

Significance
CO2-to-CO electrochemical conversion is a key step in the production of liquid fuels through dihydrogen-reductive Fischer–Tropsch chemistry. Among molecular catalysts, iron porphyrins reduced electrochemically to the Fe(0) state are particularly efficient and led to a deeper understanding of mechanisms involving coupled bond-breaking proton–electron transfer processes. The replacement of nonaqueous solvents by water should make the CO2-to-CO half-cell reaction much more attractive for applications, particularly because it would allow association with a water-oxidation anode through a proton-exchange membrane. Here it is demonstrated that electrochemical CO production catalyzed by a water-soluble iron porphyrin can occur with high catalytic efficiency. Manipulation of pH and buffering then allows conversions from those involving complete CO selectivity to ones with prescribed CO–H2 mixtures.
Keywords: CO2-to-CO conversion, contemporary energy challenges, electrochemistry, catalysis, solar fuels
Abstract
Substitution of the four paraphenyl hydrogens of iron tetraphenylporphyrin by trimethylammonio groups provides a water-soluble molecule able to catalyze the electrochemical conversion of carbon dioxide into carbon monoxide. The reaction, performed in pH-neutral water, forms quasi-exclusively carbon monoxide with very little production of hydrogen, despite partial equilibration of CO2 with carbonic acid—a low pKa acid. This selective molecular catalyst is endowed with a good stability and a high turnover frequency. On this basis, prescribed composition of CO–H2 mixtures can be obtained by adjusting the pH of the solution, optionally adding an electroinactive buffer. The development of these strategies will be greatly facilitated by the fact that one operates in water. The same applies for the association of the cathode compartment with a proton-producing anode by means of a suitable separator.
One of the most important issues of contemporary energy and environmental challenges consists of reducing carbon dioxide into fuels by means of sunlight (1–3). One route toward this ultimate goal is to first convert solar energy into electricity, which will then be used to reduce CO2 electrochemically. Direct electrochemical injection of an electron into the CO2 molecule, forming the corresponding anion radical CO2.− requires a very high energy [the standard potential of the CO2/ CO2.− couple is indeed −1.97 V vs. normal hydrogen electrode (NHE) in N,N′dimethylformamide (DMF)] (4, 5). Electrochemical conversion of CO2 to any reaction product thus requires catalytic schemes that preferably avoid this intermediate. Carbon monoxide may be an interesting step en route to the desired fuels because it can be used as feedstock for the synthesis of alkanes through the classic Fischer–Tropsch process. A number of molecular catalysts for the homogeneous electrochemical CO2-to-CO conversion have been proposed. They mainly derive from transition metal complexes by electrochemical generation of an appropriately reduced state, which is restored by the catalytic reaction. So far, nonaqueous aprotic solvents (mostly DMF and acetonitrile) have been used for this purpose (5–16). Brönsted acids have been shown to boost catalysis. However, they should not be too strong, at the risk of leading to H2 formation at the expense of the CO. Trifluoroethanol and water (possibly in large amounts) have typically played the role of a weak acid in the purpose of boosting catalysis while avoiding hydrogen evolution.
One of the most thoroughly investigated families of transition-metal complex catalysts of CO2-to-CO conversion is that of iron porphyrins brought electrochemically to the oxidation degree 0. The importance of coupling electron transfer and introduction of CO2 into the coordination sphere of iron with proton transfers required by the formation of CO, CO2 + 2e− + 2AH ↔ CO + H2O + 2A−, appeared from the very beginning of these studies. Sustained formation of CO was indeed only achieved upon addition of weak Brönsted (17–19) and Lewis acids (20, 21). Such addition of Brönsted acids, however, opens the undesired possibility that the same catalyst that converts CO2 into CO may also catalyze the reduction of the acid to dihydrogen. This is indeed what happens with Et3NH+ as the acid, limiting the set of acids used for the CO2-to-CO conversion to that of very weak acids such as propanol, water, and, the strongest one, trifluorethanol (22). Since then, the range of acidity has been extended to phenols, which proved compatible with CO faradic yields close to 100% (23, 24). Installation of phenol functionalities in the porphyrin molecules (Scheme 1) even allowed a considerable improvement of catalysis in terms of catalytic Tafel plots (turnover frequency vs. overpotential) with no degradation of the CO (vs. H2) faradaic yield (25–27). The problem should, however, resurface upon going to stronger acids. That competition with hydrogen evolution is a general issue for molecular catalysis of the CO2-to-CO conversion is confirmed by recent findings concerning catalysts derived from terpyridine complexes of first-row transition metals in (90:10, vol:vol) DMF/H2O mixtures (28).
