

Significance
The evolution of drug resistance is a major health threat. In chronic infections with rapidly mutating pathogens—including HIV, tuberculosis, and hepatitis B and C viruses—multidrug resistance can cause even aggressive combination drug treatment to fail. Oftentimes, individual drugs within a combination do not penetrate equally to all infected regions of the body. Here we present a mathematical model suggesting that this imperfect penetration can dramatically increase the chance of treatment failure by creating regions where only one drug from a combination reaches a therapeutic concentration. The resulting single-drug compartments allow the pathogen to evolve resistance to each drug sequentially, rapidly causing multidrug resistance. More broadly, our model provides a quantitative framework for reasoning about trade-offs between aggressive and moderate drug therapies.
Keywords: drug resistance, combination therapy, drug sanctuaries, spatial structure, pathogen evolution
Abstract
Infections with rapidly evolving pathogens are often treated using combinations of drugs with different mechanisms of action. One of the major goal of combination therapy is to reduce the risk of drug resistance emerging during a patient’s treatment. Although this strategy generally has significant benefits over monotherapy, it may also select for multidrug-resistant strains, particularly during long-term treatment for chronic infections. Infections with these strains present an important clinical and public health problem. Complicating this issue, for many antimicrobial treatment regimes, individual drugs have imperfect penetration throughout the body, so there may be regions where only one drug reaches an effective concentration. Here we propose that mismatched drug coverage can greatly speed up the evolution of multidrug resistance by allowing mutations to accumulate in a stepwise fashion. We develop a mathematical model of within-host pathogen evolution under spatially heterogeneous drug coverage and demonstrate that even very small single-drug compartments lead to dramatically higher resistance risk. We find that it is often better to use drug combinations with matched penetration profiles, although there may be a trade-off between preventing eventual treatment failure due to resistance in this way and temporarily reducing pathogen levels systemically. Our results show that drugs with the most extensive distribution are likely to be the most vulnerable to resistance. We conclude that optimal combination treatments should be designed to prevent this spatial effective monotherapy. These results are widely applicable to diverse microbial infections including viruses, bacteria, and parasites.
Current standard-of-care treatment for many bacterial and viral infections involves combinations of two or more drugs with unique mechanisms of action. There are two main situations in which combination therapy significantly outperforms monotherapy (treatment with a single drug). First, in clinical scenarios where precise pathogen identification is not possible before treatment begins (“empirical therapy”), or when infections are suspected to be polymicrobial, treating with multiple drugs increases the chances of targeting the virulent organism. Second, even when infections are caused by a single, precisely identified microbe, combination therapy reduces the risk of developing drug resistance. This reduced risk is believed to follow from the fact that multiple mutations are generally needed to enable pathogen growth when multiple drugs are present. In addition, the use of multiple drugs may reduce the residual population size and thus further reduce the rate of evolution of resistance. Preventing the evolution of resistance is particularly relevant to infections caused by rapidly evolving pathogens and to persistent infections that can be controlled but not cured, for which there may be a high risk of drug resistance evolving during the course of a single patient’s treatment. Despite widespread use of combination therapy, drug resistance remains a serious concern for many infections in this category, such as the human immunodeficiency virus (HIV), hepatitis B virus (HBV), hepatitis C virus (HCV), Mycobacterium tuberculosis (TB), and other chronic bacterial infections (1–5), as well as for certain cancers (6, 7). Understanding the factors that facilitate the evolution of multidrug resistance is therefore a research priority.
Combination therapy can be compromised by treatment regimes that allow resistance mutations to different drugs to be acquired progressively (i.e., in stepwise fashion) rather than concurrently. This can occur when only one drug of the combination is active during certain time periods. For example, starting patients on a single drug before adding a second drug promotes the evolution of multidrug resistance (8–11). A similar effect is seen for studies that rotate antibiotics (12–14). Even if drugs are given simultaneously but have different in vivo half-lives (15–17) or postantibiotic effects (18), periods of “effective monotherapy” with the longer-lived drug can occur, which may favor resistance evolution. HIV and TB are pathogens for which evolution of drug resistance is well studied. Surprisingly, it has been found that stepwise evolution of drug resistance is common in treated HIV- (19–21) and TB-infected individuals (18). It is unclear whether periods of effective monotherapy can explain this observation.
Whereas many recent studies have focused on the potential impact of different half-lives between drugs, much less is known about how the spatial distribution of drugs influences the evolution of multidrug resistance during combination therapy. Many treatments may involve mismatched drug penetrability—that is, there may be regions of the body where only a subset of drugs within a combination reaches a therapeutic level (22, 23). For example, many anti-HIV drugs have been observed at subclinical concentrations in the central nervous system, the genital tract, and some lymph tissue (24–26). Low concentrations in these body compartments, even when plasma concentrations are high, may allow viral replication and selection of resistance mutations (22, 27, 28), which may eventually migrate to the blood and lead to treatment failure (29). In another example, poor antibiotic penetration within biofilms (30) or certain body tissues (31) during treatment for Staphylococcus aureus infections is again associated with resistance evolution. Some medical practitioners recommend that this problem be addressed by pairing drugs with high efficacy but low penetration with other drugs of higher penetration, so that total drug coverage in the body increases (31). However, this is likely a risky strategy. We hypothesize that combination therapy with drugs that have different penetration profiles will generally be more vulnerable to resistance, as it promotes situations of effective monotherapy that may allow a migrating pathogen lineage to acquire resistance mutations in a stepwise manner.
Previous work on the effect of drug penetration on drug resistance has mainly focused on monotherapy. A mathematical model of viral infections showed that the window of drug concentrations where resistance mutations can arise and fix is greatly increased if there is a “drug-protected compartment” or “drug sanctuary”—a place where the drug level is not high enough to prevent virus replication (32). More recent theoretical work has explored the role of concentration gradients in the evolution of antibiotic resistance. This work demonstrated that when multiple mutations are needed for resistance to a single drug, either a continuous concentration gradient (33) or discrete microenvironments with differing concentrations (34) can speed up the rate of evolution. Experiments in microfluidic chambers where mobile bacteria grow in the presence of a spatial drug concentration gradient have confirmed that adaptation is accelerated (35). These results are surprisingly similar to studies that create temporal gradients in drug concentrations (36).
A few detailed simulation studies have examined resistance evolution during combination antibiotic therapy and included sources of heterogeneity in drug efficacy (37–39). These models used experimentally determined pharmacodynamic parameters and included subpopulations of slow-growing persister bacteria that may be less sensitive to one or all antibiotics in a combination. Although these studies did not specifically focus on quantifying the role of effective monotherapy due to mismatched drug distributions, they strongly suggest that it may play a role in multidrug resistance.
