3). Mammalian promoters have strongly bimodal CpG density distributions; CpG-rich promoters have a modal CpG density of 4.25 per 100-bp sites and are found at 75% of all genes; CpG-poor promoters have a modal density of 0.6 CpG sites per 100 bp and are found at the remaining 25% of genes (Fig. 1A).
Fig. 1.

CpG and m5C densities at mammalian promoters. (A) Strongly bimodal distribution of CpG densities in the region spanning –1,000 to +1,000 relative to TSS with nearly constant m5C density across all CpG densities in adult human brain DNA (7) and adult HMECs (39). (B) Nearly identical methylation densities across all promoter regions (defined as in A) in genomes from human brain and from HMECs.
It has recently become possible to obtain whole-genome methylation profiles at single CpG resolution (4–6). These data have shown that tandem and dispersed repeated sequences (especially transposons and their remnants) tend to be most heavily methylated; single-copy sequences of low-to-moderate CpG density (such as most exons, introns, and intergenic regions) tend to be heterogeneously methylated within a population of cells; and CpG-rich promoter regions (also known as promoter CpG islands) are almost exclusively unmethylated in all tissue types, with a methylation minimum and a CpG density maximum at the transcriptional start site (TSS). Enhancer sequences (as defined by their enrichment in histone H3 monomethylated at lysine 4 and by association with P300) have extremely low CpG densities, and even if methylated at all CpG sites, contain very little m5C; CpG islands that are methylated at only a few percent of all CpG sites will contain more m5C despite being largely unmethylated.
As shown in Fig. 1A, the methylation density at promoter regions is very low (∼0.6 methylated sites per 100 bp) and is surprisingly constant across a wide range of CpG densities; this methylation density does not differ between adult human brain (which expresses more genes than other tissues) and adult human mammary endothelial cells (HMECs) (Fig. 1B). The wide variation in CpG densities (much greater than any other dinucleotide) across the mammalian genome means that fractional methylation does not describe methylation status; sequences that are 100% methylated but CpG poor can contain much less m5C than CpG-rich sequences that are methylated to the extent of only a few percent. Methylation data are often presented only as fractional methylation, which is not informative in the absence of explicit CpG distributions.
The large majority of CpG-rich promoters are unmethylated in all cell types, regardless of state of expression (4–6); CpG-poor promoters can acquire variable degrees of DNA methylation when in the repressed or silent state. The variable methylation of CpG-poor promoters is unlikely to be involved in repression of the gene. The pluripotency genes Nanog and Oct4/Pou5f1 have been reported to be regulated by reversible promoter methylation (7). As shown in Fig. 2A, both genes have very CpG-poor promoters, and in adult differentiated human mammary tissue, where the genes are not expressed, a significant fraction of both genes are completely unmethylated across their promoter regions. The fact that the genes are not expressed in any cell in the population while the promoters are unmethylated in part of that population requires that DNA methylation cannot be the mechanism of repression. This might be used as a criterion for assessing the role of methylation in repression: If any appreciable fraction of the promoters for a given gene is unmethylated in a fraction of a population of nonexpressing cells, methylation is highly unlikely to be the mechanism by which that gene is repressed.
Fig. 2.

Transcription-dependent methylation transitions. (A) The NANOG and OCT4 pluripotency genes have been reported to be regulated by reversible DNA methylation of their promoters and to be completely methylated in nonexpressing tissues (10). Both genes have very CpG-poor promoters, and methylation analysis by paired-end sequencing (8) shows that a substantial number of cells from normal adult mammary tissue show completely unmethylated promoters for both genes. Terminal green CpG dinucleotides are methylated and internal green CpG dinucleotides are unmethylated, whereas terminal red CpGs are unmethylated and internal CpGs are unmethylated. None of the cells express either gene. The presence of unmethylated promoters in the nonexpressing population requires that promoter methylation cannot be the mechanism of repression. Note that the primate-specific AluSx1 retrotransposon 5′ of the NANOG TSS is heavily methylated whereas the proximal promoter is largely unmethylated. (B) Cartoon representation of transcription-dependent methylation and demethylation. (Top) CpG-poor promoters are postulated to acquire partial methylation when not expressed, as in the case of the NANOG and OCT4 genes in A; the binding of transcription factors leads to a loss of methylation when the gene is activated (12–17). Under this model, changes in methylation density are a result of transcriptional activation rather than a cause. (Bottom) Constitutive demethylation of CpG-dense/CpG island promoters in both expressing and nonexpressing cell types.