Scheme 1.

Iron-porphyrin catalysts for CO2-to-CO electrochemical conversion.
The results thus obtained in nonaqueous or partially aqueous media enabled the discovery of remarkably efficient and selective catalysts of the CO2-to-CO conversion. They were also the occasion of notable advances in terms of mechanisms and theory of concerted bond-breaking proton–electron transfer (29).
It must, however, be recognized that, from the point of view of practical applications, the use of nonaqueous solvents is not the most exciting aspect of these results. One would rather like to use water as the solvent, which would render more viable the CO2-to-CO half-cell reaction as well as its association with a water-oxidation anode through a proton-exchange membrane. Several approaches are conceivable in this respect. One of these consists of coating the electrode with a film, possibly making use of the water insolubility of the catalyst (see the three last entries of table 3 in ref. 5, refs. 30 and 31, and ref. 32 and references therein). One may also attempt to chemically attach catalyst molecules onto the electrode having in mind their use, or possible use, of the resulting films in water (33, 34).
Our approach consisted of devising a catalyst fully soluble in water and able to convert CO2 to CO selectively as well as efficiently with regard to overpotential and turnover frequency. Full solubility in water, indeed, allows an easy manipulation of pH and buffering of the system. It will also help the design of a full cell associating the cathode compartment with a proton-producing anode by means of a suitable separator (35). The challenges ahead in this endeavor were as follows. CO2 is poorly soluble in water ([CO2] = 0.0383 M) (36). Even more seriously, it is partially converted (Khydration = 1.7 × 10−3) into carbonic acid, CO3H2, which has a first ionization pKa of 3.6, that is, an apparent pKa of 6.4 (36). These features make it a priori possible that the CO2-to-CO conversion might be seriously challenged by H2 evolution from reduction of carbonic acid and/or hydrated protons (37, 38). Indeed, a previous attempt showed poor CO2/H2 selectivity and practically no catalytic effect in cyclic voltammetry (37). In the same vein, Ni cyclam seems to be a selective and efficient catalyst of the CO2-to-CO conversion in water under the condition of being operated with a mercury electrode, pointing to favorable specific interactions of the catalytic species with the mercury surface (38, 39), whereas the use of a carbon electrode leads to much less efficiency in terms of rate (maximal turnover frequency of 90 s−1) and turnover (four cycles) (15).
We have found that the iron tetraphenylporphyrin in which the four paraphenyl hydrogens have been substituted by trimethylammonio groups, hereafter designated as WSCAT (Scheme 1), fulfills these stringent requirements at neutral pHs.
As seen in Fig. 1A, a very high catalytic current is observed in cyclic voltammetry of half a millimolar solution of WSCAT saturated with CO2 at pH 6.7. In the absence of CO2 (Fig. 1B) three successive waves are observed when starting from the FeIII complex with Cl− as counter ions and presumably as axial ligand. As expected, the shape of the first, FeIII/II, wave reflects the strong axial ligation by Cl− (40). The second wave is a standard reversible FeII/I wave. The third wave is the wave of interest for catalysis. It is irreversible, as opposed to what is observed in DMF (Fig. 1C), where all three waves are one-electron reversible waves, including the third FeI/0 wave, as with the simple FeTPP (Scheme 1) (see, e.g., refs. 23 and 24). The irreversibility and somewhat increased current observed here in water presumably reflects some catalysis of H2 evolution from the reduction of water. The considerable increase in current observed in the FeI/0 potential region when CO2 is introduced into the solution is a clear indication that catalysis is taking place. It is roughly similar to what has been observed in DMF under 1 atm CO2 in the presence of a weak acid such as phenol (Fig. 1D and refs. 23 and 24).