In this paper, we examine the general role that drug penetration plays in evolution of resistance during combination therapy—thereby addressing a broad range of effective drug treatments. Specifically, we use a mathematical modeling strategy to show how the existence of anatomical compartments where only single drugs are present can drastically change the rate at which multidrug resistance emerges and leads to systemic infection despite treatment. Among several pharmacologic and genetic determinants of resistance, we find that the size of single-drug compartments is key. A simple mathematical expression describes the critical size of single-drug compartments above which drug resistance emerges at an elevated rate, due to stepwise accumulation of mutations. In addition, we discover that combination therapy strategies face a general trade-off between suppressing microbial growth throughout the entire body and preventing eventual emergence of multidrug resistance. This trade-off implies, perhaps counterintuitively, that it may be rational to allow low-level microbial growth restricted to a small compartment where no drugs penetrate, to avoid regions of mismatched drug penetration—and increased risk of resistance emerging in the entire body. We discuss implications of this work for designing optimal drug combinations to prevent spatial effective monotherapy. Finally, we use our theory to explain why stepwise evolution of resistance may occur during effective combination therapy, as is sometimes seen clinically.
Model
Our goal is to understand the role of drug penetration in the evolution of multidrug resistance. We consider an individual patient’s body to be divided into discrete and interconnected compartments where each drug either effectively suppresses pathogen growth or is completely absent (Fig. 1). We model microbial dynamics in this environment, including growth, mutation, competition between strains, and migration between compartments. For simplicity, we focus on the case of two drugs only, although extensions to combinations of three or more drugs are straightforward.
Fig. 1.

Compartment model for combination therapy with two drugs. The box represents a patient’s body and the red and blue shaded areas indicate the presence of drug 1 and drug 2, respectively. Mismatched drug penetration creates regions in the body where only one drug from the combination is present. We refer to these regions as single-drug compartments. Colored circles represent the pathogen genotypes: wild type (light gray), mutant resistant to drug 1 (blue), mutant resistant to drug 2 (red), and double-drug-resistant mutant (purple). In the sanctuary all of the pathogen genotypes can grow because none of the drugs is present. In the single-drug compartments only pathogens carrying a resistance mutation against the active drug can grow; that is, each drug alone suppresses pathogen growth. Finally, in the double-drug compartment only the double-drug-resistant mutant can grow. All of the compartments are connected by migration as indicated by the black arrows. Treatment failure occurs when the double-drug compartment, which always composes the majority of the body, is colonized by the double mutant. Note that we do not always require that both single-drug compartments exist, and the compartment sizes may not follow this particular geometric relationship.
To describe population dynamics of the pathogen in this scenario, we use a viral dynamics model (40) (SI Appendix) that tracks infected and uninfected cells. We analyze the model, using a fully stochastic simulation (SI Appendix), and derive approximate analytic formulas to describe the dominant processes. Other ways of modeling pathogen growth with limited resources, such as the logistic model, could be used instead and we expect this would have little influence on the results. In this model, pathogen fitness can be measured in terms of the basic reproductive ratio , the number of new infections generated by a single infected cell before it dies, when target cells are in excess. A strain can lead to a sustainable infection in a compartment only if (i.e., growth is positive). When this occurs, the pathogen population can reach an equilibrium level that we refer to as the carrying capacity (K).
We consider at most four compartments within a single patient (Fig. 1): one compartment where no drugs are present (the sanctuary), two compartments where only one of the drugs is present (single-drug compartments 1 and 2), and one compartment where both drugs are present (the double-drug compartment, which we always take to be by far the largest compartment). The pathogen population within each compartment is assumed to be well mixed and follows the viral dynamics model. The size of each compartment j is given by the number of target cells that it contains when infection is absent. The carrying capacity of pathogen strain i infecting compartment j increases monotonically with pathogen fitness and is always less than the compartment size ( for all i), assuming that the death rate of infected cells exceeds that of uninfected cells. In the absence of mutation or migration, there is competitive exclusion between strains within a compartment, and the strain with the highest fitness goes to fixation. With migration or mutation, multiple strains may coexist within a compartment, although the locally suboptimal strains generally occur at much lower frequencies.
The four compartments are connected by migration of pathogens (but not drugs), and every strain in the body migrates from compartment j to compartment k at a rate per time. We use a simple and biologically realistic migration scheme in which each pathogen migrates out of its home compartment at the same rate m. Migrants from a given compartment are then distributed into all four compartments (including the one they came from) proportionally to the compartment sizes, so that larger compartments get more migrants.
A single mutation is needed for resistance to each drug. Mutations conferring resistance to drug i occur at a rate (and can revert at the same rate). Resistance to two drugs requires that both mutations occur, and we do not allow recombination or any other form of lateral gene transfer to break linkage between the two mutations. We design our fitness landscape so that four assumptions are met: (i) Each drug alone suppresses pathogen growth, (ii) a wild-type pathogen in the sanctuary has the highest possible fitness, (iii) a doubly resistant pathogen is always viable, and (iv) in the single-drug compartments, the strain with resistance only to the drug present is the fittest. Formally, if a strain is not resistant to a drug i present in the compartment where it resides, its fitness is reduced by a factor of , where is the drug efficacy. Resistance mutations come with a fitness cost . The fitness of resistant strains is completely unaffected by the presence of the drug. The fitness values for each genotype in each compartment, relative to that of a wild-type strain in the sanctuary , are shown in Table 1. To satisfy condition i, we constrain , and to meet condition iii, we require . At the start of treatment, we suppose the wild-type pathogen to be present in all compartments; we first focus on the case without preexisting resistance mutations and later consider how preexisting resistance alters results.
Table 1.
The fitness of each pathogen strain in each compartment relative to the fitness of the wild-type strain in the absence of the drug
| Sanctuary | SDC 1 | SDC 2 | DDC | |
| Wild type | 1 | |||
| Single mutant 1 | ||||
| Single mutant 2 | ||||
| Double mutant |
The efficacy of drug i is and the fitness cost of resistance to drug i is . DDC, double-drug compartment; SDC, single-drug compartment.
We apply this model to a physiologic scenario where the double-drug compartment occupies the vast majority of the body and where isolated infections within the small sanctuary or single-drug sites are not life threatening on their own. Therefore, treatment failure is said to occur when the multidrug-resistant mutant colonizes the double-drug compartment. We define colonization as a pathogen load high enough such that the probability of chance extinction is negligible. We investigate how the presence and size of single-drug compartments—created by combinations of drugs with mismatched penetration profiles—determine two clinical outcomes: the rate at which treatment failure occurs and the evolutionary path by which the multidrug-resistant mutant emerges. Under the direct evolutionary path, multiple-resistance mutations are acquired near simultaneously [this is sometimes referred to as “stochastic tunneling” (41, 42)]; under stepwise evolution, a single-drug compartment is colonized with a single-resistant strain before the emergence of multidrug resistance.