Earlier claims of very large changes in genomic methylation patterns with age (8) have not been substantiated by the results of whole-genome methylation profiling. As shown in the example of Fig. 3, the methylation status of the genomic region that contains the imprinted genes H19 and Igf2 does not notably differ between fetal and aged mouse brain, even though expression of H19 and Igf2 ceases soon after birth. The sex-specific methylation difference established during gametogenesis at the imprinting control region/differentially methylated region persists throughout the lifespan of the individual, and pronounced age-related changes in methylation patterns are not apparent in whole-genome methylation data.
Fig. 3.
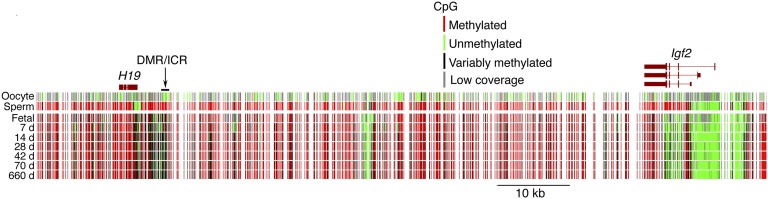
Faithful transmission of genomic methylation patterns with advancing age in mouse brain DNA. The paternal differentially methylated region or imprinting control region (DMR/ICR) undergoes de novo methylation in male but not female germ cells; this results of the paternal allele of Igf2 and the maternal allele of H19 in mesodermal tissues of embryonic and fetal mice. Both genes are silenced in adult tissues but the Igf2 promoters remain unmethylated. Methylation patterns across the region are very similar throughout the lifespan of the animals, and this trend holds true for the large majority of the genome. Methylation patterns are from the data described in ref. 7.
Transcriptional Activation Causes Loss of Methylation
Promoter sequences show a strong bias against CpG dinucleotides that lie with transcription factor binding sites (9). The many published correlations between the loss of methylation and transcriptional activation (almost all at CpG-poor promoters or at CpG sites not actually within promoters) during development are likely to reflect a consequence of transcriptional activation rather than a cause. This is consistent with numerous reports of a loss of methylation from local CpG sites after binding of transcription factors such as Sp1 (10–13); DNA methylation is even reduced around methylated lac operator transgenes upon the binding of the lac repressor in cultured mammalian cells (14). The transcription-induced demethylation hypothesis is depicted in Fig. 2B, where CpG-rich promoters remain largely unmethylated regardless of state of expression, whereas CpG-poor promoters drift toward the partially methylated state during prolonged inactivity and the methylation is removed upon the activation of transcription. In the case of the inducible IL4 gene, before induction of expression in primary human T lymphocytes, the gene bears a largely unmethylated CpG-poor promoter but methylated coding regions; after induction of transcription, the unmethylated domain progressively extends from the promoter through the gene body (15). Examination of CpG sites downstream of the promoter reveal a strong demethylation–activation correlation, but there is a much weaker correlation at the promoter, which is largely unmethylated in both expressing and nonexpressing cells. As stated previously, this and many of the other reported correlations between expression and demethylation are likely represent a consequence of transcriptional activation rather than a cause.
Genome-Wide Demethylation and Aberrant Gene Expression
If DNA methylation mediates tissue-specific gene expression, global demethylation of the genome should induce ectopic gene expression in the form of coexpression of genes that are normally expressed only in different cells types. In 1984, Weinstein and coworkers treated cultured fibroblastoid C3H10T1/2 cells with the DNA methyltransferase inhibitor 5-azacytidine and observed demethylation of the β-globin genes (16) whose in vivo expression had been shown in 1978 to be correlated with a loss of DNA methylation during erythropoiesis (17). However, 5-azacytidine–induced demethylation did not activate β-globin expression in these nonerythroid cells, and the authors concluded “that loss of DNA methylation is not sufficient per se to induce the expression of certain loci. Presumably, the expression of these loci requires additional factors, some of which may be related to cell lineage and differentiation.”