Fig. 1.

Cyclic voltammetry of 0.5 mM WSCAT at 0.1 V/s, temperature 21 °C. (A) In water + 0. 1 M KCl brought to pH 6.7 by addition of KOH under 1 atm CO2. (B) Same as A but in the absence of CO2. (C) Same as B but in DMF + 0.1 n-Bu4NBF4. (D) Same as C but under 1 atm CO2, and presence of 3 M phenol.
What is the nature of the catalysis observed in the FeI/0 potential region? Does it involve acid reduction (CO3H2 and/ or H+) or conversion of CO2 into one of the reduction products? The answer is given by the results of preparative-scale electrolyses.
A first series of preparative-scale electrolyses was performed at −0.97 V vs. NHE over electrolysis times between 1 and 4 h. The pH was brought to 6.7 by addition of KOH. The current density was ca. 0.1 mA/cm2 in all cases. CO was found to be largely predominant with formation of only a very small amount of hydrogen. Over five of these experiments the average faradaic yields of detected products were as follows: CO, 90%; H2, 7%; acetate, 1.4%; formate, 0.7%; and oxalate, 0.5%. The catalyst was quite stable during these periods of time. The variation of pH during electrolysis was small, passing from 6.7 to 6.9. The decrease in peak current registered before and after electrolysis was less than 5%.
A longer-duration electrolysis (Fig. 2) was carried out at a somewhat less negative potential, −0.86 V vs. NHE, leading to a quasi-quantitative formation of CO (faradaic yield between 98% and 100%). After 72 h of electrolysis, the current was decreased by approximately half but CO continued to be the only reaction product.
Fig. 2.

Electrolysis of a 0.5 mM WSCAT solution in water + 0.1 M KCl brought to pH 6.7 by addition of KOH under 1 atm CO2 at −0.86 V vs. NHE. Temperature 21 °C. The reaction products were analyzed at the end of each day. Charge passed (Top); current density (Bottom).
As an example of pH manipulation, the addition of a 0.1 M formic acid buffer at pH 3.7 resulted in the exclusive formation of hydrogen. With a 0.1 M phosphate buffer adjusted at the same pH, 6.7, as that where the electrolyses with no additional buffer were carried out, a 50–50 CO–H2 mixture was obtained.
Although deserving a precise kinetic analysis, a likely interpretation of the factors favorable to CO formation against H2 evolution is summarized in Scheme 2. Despite Fe0 porphyrins’ being good catalysts of H2 evolution (22), the fact that the reaction that converts CO2 into CO3H2 is thermodynamically uphill and kinetically slow favors the CO-formation pathway, making it able to resist the fast H2 evolution pathway in pH-neutral media. Another favorable factor is the wealth of H-bonding possibilities offered by water, a factor that has been shown to be quite important in stabilizing the primary Fe0–CO2 adduct and therefore in boosting catalysis as transpires from the results obtained with OH-substituted tetraphenylporphyrins CAT and FCAT (Scheme 1) (25–27). Addition of a buffer, such as phosphate in 0.1 M concentration, opens an additional catalytic pathway for H2 evolution able to compete with the CO-formation pathway even if the pH is kept at the same neutral value. Indeed, along this pathway, the delivery of protons (by PO4H2−) is not under a stringent kinetic limitation as it is in the pathway represented in Scheme 2. It is nevertheless clear that a future detailed mechanistic and kinetic investigation of the effect of pH changes in buffered and nonbuffered media is clearly warranted, based on cyclic voltammetry and determination of the CO and H2 faradaic yields.
Scheme 2.

Competition between CO formation and H2 evolution.