Results
Mismatched Drug Penetration Can Speed Up Emergence of Resistance.
Using parameter values appropriate for HIV treatment (SI Appendix), we simulate pathogen evolution according to the model described above. For simplicity, we first consider the presence of only one single-drug compartment (containing drug 1). The probability of treatment failure via double-drug resistance after 1 year (Fig. 2A) or 10 years (Fig. 2B) increases dramatically with the size of the single-drug compartment, even when this region is two to three orders of magnitude smaller than the area covered by both drugs. This demonstrates that imperfect drug penetration can be highly detrimental to treatment outcomes.
Fig. 2.

Resistance evolution in the presence of a single-drug compartment. Even a small single-drug compartment can considerably speed up the evolution of double-drug resistance. (A and B) The shaded area gives the fraction of simulated patients that failed treatment after 1 year or 10 years as a function of the size of the single-drug compartment containing drug 1 (SDC1) relative to the size of the double-drug compartment (DDC). We further indicate whether treatment failure occurred via direct (gray circles) or stepwise (pink circles) evolution. Solid lines are analytic calculations (SI Appendix, sections 5 and 6). The vertical dotted lines are further simplified, closed-form analytical expressions for the point where the stepwise path to resistance becomes more important than the direct path (SI Appendix, sections 4.2 and 7). (C and D) Evolution of drug resistance over time for a simulated patient in the absence (C) or presence (D) of SDC1. When there are no single-drug compartments, mutants resistant to drug 1 go to extinction recurrently by competition with the wild type in the sanctuary, whereas in the presence of SDC1, mutants resistant to drug 1 can escape competition and establish a continuous population (blue line) from which a doubly resistant strain can evolve (purple). Parameters: cells. changes along the x axis for A and B and for each value of treatment has failed in at least 2,000 simulated patients. for C and for D.
Mismatched drug penetration hastens the emergence of multidrug resistance by allowing for stepwise evolution (Fig. 2 C and D). Specifically, single-resistant mutants can evade competition with wild-type strains by migrating to the single-drug compartment, which serves as a platform from which resistance to the second drug may evolve (Fig. 2D). When drugs have identical penetration, there are only two compartments—the sanctuary and the double-drug compartment. In typical simulations (Fig. 2C), single-resistant mutants arising in the sanctuary are driven recurrently to extinction by the fitter wild type. As a result, the only way that double-drug resistance can emerge is by appearance of both mutations nearly simultaneously, enabling successful migration to the double-drug compartment (direct evolution). This slow process increases time to treatment failure.
Consistent with the above explanation, the prevalence of failure by direct evolution depends weakly on single-drug compartment size, only decreasing slightly with compartment size as the competing stepwise path occurs first (Fig. 2 A and B). Failure by stepwise evolution, however, increases substantially with the size of the single-drug compartment, and it is the dominant path if the single-drug compartment exceeds a critical size, investigated below.
Stepwise vs. Direct Evolution.
Using a simplified model of colonization of each compartment, we can approximate the critical single-drug compartment size above which stepwise evolution becomes the dominant process. Specifically, we approximate the colonization process by transitions between discrete states of the population, where each state is described by the presence or absence of each strain in each compartment. For brevity, we assume that the mutation rate and fitness cost are the same for both mutational steps and that there is only one single-drug compartment. In state 0, only the sanctuary is colonized (by the wild-type strain); in state 1, the single-drug compartment is also colonized (by the single-resistant mutant); and state 2 is the end state where the double-drug compartment is colonized (by the double-resistant strain). Rates of treatment failure can be computed exactly in this simplified model (SI Appendix, sections 4–6), which provides an excellent approximation to the full stochastic simulation (Fig. 2 A and B).
Using this model, we can obtain simple approximate expressions for the size of the single-drug compartment (SDC) where the stepwise path starts to overtake the direct path (detailed in SI Appendix, section 7). The SDC becomes colonized (transition from state 0 to state 1) by one of two events. Either a mutation occurs within the sanctuary, and then that strain migrates to the SDC, or a wild-type strain migrates from the sanctuary to the SDC, where it manages to replicate and mutate despite the presence of the drug. In both cases the mutant must escape extinction to establish an infection in the SDC. In the limit where mutation cost is small but drug efficacy is high , mutation typically precedes migration, and the rate of invasion of the single-drug compartment is approximately
Here is the number of single mutants in the sanctuary (“SAN,” at mutation–selection equilibrium), is the migration rate to the single-drug compartment, and is the establishment probability of a resistant mutant in the single-drug compartment (see SI Appendix for full derivation with respect to the viral dynamics model). If invasion is successful, we assume that the population in the newly invaded compartment reaches its carrying capacity instantaneously. Doing so relies on a separation of timescales between the slow processes of mutation and migration and the faster process of growth to equilibrium.
Similarly, once the single-drug compartment is colonized, the double-drug compartment (DDC) can be invaded. Again, the mutation–migration path is most likely, with rate approximately
The double-drug compartment can also be invaded directly from the sanctuary. There are three paths by which this can happen, depending on whether none, one, or both of the necessary mutational steps occur before migration. By the same logic as above, the mutation–mutation–migration path is most likely, and the rate is approximately
In the scenario under consideration, the mutation rate is much smaller than the cost of mutations so that . We also assume that both drugs penetrate in a large part of the body so that the double-drug compartment is always much larger than the single-drug compartment . It is therefore likely that and . Using these expressions, we can determine the overall rate at which the DDC becomes colonized via the SDC (stepwise evolution) and compare it to the rate of direct evolution.
First, we consider treatment outcomes when a short enough time (t) has passed so that drug resistance is rare and all steps are rate limited (, , ). In this regime, the minimum size of the SDC at which stepwise evolution outpaces the direct path (lines cross in Fig. 2 A and B) increases with the pathogen virulence , but decreases with the migration rate (m) and (weakly) with the fitness of the single mutant . It also decreases with the time of observation (t) because the stepwise path requires two steps, so that for very small t, the SDC needs to be larger for it to be possible that both steps are completed. This approximation (SI Appendix, section 7, Approximation 1) describes the cross point after 1 year of treatment well (Fig. 2A).