Other lines of evidence indicate that genome-wide demethylation does not cause gene dysregulation of the sort that would be expected after ablation of a putative global system of developmental gene control. Deletion of the DNA methyltransferase 1 (Dnmt1) gene in mouse embryonic stem (ES) cells leads to the loss of nearly all genomic m5C, and mouse embryos homozygous for Dnmt1 mutations lose most m5C. Dnmt1 mutant mouse embryos did not show activation of genes that had been reported to be repressed by methylation, even though the genes were severely demethylated (18). ES cells retain a normal phenotype in the absence of DNA methylation, but demethylation induces cell-autonomous apoptosis in all differentiated cells via an unknown pathway (19). Gene expression profiling in short-term cultures of primary fibroblasts that lacked TP53 and DNMT1 after excision of a conditional allele of Dnmt1 showed that fewer than 10% of tested genes changed expression level under conditions where many of the cells in the population were undergoing apoptosis, and except for intracisternal A-particle retrotransposons the magnitude of the expression changes was generally small (20). Kono and coworkers (21) analyzed gene expression in growing oocytes that had severe undermethylation of single-copy DNA sequences as a result of removal of DNMT3L, a factor that targets DNA methyltransferase complexes to DNA bound by histone H3 unmethylated at lysine 4 (22). Gene expression was essentially identical in the unmethylated and normally methylated oocytes. When Dnmt3L-null oocytes that are largely free of DNA methylation at single-copy sequences are fertilized by wild-type sperm, developmental abnormalities in the resulting embryos are largely attributable to dysregulation of imprinted genes rather than ectopic derepression of tissue-specific genes (23, 24).
5-Azacytidine–induced genome-wide demethylation does induce myogenic differentiation of C3H10t1/2 cells (25) as a result of demethylation and activation of the MyoD gene (26), but this is an artifact of cell culture; the MyoD gene is unmethylated in nonmuscle tissues (27). The promoters of many tissue-specific genes that are not methylated in nonexpressing tissues acquire ectopic de novo methylation in established lines of cultured cells (27, 28), and this has led to an exaggerated estimate of the number of promoters that are silenced by promoter methylation.
Genetic Evidence
A large and increasing number of mutations that cause developmental abnormalities in mice and humans have been reported, but very few of these mutations affect genes involved in the establishment or maintenance of genomic methylation patterns. Dominant mutations in human DNMT1 that affect the domain of DNMT1 that interacts with UHRF1 cause a spectrum of sensory and autonomic abnormalities, with ataxia, dementia, sensorineural deafness, and other neurological defects (29). The DNMT1 syndromes are of adult onset without early developmental abnormalities, and postmitotic neurons are unlikely to require maintenance methylation; conditional deletion of Dnmt1 in postmitotic mouse neurons has been reported to have no discernable effect (29). The neurological abnormalities caused by mutations in DNMT1 are probably not caused by methylation abnormalities but rather more likely to be due to intracellular aggregation of the mutant protein, which reportedly having an abnormal intracellular distribution (30). This is consistent with their adult onset and their dominant nature. Individuals with homozygous loss-of-function mutations in Dnmt3B have immunodeficiency, centromere instability, and facial anomalies syndrome (31), with demethylation of satellite 1 and 2 DNA and of other sequences. A variable combined immunodeficiency of unknown cause is the most prominent abnormality, and development of affected individuals is essentially normal; the facial dysmorphia characteristic of the disorder is mild in nature. The TET1 and TET2 proteins, which oxidize the methyl group of m5C, have been attributed important roles in genome demethylation, but mice that lack both proteins are largely viable and fertile (32). This would not be the case if TET1 and TET2 play important roles in programmed DNA demethylation. There are few incipient developmental abnormalities in embryos that bear severely demethylated genomes as a result of homozygous loss of Dnmt1 expression (19); death results from cell-autonomous apoptosis rather than from the gross morphological abnormalities that would be expected to occur if DNA methylation were a major regulator of gene expression. Mice that contain hypomorphic alleles of Dnmt1 have partially demethylated genomes and are small and prone to retrotransposon-induced lymphoma, but lack prominent developmental abnormalities (33). In short, genetic analysis has failed to implicate programmed methylation and demethylation as important regulators of cellular differentiation during embryonic development.