In view of the paucity of data concerning homogeneous molecular catalysis of the CO2-to-CO conversion in water, benchmarking with other catalysts in terms of overpotential and turnover frequency does not seem possible at the moment. We may just note that a preliminary estimation of the maximal turnover frequency through the foot-of-the-wave analysis (discussed below) leads to the exceptionally high figure of 107 s−1 (i.e., a second-order rate constant of 2.5 × 108 M−1⋅s−1).
We are thus led to make the comparison with the characteristics of other catalysts obtained in an aprotic solvent such as DMF or acetonitrile, starting from the results shown in Fig. 1 C and D. The standard potential of the FeI/Fe0 couple in DMF (Fig. 1C) is −1.23 V vs. NHE. A systematic analysis of the wave obtained under 1 atm CO2 and presence of 3 M phenol was carried out according to the foot-of-the wave approach, which aims at minimizing the effects of side phenomena that interfere at large catalytic currents. This technique has been previously described in detail and successfully applied in several instances (23–27). It was applied here, assuming that the reaction mechanism is of the same type as for FeTPP in the presence of phenol (Scheme 3) (24).
Scheme 3.
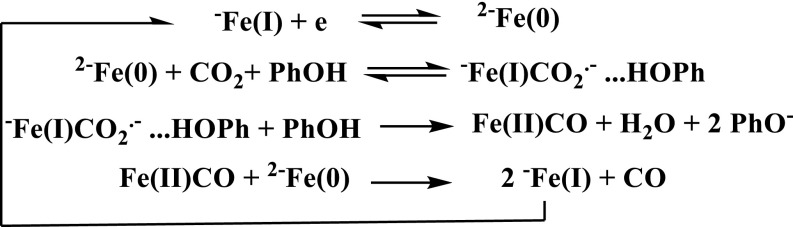
Likely mechanism of the CO2-to-CO conversion pathway.
Combination of the foot-of-the wave analysis with increasing scan rates, which both minimize the effect of side phenomena (23–27), allowed the determination of the turnover frequency as a function of the overpotential, leading to the catalytic Tafel plot for the WSCAT catalyst shown as the red curve in Fig. 3. The turnover frequency, TOF, takes into account that the molecules that participate in catalysis are only those contained in the thin reaction-diffusion layer adjacent to the electrode surface in pure kinetic conditions (23, 24). The overpotential, η, is the difference between the standard potential of the reaction to be catalyzed and the electrode potential. Correlations between TOF and η provide catalytic Tafel plots that are able to benchmark the intrinsic properties of the catalyst independently of parameters such as cell configuration and size. Good catalysts appear in the upper left corner and bad catalysts in the bottom right corner of Fig. 3. These plots allow one to trade between the rapidity of the catalytic reaction and the energy required to run it. The other Tafel plots shown in Fig. 3 are simply a repetition of what has been established in detail in ref. 24.
Fig. 3.

Catalytic Tafel plots in DMF or acetonitrile. See ref. 27 for details and references.
It clearly appears that WSCAT is the best catalyst of the set of molecules represented in Fig. 3. It is expected that the electron-withdrawing properties of the para-N-trimethylammonium groups leads to a positive shift of the FeI/Fe0 couple, being thus a favorable factor in terms of overpotential (positive shifts of 200 mV vs. FeTPP, 100 mV vs. CAT, and 40 mV vs. FCAT). What is more surprising is that this effect, which tends to decrease the electron density on the iron and porphyrin ring at the oxidation state 0, does not slow down the catalytic reaction to a large extent. Systematic analysis of the factors that control the standard potential and the catalytic reactivity as a function of the substituents installed on the porphyrin ring is a clearly warranted future task.