Alternatively, if the treatment time is long enough so that most individuals who developed single-drug resistance progressed to treatment failure , but the other (slower) steps remain rate limiting, then a simpler and more intuitive result emerges: The stepwise path is more important than the direct path if
The single-drug compartment therefore plays an important role if its size, relative to that of the double-drug compartment, is at least equal to the mutation-to-cost ratio. Intuitively, if mutations are rare and costly, then double mutants occur infrequently and the stepwise path to multidrug resistance is relatively more important. Even if mutations are rather common (say, ) and not very costly , the stepwise path is still dominant if the single-drug compartment is at least 1/100th the size of the double-drug compartment. For the parameters used in the figures, this approximation describes the cross point after 10 years of treatment well (Fig. 2B).
Trade-Off Between Halting Pathogen Growth and Preventing Resistance.
The choice of antimicrobial therapy generally presents a trade-off between maximizing clinical efficacy and minimizing the chance that drug resistance emerges (43). The spatial setting introduces new dimensions to this trade-off. In this setting, minimizing the size of single-drug compartments can impede the stepwise evolution of resistance. Pursuing this goal, however, involves choosing drugs that penetrate the same anatomical regions, potentially reducing the portion of the body that receives any drug at all. The physician therefore may sometimes be faced with a trade-off: to halt wild-type growth immediately (smaller sanctuary) or to prevent stepwise evolution of resistance (smaller single-drug compartments).
To investigate this trade-off, we vary single-drug compartment size relative to the sanctuary, keeping double-drug compartment and total system size constant (Fig. 3). We do this analysis imagining that one drug of the combination is fixed, and another can be chosen that has either equal or greater penetration. Consistent with the above findings, the rate of treatment failure by double-drug resistance increases dramatically as single-drug compartment size increases from zero. At the same time, however, the sanctuary shrinks from its maximum size, reducing total pathogen load in the body before failure.
Fig. 3.

Trade-off between total drug coverage and the presence of single-drug compartments. (A) The adaptation rate (purple circles, left y axis) and time-averaged infection size (orange circles, right y axis) are plotted as a function of the size of the single-drug compartment with drug 1 , assuming that the sum of the sizes of the sanctuary and SDC1 is constant. Diagrams below the x axis illustrate the changes in compartment sizes, following the style of Fig. 1. The adaptation rate is defined as the inverse of the mean time to treatment failure and is plotted relative to the rate when . We show adaptation rate only from acquired genetic variation (solid circles) and from both acquired and standing genetic variation (i.e., preexisting resistance, open circles); the difference is shown by the gray area and the vertical lines. The infection size is calculated as the mean of the time-averaged number of infected cells in all compartments before treatment failure occurs. Increasing the size of the single-drug compartment provides better control of the infection before treatment fails, but strongly favors resistance evolution if the reduction of the sanctuary is not large enough. (B) Ratio of the rate of adaptation from standing and acquired genetic variation to the rate of adaptation only from acquired genetic variation . The relative contribution of standing genetic variation to treatment failure increases with the size of the SDC. Parameters: cells. Each point is an average over at least 30,000 simulated patients.
The trend in treatment failure reverses, however, as the sanctuary is further reduced (right half of Fig. 3A). Even when stepwise evolution is still the dominant mode of treatment failure, a small sanctuary limits the rate at which single mutations are generated and therefore decreases the overall rate of emergence of multidrug resistance. In the complete absence of a sanctuary, treatment failure can occur only if preexisting resistance is selected or if resistance is generated very quickly after treatment starts. Because these events are not guaranteed, cure becomes a possible outcome (SI Appendix, Fig. S1) and the rate of resistance evolution is dramatically reduced. The rate of treatment failure is greatest when the sanctuary and single-drug compartment are similar in size, highlighting the fact that stepwise evolution is driven by interaction between a sanctuary and single-drug compartments.
These findings suggest that eliminating all sanctuary sites should be a primary goal (moving toward far right in Fig. 3A), because this reduces pathogen load and the risk that resistance evolves. If this is not feasible (for example, if the pathogen has a latent phase not targeted by treatment), then preventing any zones of single-drug coverage should take precedence to keep the rate of evolution of drug resistance as low as possible (moving toward far left in Fig. 3A).
Accounting for Preexisting Mutations.
To focus clearly on the processes by which resistance is acquired during combination therapy, we have so far ignored the contribution of preexisting mutants (known in the population genetics literature as “standing genetic variation”). To instead include this factor, we simulate the model for a period before the introduction of treatment, allowing both single- and double-resistant mutants to occur along with the wild-type strain in each compartment. Previous work has focused extensively on comparing the relative roles of preexisting and acquired resistance in viral dynamics models (21, 44–46), and here we simply summarize the trends in our model.
The addition of preexisting resistance acts to increase the overall rate of treatment failure, and this increase is more prominent for certain parameter values and for smaller treatment times (compare SI Appendix, Fig. S2A with Fig. 2A). However, the inclusion of preexisting resistance does not affect any of the general trends, such as the dominant path to resistance (SI Appendix, Fig. S2) or the trade-off between the size of the sanctuary and the single-drug compartment (Fig. 3). Importantly, the role of preexisting resistance—defined as the percentage of failures attributable to standing genetic variation—increases dramatically with single-drug compartment size (Fig. 3B). Therefore, the presence of compartments where only single drugs penetrate can increase the rate of treatment failure both by making it quicker to acquire multiple-resistance mutations and by selecting for preexisting single-drug-resistant mutants.
Preexisting mutations are particularly relevant for curable infections, as opposed to chronic ones. In such infections, either a sanctuary zone does not exist or it is small enough to be eradicated by immune responses. As treatment duration is limited, treatment failure can occur only if there are preexisting resistance mutations or if the pathogen acquires resistance shortly after treatment starts. In this limit, the dynamics are a classic “race to rescue” described by Orr and Unckless (47), which results in either cure or treatment failure. We find that zones of mismatched penetration reduce the probability of curing the infection (SI Appendix, Fig. S1) by selecting for single-drug-resistant mutations that would otherwise become extinct under combination therapy. In a scenario where sanctuary regions exist initially but eventually decay (for example, if they are caused by long-lived persister cells), we expect that mismatched drug penetration will both decrease the probability of cure and decrease the time to resistance in those patients in whom cure is not achieved.
Order of Mutations.
Because pharmacological factors determining penetration of anatomical compartments vary widely among drugs (23, 25, 30, 48), we generally expect that each drug in a combination has its own single-drug compartment. In this general case, we can ask, To which drug does the pathogen become resistant first? More precisely, if stepwise evolution occurs, is it likely to be through the path or ? Examining the rate of each path as a function of the size of each single-drug compartment (Fig. 4A) shows that resistance is more likely to emerge first to the drug with the highest coverage (and therefore largest SDC) and that the odds of resistance occurring to one drug before another are proportional to the ratio of the corresponding SDCs over a large parameter range.