Functions of Genomic Methylation Patterns
The conservation of regulatory pathways among organisms that bear either methylated or unmethylated genomes argues against a major role for DNA methylation in mammalian development, as do the low levels of promoter methylation and lack of ectopic gene activation after reduction or removal of genomic methylation patterns. The many reported correlations between transcriptional activation and demethylation of local sequences can be most parsimoniously explained by a transcription-dependent loss of methylation; this is supported by the ability of common transcription factors to induce a loss of methylation at CpG sites that are in the vicinity of their binding sequences (10–12, 14). Although it is impossible to totally exclude a regulatory function for dynamic DNA methylation in embryonic development [and such regulation may occur during gametogenesis, where methylation patterns are subject to sweeping changes, and as in the case of cancer–testis antigen genes (34)], an assessment of the available data does not support the existence of a biochemical system that regulates embryogenesis by programmed methylation and demethylation of regulatory sequences. Most of the evidence is entirely correlative in nature, and robust criteria that could identify genes regulated by dynamic programmed DNA methylation and demethylation during development are seldom applied. It is not clear that any gene has been shown to be repressed in a cell type normally capable of transcribing the gene by a methylation pattern present on that gene in a nonexpressing tissue.
Strong evidence implicates DNA methylation in irreversible gene silencing. DNA methylation is essential for the monoallelic expression of imprinted genes; a failure to establish methylation at imprinting control regions in growing oocytes results in offspring that show biallelic expression of imprinted genes that are normally expressed only from the paternal allele (23). DNA methylation is also required for the transcriptional silencing of endogenous retroviruses and other retrotransposons in both germ cells and somatic tissues (35, 36). Lastly, demethylation of the Xist locus activates Xist expression on all X chromosomes and causes inactivation of all X chromosomes present in the cell (37). Genomic imprints and transposon methylation are established in germ cells, whereas X inactivation occurs during the periimplantation stages. In all three cases the epigenetic decision to methylate is essentially irreversible over the lifetime of the organism, and in all three cases all of the factors necessary for activation of transcription are present in the cell, as shown by their reactivation upon demethylation. It is suggested that mammalian genomic methylation patterns represent an evolutionary adaptation of a genome defense system that endows genomes with the ability to inactivate specific genomic regions in a self-perpetuating manner which is essentially irreversible over the lifespan of the organism (38), whereas dynamic gene activation and repression during development are controlled by conserved protein- and RNA-based pathways that are largely common to both methylating and nonmethylating organisms.
Acknowledgments
We thank K. Anderson for comments on the manuscript and K. Weinberger for advice. This work was supported by grants from the NIH (to T.H.B. and J.R.E.) and by a grant from the Department of Defense (to J.R.E.).
Footnotes
The authors declare no conflict of interest.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Epigenetic Changes in the Developing Brain: Effects on Behavior,” held March 28–29, 2014, at the National Academy of Sciences in Washington, DC. The complete program and video recordings of most presentations are available on the NAS website at www.nasonline.org/Epigenetic_changes.
This article is a PNAS Direct Submission.