For the moment, we may conclude that substitution of the four parahydrogens of FeTPP by trimethylammonium groups has produced a water-soluble catalyst that is able, for the first time to our knowledge, to catalyze quantitatively the conversion of carbon dioxide into carbon monoxide in pH-neutral water with very little production of hydrogen. This seems a notable result in view of the hydration of CO2, producing carbonic acid—a low pKa acid—the catalytic reduction of which, and/or of the hydrated protons it may generate, into hydrogen might have been a serious competing pathway. Although a number of mechanistic details should be worked out, this notable result seemingly derives not only from the relatively small value of the hydration constant but also from the slowness of this reaction. As judged from its performances in DMF, this catalyst moreover seems particularly efficient in terms of catalytic Tafel plots relating the turnover frequency to the overpotential. Manipulation of the pH under unbuffered conditions or by introduction of an additional buffer should open the way to the production of CO–H2 mixtures in prescribed proportions. The substitution by the four trimethylammonium groups ensured the water solubility of the catalyst. It may also be an adequate way of immobilizing it onto the electrode surface by means of a negatively charged polymer to be associated with a water-oxidation anode through a proton-exchange membrane.
Materials and Methods
Cyclic voltammograms were obtained in a three-electrode cell, the working electrode being a 3-mm-diameter glassy carbon disk. The experiments were carried out either under argon or carbon dioxide atmosphere at 21 °C, with a careful compensation of the ohmic drop. Electrolyses were performed with a carbon crucible as working electrode, the volume of the solution was 5 mL, and the active surface area was ca. 16 cm2. In this case the ohmic drop was minimized by immersing the reference electrode directly into the solution as close as possible to the working electrode. Then, the quantification of the products (CO and H2) was performed by chromatography analyses of the gas evolved in the headspace, with calibration curves for both H2 and CO being determined separately by injecting known quantities of pure gas. Experimental details concerning the synthesis of WSCAT and full complementary experimental details can be found in Supporting Information.
Supplementary Material
Acknowledgments
Partial financial support from the Société d’accélération du transfert de technologies Ile-de-France Innov (Project 054) is gratefully acknowledged.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507063112/-/DCSupplemental.
References
- 1.Lewis NS, Nocera DG. Powering the planet: Chemical challenges in solar energy utilization. Proc Natl Acad Sci USA. 2006;103(43):15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray HB. Solar fuel II. The quest for catalysts. Eng Sci. 2009;2:27–31. [Google Scholar]
- 3.Aresta M, Dibenedetto A, Angelini A. Catalysis for the valorization of exhaust carbon: From CO2 to chemicals, materials, and fuels. Technological use of CO2. Chem Rev. 2014;114(3):1709–1742. doi: 10.1021/cr4002758. [DOI] [PubMed] [Google Scholar]
- 4.Savéant J-M. 2006. Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry (Wiley, Hoboken, NJ), pp 152–154.
- 5.Savéant J-M. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem Rev. 2008;108(7):2348–2378. doi: 10.1021/cr068079z. [DOI] [PubMed] [Google Scholar]
- 6.Benson EE, Kubiak CP, Sathrum AJ, Smieja JM. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem Soc Rev. 2009;38(1):89–99. doi: 10.1039/b804323j. [DOI] [PubMed] [Google Scholar]