Fig. 4.
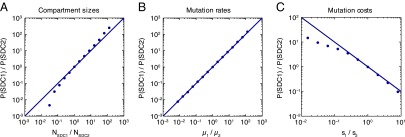
Stepwise resistance evolution in the presence of two single-drug compartments. (A–C) Fraction of simulated patients that failed via the path where the single-drug compartment with drug 1 is colonized before treatment failure (P(SDC1): SAN SDC1 DDC) relative to the fraction that failed via the path where the single-drug compartment with drug 2 is colonized before (P(SDC2): SAN SDC2 DDC) as a function of (A) compartment sizes, (B) mutation rates, and (C) mutation costs. (A) The x axis corresponds to the ratio of the size of the single-drug compartment with drug 1 to the size of the single-drug compartment with drug 2 . (B) The x axis corresponds to the ratio of the mutation rate for resistance to drug 1 to the mutation rate for resistance to drug 2 . (C) The x axis corresponds to the ratio of the cost of a resistance mutation to drug 1 to the cost of a resistance mutation to drug 2 . Simulation results (circles) are overlaid with the lines (A and B) or (C). Parameters: cells. changes along the x axis (A), changes along the x axis (B), and changes along the x axis (C). The total number of simulated patients for each point is at least 6,000.
Moreover, the mutation rates and costs associated with resistance to each drug may differ, also influencing the likelihood of a particular path to resistance. Resistance is more likely to emerge first for the drug associated with the highest mutation rate (Fig. 4B) and lowest fitness cost (Fig. 4C), with the relative rates again being approximated by the ratios of the parameters. Drug efficacy may also vary, although in the regime where each drug individually suppresses wild-type pathogen growth and the cost of mutations is not too high , drug efficacy barely influences the order in which resistance mutations are acquired (SI Appendix, Fig. S3).
Discussion
Antimicrobial drugs fail to reach effective concentrations in many tissues and body organs, allowing pathogen replication and potential evolution of resistance (26, 28, 31, 48, 49). We studied the role of imperfect drug penetration in the development of drug resistance during combination therapy, using a model of within-host pathogen evolution. In particular, we focused on the consequences of mismatched drug penetration, which may be common during combination therapy (22, 23, 25, 31). Our findings are summarized in Fig. 5.
Fig. 5.

Summary of the evolution of resistance with imperfect drug coverage. (A) When both drugs have high, matched penetration throughout the body, the evolution of multidrug resistance is slow, because it requires either preexisting multidrug resistance or near-simultaneous acquisition of both mutations along with migration out of a sanctuary site. If one drug (B) or both drugs (C) have a lower penetration, treatment outcomes may suffer in different ways. (B) If there are regions where only one drug reaches an effective concentration, then the evolution of multidrug resistance speeds up, because mutations may emerge in a stepwise fashion via single-drug compartments. Single mutations can arise de novo from a wild-type pathogen in the sanctuary or be selected from preexisting mutations in the single-drug compartment when treatment is started. (C) If the sanctuary is larger but both drugs reach the same regions of the body, then resistance still evolves slowly, but the infection size before treatment failure will be larger. Therefore, if high penetration of all drugs is impossible, there is a trade-off when choosing which drugs to pair in combinations: halting growth of the wild-type pathogen immediately (B) or preventing the sequential accumulation of resistance mutations (C).
In this model, mismatched penetration of two drugs into anatomical compartments sped up the evolution of multidrug resistance dramatically by creating zones of spatial monotherapy where only one drug from a combination regime is at therapeutic concentration. These zones, or “single-drug compartments,” positively select for single-drug-resistant mutants, thereby favoring the fast stepwise accumulation of resistance mutations (Figs. 2 A, B, and D and 5B). Stepwise resistance evolution is hindered when drugs have identical penetration profiles, because in that case single-drug-resistant mutants compete with the fitter wild type in the sanctuary and they therefore suffer recurrent extinction (Figs. 2C and 5 A and C). Without access to the stepwise path, resistance mutations must be acquired near simultaneously; the system thus takes a far slower “direct” path to treatment failure. Even slight differences in penetration of coadministered drugs lead to a high risk of multidrug resistance, because the stepwise path dominates the direct path even for very small single-drug compartments (Fig. 2 A and B).
The effects of single-drug compartments are most severe for chronic infections, during which pathogen replication persists to generate de novo resistance mutations, and treatment creates a long-term selective advantage for resistant strains. However, we have also demonstrated that mismatched penetration can speed up the development of resistance from preexisting mutations (Fig. 3) and can reduce the probability of cure for infections without a sanctuary (SI Appendix, Fig. S1), suggesting that these results may have applications to acute infections as well.
Although mismatched drug penetration generally favors resistance evolution and should be avoided, this may not always be possible. Immediate clinical efficacy may at times be more important than the prevention of resistance. It may therefore be advantageous, in some cases, to select a combination of drugs with different penetration profiles, if doing so eliminates sanctuary sites in the body. With no sanctuary sites, the total pathogen load will be as low as possible during treatment and few new mutations will be created. This slows the rate of evolution of drug resistance (Fig. 3) and makes complete eradication of the infection (cure) possible. If elimination of sanctuaries is not possible, however, then avoiding single-drug compartments due to mismatched penetration in a combination regime should be the main strategy for preventing multidrug resistance. If there are several single-drug compartments, eliminating one may have little effect if another remains. If neither single-drug compartments nor sanctuaries can be eliminated, then the optimal solution to reduce resistance is not obvious without some knowledge of the relevant parameters. Some insight as to where a particular treatment regime falls along this trade-off curve may be gained by observing the patterns of resistance acquisition. These include the overall prevalence of single-resistant strains before multidrug resistance emerges and the relative order in which different single-resistant strains appear. Previous work has questioned the orthodoxy that “aggressive” antimicrobial chemotherapy is optimal for preventing resistance (43, 50). If we consider that one aspect of treatment aggressiveness is the extent of drug penetration, then our model demonstrates the complexities involved in answering this question and motivates further work aimed at estimating the size of drug-protected compartments for relevant combination therapies.