References
- 1.Stein R, Razin A, Cedar H. In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc Natl Acad Sci USA. 1982;79(11):3418–3422. doi: 10.1073/pnas.79.11.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Busslinger M, Hurst J, Flavell RA. DNA methylation and the regulation of globin gene expression. Cell. 1983;34(1):197–206. doi: 10.1016/0092-8674(83)90150-2. [DOI] [PubMed] [Google Scholar]
- 3.Louie E, Ott J, Majewski J. Nucleotide frequency variation across human genes. Genome Res. 2003;13(12):2594–2601. doi: 10.1101/gr.1317703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards JR, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20(7):972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stadler MB, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480(7378):490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 7.Li JY, et al. Synergistic function of DNA methyltransferases Dnmt3a and Dnmt3b in the methylation of Oct4 and Nanog. Mol Cell Biol. 2007;27(24):8748–8759. doi: 10.1128/MCB.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medvedeva YA, et al. FANTOM consortium Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014;15:119. doi: 10.1186/1471-2164-15-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8(19):2282–2292. doi: 10.1101/gad.8.19.2282. [DOI] [PubMed] [Google Scholar]
- 11.Brandeis M, et al. Sp1 elements protect a CpG island from de novo methylation. Nature. 1994;371(6496):435–438. doi: 10.1038/371435a0. [DOI] [PubMed] [Google Scholar]
- 12.Matsuo K, et al. An embryonic demethylation mechanism involving binding of transcription factors to replicating DNA. EMBO J. 1998;17(5):1446–1453. doi: 10.1093/emboj/17.5.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mummaneni P, Yates P, Simpson J, Rose J, Turker MS. The primary function of a redundant Sp1 binding site in the mouse aprt gene promoter is to block epigenetic gene inactivation. Nucleic Acids Res. 1998;26(22):5163–5169. doi: 10.1093/nar/26.22.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han L, Lin IG, Hsieh CL. Protein binding protects sites on stable episomes and in the chromosome from de novo methylation. Mol Cell Biol. 2001;21(10):3416–3424. doi: 10.1128/MCB.21.10.3416-3424.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee DU, Agarwal S, Rao A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity. 2002;16(5):649–660. doi: 10.1016/s1074-7613(02)00314-x. [DOI] [PubMed] [Google Scholar]
- 16.Hsiao WL, Gattoni-Celli S, Kirschmeier P, Weinstein IB. Effects of 5-azacytidine on methylation and expression of specific DNA sequences in C3H 10T1/2 cells. Mol Cell Biol. 1984;4(4):634–641. doi: 10.1128/mcb.4.4.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waalwijk C, Flavell RA. DNA methylation at a CCGG sequence in the large intron of the rabbit beta-globin gene: Tissue-specific variations. Nucleic Acids Res. 1978;5(12):4631–4634. doi: 10.1093/nar/5.12.4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev. 1999;13(1):26–34. doi: 10.1101/gad.13.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 20.Jackson-Grusby L, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27(1):31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi H, et al. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8(1):e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ooi SK, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448(7154):714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294(5551):2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 24.Proudhon C, et al. Protection against de novo methylation is instrumental in maintaining parent-of-origin methylation inherited from the gametes. Mol Cell. 2012;47(6):909–920. doi: 10.1016/j.molcel.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Constantinides PG, Jones PA, Gevers W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature. 1977;267(5609):364–366. doi: 10.1038/267364a0. [DOI] [PubMed] [Google Scholar]
- 26.Lassar AB, Paterson BM, Weintraub H. Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell. 1986;47(5):649–656. doi: 10.1016/0092-8674(86)90507-6. [DOI] [PubMed] [Google Scholar]
- 27.Jones PA, et al. De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proc Natl Acad Sci USA. 1990;87(16):6117–6121. doi: 10.1073/pnas.87.16.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;62(3):503–514. doi: 10.1016/0092-8674(90)90015-7. [DOI] [PubMed] [Google Scholar]
- 29.Fan G, et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21(3):788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein CJ, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43(6):595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu GL, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402(6758):187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 32.Dawlaty MM, et al. Combined deficiency of Tet1 and Tet2 causes epigenetic abnormalities but is compatible with postnatal development. Dev Cell. 2013;24(3):310–323. doi: 10.1016/j.devcel.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaudet F, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300(5618):489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 34.De Smet C, Loriot A. DNA hypomethylation and activation of germline-specific genes in cancer. In: Karpf AR, editor. Advances in Exp Med Biol. 2013. (Springer Science+Business Media, New York) [DOI] [PubMed] [Google Scholar]
- 35.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20(2):116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 36.Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431(7004):96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 37.Panning B, Jaenisch R. DNA hypomethylation can activate Xist expression and silence X-linked genes. Genes Dev. 1996;10(16):1991–2002. doi: 10.1101/gad.10.16.1991. [DOI] [PubMed] [Google Scholar]
- 38.Bestor TH. Cytosine methylation mediates sexual conflict. Trends Genet. 2003;19(4):185–190. doi: 10.1016/S0168-9525(03)00049-0. [DOI] [PubMed] [Google Scholar]
- 39.Hon GC, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22(2):246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]