- 7.Rakowski DuBois M, DuBois DL. Development of molecular electrocatalysts for CO2 reduction and H2 production/oxidation. Acc Chem Res. 2009;42(12):1974–1982. doi: 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- 8.Windle CD, Perutz RN. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord Chem Rev. 2012;256(21–22):2562–2570. [Google Scholar]
- 9.Schneider J, Jia H, Muckerman JT, Fujita E. Thermodynamics and kinetics of CO2, CO, and H+ binding to the metal centre of CO2 reduction catalysts. Chem Soc Rev. 2012;41(6):2036–2051. doi: 10.1039/c1cs15278e. [DOI] [PubMed] [Google Scholar]
- 10.Costentin C, Robert M, Savéant J-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem Soc Rev. 2013;42(6):2423–2436. doi: 10.1039/c2cs35360a. [DOI] [PubMed] [Google Scholar]
- 11.Berardi S, et al. Molecular artificial photosynthesis. Chem Soc Rev. 2014;43(22):7501–7519. doi: 10.1039/c3cs60405e. [DOI] [PubMed] [Google Scholar]
- 12.Bourrez M, Molton F, Chardon-Noblat S, Deronzier A. [Mn(bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew Chem Int Ed Engl. 2011;50(42):9903–9906. doi: 10.1002/anie.201103616. [DOI] [PubMed] [Google Scholar]
- 13.Bourrez M, et al. Pulsed-EPR evidence of a manganese(II) hydroxycarbonyl intermediate in the electrocatalytic reduction of carbon dioxide by a manganese bipyridyl derivative. Angew Chem Int Ed Engl. 2014;53(1):240–243. doi: 10.1002/anie.201306750. [DOI] [PubMed] [Google Scholar]
- 14.Smieja JM, et al. Manganese as a substitute for rhenium in CO2 reduction catalysts: The importance of acids. Inorg Chem. 2013;52(5):2484–2491. doi: 10.1021/ic302391u. [DOI] [PubMed] [Google Scholar]
- 15.Froehlich JD, Kubiak CP. Homogeneous CO2 reduction by Ni(cyclam) at a glassy carbon electrode. Inorg Chem. 2012;51(7):3932–3934. doi: 10.1021/ic3001619. [DOI] [PubMed] [Google Scholar]
- 16.Sampson MD, et al. Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: Eliminating dimerization and altering catalysis. J Am Chem Soc. 2014;136(14):5460–5471. doi: 10.1021/ja501252f. [DOI] [PubMed] [Google Scholar]
- 17.Hammouche M, Lexa D, Savéant J-M, Momenteau M. Catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. J Electroanal Chem. 1988;249:347–351. [Google Scholar]
- 18.Bhugun I, Lexa D, Saveant J-M. Ultraefficient selective homogeneous catalysis of the electrochemical reduction of carbon dioxide by an iron(0) porphyrin associated with a weak Brönsted acid co-catalyst. J Am Chem Soc. 1994;116(11):5015–5016. [Google Scholar]
- 19.Bhugun I, Lexa D, Saveant JM. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins: Synergystic effect of weak Brönsted acids. J Am Chem Soc. 1996;118(7):1769–1776. [Google Scholar]
- 20.Hammouche M, Lexa D, Momenteau M, Savéant J-M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron (“0”) porphyrins. Role of the addition of magnesium cations. J Am Chem Soc. 1991;113:8455–8466. [Google Scholar]
- 21.Bhugun I, Lexa D, Saveant JM. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins. Synergistic effect of Lewis acid cations. J Phys Chem. 1996;100:19981–19985. [Google Scholar]
- 22.Bhugun I, Lexa D, Savéant J-M. Homogeneous catalysis of electrochemical hydrogen evolution by iron(0) porphyrins. J Am Chem Soc. 1996;118(16):3982–3983. [Google Scholar]
- 23.Costentin C, Drouet S, Robert M, Savéant J-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J Am Chem Soc. 2012;134(27):11235–11242. doi: 10.1021/ja303560c. [DOI] [PubMed] [Google Scholar]
- 24.Costentin C, Drouet S, Passard G, Robert M, Savéant J-M. Proton-coupled electron transfer cleavage of heavy-atom bonds in electrocatalytic processes. Cleavage of a C-O bond in the catalyzed electrochemical reduction of CO2. J Am Chem Soc. 2013;135(24):9023–9031. doi: 10.1021/ja4030148. [DOI] [PubMed] [Google Scholar]