In particular, our model offers an explanation for why the strategy suggested for some antibiotic treatments of pairing a broadly penetrating drug (e.g., rifampicin) with a narrowly penetrating one (e.g., vancomycin) to increase total drug coverage (51) might fail frequently due to the rapid evolution of resistance against the drug with higher penetration (51–53). It also offers an alternative explanation of why certain drugs are more vulnerable to resistance. This vulnerability is usually explained by a low genetic barrier to resistance (i.e., only one mutation needed) or by their long half-life. Our model suggests that broad penetration may also make a drug vulnerable to the evolution of resistance, if the drug is paired with drugs with lower penetration (Fig. 4).
Our model also offers an explanation of stepwise evolution of resistance in HIV infection, the commonly observed pattern whereby the virus gains one resistance mutation at a time (19, 54, 55). As treatment regimes are designed so that each drug is active against mutants resistant to the others, single-resistant mutants should be driven to extinction both in sanctuary zones (by competition with fitter wild type) and where all drugs are active (by sensitivity to all drugs save one). It has been hypothesized that either nonadherence to treatment or different drug half-lives cause “effective temporal monotherapy,” which is to blame for the appearance of single-drug-resistant viruses (16, 17). We propose that mismatched penetration of drugs in a combination treatment offers an alternative explanation for this stepwise evolution of resistance, via “effective spatial monotherapy.” Very small single-drug compartments are sufficient to cause this effect, suggesting that these regions may be very hard to detect and could remain overlooked.
In this study, we focused on the case of treatment with two drugs, but we expect that our results could generalize to three or more different drugs. Adding a third drug to a regimen may reduce the size of the sanctuaries and/or the size of single-drug compartments and should therefore reduce the rate at which multidrug resistance evolves.
Several extensions to our model can be considered in future studies. First, we assume that drug compartments are discrete and have a fixed size; however, drug concentrations can be continuous in space and the pharmacokinetics of individual drugs can modify the size of the different compartments over time. Second, we have assumed that treatment fails when the double-drug compartment is invaded, but depending on the size and location of drug compartments in the body, treatment may fail when a single-drug compartment is invaded. Also, we assume a very specific migration model between the compartments [known in population genetics as the island model (56, 57)], but other migration models may be possible. Specifically, not all compartments may be connected by migration and the migration rates may be independent of the size of the target compartment.
Throughout this paper we have considered the fitness effect of multiple drugs or multiple mutations to be independent (Table 1), reducing the number of parameters in our model. Actual fitness landscapes may be more complex than this assumption allows. First, drugs may interact, so that their combined efficacy deviates from the product of their independent effects (58). Interactions may be synergistic, leading to greater reductions in pathogen fitness, or antagonistic, leading to smaller reductions (38, 39, 59–61). In the case where resistance mutations accumulate in compartments where both drugs are present, previous modeling and experimental studies have shown that extreme drug antagonism may hinder evolution of multidrug resistance (62, 63). In contrast to that case, we have shown here that when mutational costs are low (small s) and drugs are effective , treatment failure is far more likely to be caused by mutations generated in the absence of a drug that later migrate to a region where the drug penetrates. Therefore, for the scenarios considered in this study, we believe that these interactions have minimal effects as long as each drug is suppressive alone and in combination.
A second possible complication in the fitness landscape is that resistance mutations may interact, so that their combined fitness effects are not multiplicative, instead displaying patterns of epistasis. One type of interaction is cross-resistance, by which gaining resistance to one drug makes a pathogen strain either more or less susceptible to the other. Because positive cross-resistance (reduced susceptibility to the other drug) reduces the fitness gap between the single and double mutant in the presence of drugs, we would expect it to increase the rate of the stepwise path more than that of the direct path, hence amplifying the effect of single-drug compartments. Negative cross-resistance (increased susceptibility) conversely would diminish the stepwise path. A second type of interaction arises where costs of the resistance mutations are not independent, affecting their frequency (mutation–selection balance) in compartments lacking the drug. In many viral infections, the combined costs are lower than the product of individual costs (positive epistasis) (64–66). This scenario confers an advantage to double mutants, accelerating both paths to treatment failure, whereas negative epistasis would impede both paths.
Throughout this paper, we have assumed that no recombination (or any other form of lateral gene transfer) occurs between the two resistance loci. In general, recombination may increase or decrease the rate at which multiple-drug resistance develops (67, 68). However, two features of the clinical setting envisioned here minimize its importance to treatment failure. First, without epistasis, recombination will not meaningfully affect the individual gene frequencies (67). Second, in our model, there is no single compartment where the two resistance mutations are each beneficial individually, meaning that there is never a situation where both single mutants are common. As the two single mutants rarely contact one another, recombination cannot speed up the appearance of the double mutant beyond the action of mutation alone (68, 69).
Drug compartments are commonly described as specific anatomical locations in the body like organs or tissues. For instance, not all antimicrobial drugs penetrate to therapeutic concentrations in the central nervous system (22, 27, 28, 70), the genital tract (22, 25), the lymphoid tissue (22, 26), or other infected tissues (23, 48, 49). However, the compartments in our model could be interpreted in many ways. For example, they could represent different cell types, such as cells in a tumor that are not reached by anticancer drugs (71, 72), or phenotypically resistant subpopulations of bacteria that have low permeability to antibiotics (73) or replicate slowly (18, 37). The latter scenario was explored in a computational model of TB treatment (37) that analyzed the combined effect of noncompliance to treatment and heterogeneity in drug sensitivity due to differences in cell turnover rates. Overall this model is consistent with our results, finding that when patients were compliant to treatment, larger single-drug compartments led to more resistance. However, when patients followed a particular pattern of imperfect compliance to treatment—by stopping drugs once bacterial loads were below a threshold value—larger single-drug compartments actually slowed down resistance evolution. This occurred because the very slow antibiotic-mediated killing of cells in this compartment, due to the low cell turnover, meant that patients with larger compartments had to take drugs for much longer to reduce bacterial loads. Higher time-averaged drug loads understandably led to lower resistance risk. This comparison points out the importance of particular assumptions in determining outcomes and motivates further studies aimed at understanding the combined effect of spatial and temporal monotherapy.
Compartments could also exist at a population level, caused by interindividual differences in pharmacokinetic parameters (74) or differential targeting of geographic regions with insecticides, herbicides, or therapeutics. Finally, this model might be relevant to other evolutionary processes where multiple adaptations are ultimately needed for survival and to the study of the role of spatial heterogeneity in adaptation.
Materials and Methods
We use a basic viral dynamics model (40) to simulate the infection within each compartment and we include stochastic mutation and stochastic migration among all of the compartments. We perform exact stochastic simulations, tracking the genotype and location of every infected cell in the body and explicitly simulating all of the events that might occur to a cell: replication (representing either division of a bacterial cell or infection of a new cell by a virus), mutation (upon replication), death, and migration among different compartments. Simulations are performed using the Gillespie algorithm. Details of the model, analytic approximations, and simulation methods are provided in SI Appendix.
Supplementary Material
Acknowledgments
The authors thank A. Harpak, P. Altrock, J. Wakeley, J. Hermisson, D. Weissman, H. Uecker, H. Alexander, B. Waclaw, S. van Doorn, and P. Abel zur Wiesch for helpful feedback on the project and the manuscript at various stages. S.M.-G. was funded by the Erasmus Mundus Masters Programme in Evolutionary Biology and by the European Research Council (Starting Grant 30955). A.L.H. and D.I.S.R. were supported by Bill and Melinda Gates Foundation Grand Challenges Explorations Grant OPP1044503. A.L.H. was also supported by National Institutes of Health (NIH) Grant DP5OD019851. D.A.P. was supported by NIH Grants RO1GM100366 and RO1GM097415. M.A.N. was supported by the John Templeton Foundation. P.S.P. received funding from the Human Frontiers Science Program (LT000591/2010-L).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424184112/-/DCSupplemental.
References
- 1.Chernish RN, Aaron SD. Approach to resistant gram-negative bacterial pulmonary infections in patients with cystic fibrosis. Curr Opin Pulm Med. 2003;9(6):509–515. doi: 10.1097/00063198-200311000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Gupta RK, et al. Global trends in antiretroviral resistance in treatment-naive individuals with HIV after rollout of antiretroviral treatment in resource-limited settings: A global collaborative study and meta-regression analysis. Lancet. 2012;380(9849):1250–1258. doi: 10.1016/S0140-6736(12)61038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tana MM, Ghany MG. Hepatitis B virus treatment: Management of antiviral drug resistance. Clin Liver Dis. 2013;2(1):24–28. doi: 10.1002/cld.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawlotsky J-M. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology. 2011;53(5):1742–1751. doi: 10.1002/hep.24262. [DOI] [PubMed] [Google Scholar]
- 5.Zumla A, et al. Drug-resistant tuberculosis—current dilemmas, unanswered questions, challenges, and priority needs. J Infect Dis. 2012;205(Suppl 2):S228–S240. doi: 10.1093/infdis/jir858. [DOI] [PubMed] [Google Scholar]
- 6.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: An evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 7.Bozic I, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife. 2013;2(June):e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nijhuis M, et al. Lamivudine-resistant human immunodeficiency virus type 1 variants (184V) require multiple amino acid changes to become co-resistant to zidovudine in vivo. J Infect Dis. 1997;176(2):398–405. doi: 10.1086/514056. [DOI] [PubMed] [Google Scholar]
- 9.Gulick RM, et al. Simultaneous vs sequential initiation of therapy with indinavir, zidovudine, and lamivudine for HIV-1 infection: 100-week follow-up. JAMA. 1998;280(1):35–41. doi: 10.1001/jama.280.1.35. [DOI] [PubMed] [Google Scholar]
- 10.Yim HJ, et al. Evolution of multi-drug resistant hepatitis B virus during sequential therapy. Hepatology. 2006;44(3):703–712. doi: 10.1002/hep.21290. [DOI] [PubMed] [Google Scholar]
- 11.Zoulim F. Hepatitis B virus resistance to antiviral drugs: Where are we going? Liver Int. 2011;31(Suppl 1):111–116. doi: 10.1111/j.1478-3231.2010.02399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedrick TL, et al. Outbreak of resistant Pseudomonas aeruginosa infections during a quarterly cycling antibiotic regimen. Surg Infect. 2008;9(2):139–152. doi: 10.1089/sur.2006.102. [DOI] [PubMed] [Google Scholar]
- 13.van Loon HJ, et al. Antibiotic rotation and development of gram-negative antibiotic resistance. Am J Respir Crit Care Med. 2005;171(5):480–487. doi: 10.1164/rccm.200401-070OC. [DOI] [PubMed] [Google Scholar]
- 14.Abel zur Wiesch P, Kouyos R, Abel S, Viechtbauer W, Bonhoeffer S. Cycling empirical antibiotic therapy in hospitals: Meta-analysis and models. PLoS Pathog. 2014;10(6):e1004225. doi: 10.1371/journal.ppat.1004225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hastings IM, Watkins WM, White NJ. The evolution of drug-resistant malaria: The role of drug elimination half-life. Philos Trans R Soc Lond B Biol Sci. 2002;357(1420):505–519. doi: 10.1098/rstb.2001.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bangsberg DR, Kroetz DL, Deeks SG. Adherence-resistance relationships to combination HIV antiretroviral therapy. Curr HIV/AIDS Rep. 2007;4(2):65–72. doi: 10.1007/s11904-007-0010-0. [DOI] [PubMed] [Google Scholar]
- 17.Rosenbloom DI, Hill AL, Rabi SA, Siliciano RF, Nowak MA. Antiretroviral dynamics determines HIV evolution and predicts therapy outcome. Nat Med. 2012;18(9):1378–1385. doi: 10.1038/nm.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchison DA. How drug resistance emerges as a result of poor compliance during short course chemotherapy for tuberculosis. Int J Tuberc Lung Dis. 1998;2(1):10–15. [PubMed] [Google Scholar]
- 19.Harrigan PR, et al. Predictors of HIV drug-resistance mutations in a large antiretroviral-naive cohort initiating triple antiretroviral therapy. J Infect Dis. 2005;191(3):339–347. doi: 10.1086/427192. [DOI] [PubMed] [Google Scholar]
- 20.Bacheler LT, et al. Human immunodeficiency virus type 1 mutations selected in patients failing efavirenz combination therapy. Antimicrob Agents Chemother. 2000;44(9):2475–2484. doi: 10.1128/aac.44.9.2475-2484.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pennings PS. Standing genetic variation and the evolution of drug resistance in HIV. PLoS Comput Biol. 2012;8(6):e1002527. doi: 10.1371/journal.pcbi.1002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solas C, et al. Discrepancies between protease inhibitor concentrations and viral load in reservoirs and sanctuary sites in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2003;47(1):238–243. doi: 10.1128/AAC.47.1.238-243.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dartois V. The path of anti-tuberculosis drugs: From blood to lesions to mycobacterial cells. Nat Rev Microbiol. 2014;12(3):159–167. doi: 10.1038/nrmicro3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varatharajan L, Thomas SA. The transport of anti-HIV drugs across blood-CNS interfaces: Summary of current knowledge and recommendations for further research. Antiviral Res. 2009;82(2):A99–A109. doi: 10.1016/j.antiviral.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Else LJ, Taylor S, Back DJ, Khoo SH. Pharmacokinetics of antiretroviral drugs in anatomical sanctuary sites: The male and female genital tract. Antivir Ther. 2011;16(8):1149–1167. doi: 10.3851/IMP1919. [DOI] [PubMed] [Google Scholar]