- 25.Costentin C, Drouet S, Robert M, Savéant J-M. A local proton source enhances CO2 electroreduction to CO by a molecular Fe catalyst. Science. 2012;338(6103):90–94. doi: 10.1126/science.1224581. [DOI] [PubMed] [Google Scholar]
- 26.Costentin C, Passard G, Robert M, Savéant J-M. Pendant acid-base groups in molecular catalysts: H-bond promoters or proton relays? Mechanisms of the conversion of CO2 to CO by electrogenerated iron(0)porphyrins bearing prepositioned phenol functionalities. J Am Chem Soc. 2014;136(33):11821–11829. doi: 10.1021/ja506193v. [DOI] [PubMed] [Google Scholar]
- 27.Costentin C, Passard G, Robert M, Savéant J-M. Ultraefficient homogeneous catalyst for the CO2-to-CO electrochemical conversion. Proc Natl Acad Sci USA. 2014;111(42):14990–14994. doi: 10.1073/pnas.1416697111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elgrishi N, Chambers MB, Artero V, Fontecave M. Terpyridine complexes of first row transition metals and electrochemical reduction of CO2 to CO. Phys Chem Chem Phys. 2014;16(27):13635–13644. doi: 10.1039/c4cp00451e. [DOI] [PubMed] [Google Scholar]
- 29.Costentin C, Robert M, Savéant J-M, Tard C. Breaking bonds with electrons and protons. Models and examples. Acc Chem Res. 2014;47(1):271–280. doi: 10.1021/ar4001444. [DOI] [PubMed] [Google Scholar]
- 30.Furuya N, Matsui K. Electroreduction of carbon dioxide on gas-diffusion electrodes modified by metal phthalocyanines. J Electroanal Chem. 1989;271:181–191. [Google Scholar]
- 31.Sonoyama N, Kirii M, Sakata T. Electrochemical reduction of CO2 at metal-porphyrin supported gas diffusion electrodes under high pressure CO2. Electrochem Commun. 1999;1:213–216. [Google Scholar]
- 32.Walsh JJ, Neri G, Smith CL, Cowan AJ. Electrocatalytic CO2 reduction with a membrane supported manganese catalyst in aqueous solution. Chem Commun (Camb) 2014;50(84):12698–12701. doi: 10.1039/c4cc06404f. [DOI] [PubMed] [Google Scholar]
- 33.Blakemore JD, Gupta A, Warren JJ, Brunschwig BS, Gray HB. Noncovalent immobilization of electrocatalysts on carbon electrodes for fuel production. J Am Chem Soc. 2013;135(49):18288–18291. doi: 10.1021/ja4099609. [DOI] [PubMed] [Google Scholar]
- 34.Elgrishi N, Griveau S, Chambers MB, Bedioui F, Fontecave M. Versatile functionalization of carbon electrodes with a polypyridine ligand: Metallation and electrocatalytic H(+) and CO2 reduction. Chem Commun (Camb) 2015;51(14):2995–2998. doi: 10.1039/c4cc10027a. [DOI] [PubMed] [Google Scholar]
- 35.Delacourt C, Ridgway PL, Kerr JB, Newman J. Design of an electrochemical cell making syngas (CO + H2) from CO2 and H2O reduction at room temperature. J Electrochem Soc. 2008;155:B42–B49. [Google Scholar]
- 36. Lide, DR, ed (2000) Handbook of Chemistry and Physics (CRC, Boca Raton, FL), 81st Ed.
- 37.Kang P, Chen Z, Nayak A, Zhang S, Meyer TJ. Single catalyst electrocatalytic reduction of CO2 in water to H2+CO syngas mixtures with water oxidation to O2. Energy Environ. Sci. 2014;7:4007–4012. [Google Scholar]
- 38.Beley M, Collin JP, Ruppert R, Sauvage JP. Nickel(II)-cyclam: An extremely selective electrocatalyst for reduction of carbon dioxide in water. Chem Commun. 1984;1984:1315–1316. [Google Scholar]
- 39.Beley M, Collin JP, Ruppert R, Sauvage JP. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: Study of the factors affecting the efficiency and the selectivity of the process. J Am Chem Soc. 1986;108(24):7461–7467. doi: 10.1021/ja00284a003. [DOI] [PubMed] [Google Scholar]
- 40.Gueutin C, Lexa D, Momenteau M, Savéant J-M, Xu F. Molecular environment effects in redox and coordination chemistry. Protection against solvation, local solvation and steric hindrance to ligation in the electrochemistry of basket-handle iron porphyrins. Inorg Chem. 1986;25(23):4294–4307. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.