- 26.Fletcher CV, et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci USA. 2014;111(6):2307–2312. doi: 10.1073/pnas.1318249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antinori A, et al. Efficacy of cerebrospinal fluid (CSF)-penetrating antiretroviral drugs against HIV in the neurological compartment: Different patterns of phenotypic resistance in CSF and plasma. Clin Infect Dis. 2005;41(12):1787–1793. doi: 10.1086/498310. [DOI] [PubMed] [Google Scholar]
- 28.Edén A, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis. 2010;202(12):1819–1825. doi: 10.1086/657342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Lelyveld SF, et al. Therapy failure following selection of enfuvirtide-resistant HIV-1 in cerebrospinal fluid. Clin Infect Dis. 2010;50(3):387–390. doi: 10.1086/649874. [DOI] [PubMed] [Google Scholar]
- 30.Singh R, Ray P, Das A, Sharma M. Penetration of antibiotics through Staphylococcus aureus and Staphylococcus epidermidis biofilms. J Antimicrob Chemother. 2010;65(9):1955–1958. doi: 10.1093/jac/dkq257. [DOI] [PubMed] [Google Scholar]
- 31.Deresinski S. Vancomycin in combination with other antibiotics for the treatment of serious methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis. 2009;49(7):1072–1079. doi: 10.1086/605572. [DOI] [PubMed] [Google Scholar]
- 32.Kepler TB, Perelson AS. Drug concentration heterogeneity facilitates the evolution of drug resistance. Proc Natl Acad Sci USA. 1998;95(20):11514–11519. doi: 10.1073/pnas.95.20.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greulich P, Waclaw B, Allen RJ. Mutational pathway determines whether drug gradients accelerate evolution of drug-resistant cells. Phys Rev Lett. 2012;109(8):088101. doi: 10.1103/PhysRevLett.109.088101. [DOI] [PubMed] [Google Scholar]
- 34.Hermsen R, Deris JB, Hwa T. On the rapidity of antibiotic resistance evolution facilitated by a concentration gradient. Proc Natl Acad Sci USA. 2012;109(27):10775–10780. doi: 10.1073/pnas.1117716109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, et al. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science. 2011;333(6050):1764–1767. doi: 10.1126/science.1208747. [DOI] [PubMed] [Google Scholar]
- 36.Toprak E, et al. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2012;44(1):101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipsitch M, Levin BR. Population dynamics of tuberculosis treatment: Mathematical models of the roles of non-compliance and bacterial heterogeneity in the evolution of drug resistance. Int J Tuberc Lung Dis. 1998;2(3):187–199. [PubMed] [Google Scholar]
- 38.Ankomah P, Levin BR. Two-drug antimicrobial chemotherapy: A mathematical model and experiments with Mycobacterium marinum. PLoS Pathog. 2012;8(1):e1002487. doi: 10.1371/journal.ppat.1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ankomah P, Johnson PJT, Levin BR. The pharmaco-, population and evolutionary dynamics of multi-drug therapy: Experiments with S. aureus and E. coli and computer simulations. PLoS Pathog. 2013;9(4):e1003300. doi: 10.1371/journal.ppat.1003300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nowak MA, May RMC. Virus Dynamics: Mathematical Principles of Immunology and Virology. Oxford Univ Press; New York: 2000. [Google Scholar]
- 41.Iwasa Y, Michor F, Nowak MA. Stochastic tunnels in evolutionary dynamics. Genetics. 2004;166(3):1571–1579. doi: 10.1534/genetics.166.3.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weissman DB, Desai MM, Fisher DS, Feldman MW. The rate at which asexual populations cross fitness valleys. Theor Popul Biol. 2009;75(4):286–300. doi: 10.1016/j.tpb.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kouyos RD, et al. 2014. The path of least resistance: Aggressive or moderate treatment? Proc R Soc B Biol Sci 281(1794):20140566.
- 44.Bonhoeffer S, Nowak MA. Pre-existence and emergence of drug resistance in HIV-1 infection. Proc Biol Sci. 1997;264(1382):631–637. doi: 10.1098/rspb.1997.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ribeiro RM, Bonhoeffer S, Nowak MA. The frequency of resistant mutant virus before antiviral therapy. AIDS. 1998;12(5):461–465. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 46.Alexander HK, Bonhoeffer S. Pre-existence and emergence of drug resistance in a generalized model of intra-host viral dynamics. Epidemics. 2012;4(4):187–202. doi: 10.1016/j.epidem.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 47.Orr HA, Unckless RL. Population extinction and the genetics of adaptation. Am Nat. 2008;172(2):160–169. doi: 10.1086/589460. [DOI] [PubMed] [Google Scholar]
- 48.Müller M, dela Peña A, Derendorf H. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: Distribution in tissue. Antimicrob Agents Chemother. 2004;48(5):1441–1453. doi: 10.1128/AAC.48.5.1441-1453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Joukhadar C, et al. Impaired target site penetration of beta-lactams may account for therapeutic failure in patients with septic shock. Crit Care Med. 2001;29(2):385–391. doi: 10.1097/00003246-200102000-00030. [DOI] [PubMed] [Google Scholar]
- 50.Read AF, Day T, Huijben S. 2011. The evolution of drug resistance and the curious orthodoxy of aggressive chemotherapy. Proc Natl Acad Sci USA 108(Suppl 2):10871–10877.
- 51.Jung YJ, et al. Effect of vancomycin plus rifampicin in the treatment of nosocomial methicillin-resistant Staphylococcus aureus pneumonia. Crit Care Med. 2010;38(1):175–180. doi: 10.1097/ccm.0b013e3181b9ecea. [DOI] [PubMed] [Google Scholar]
- 52.Simon GL, Smith RH, Sande MA. Emergence of rifampin-resistant strains of Staphylococcus aureus during combination therapy with vancomycin and rifampin: A report of two cases. Rev Infect Dis. 1983;5(Suppl 3):S507–S508. doi: 10.1093/clinids/5.supplement_3.s507. [DOI] [PubMed] [Google Scholar]
- 53.Mwangi MM, et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci USA. 2007;104(22):9451–9456. doi: 10.1073/pnas.0609839104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pennings PS, Kryazhimskiy S, Wakeley J. Loss and recovery of genetic diversity in adapting populations of HIV. PLoS Genet. 2014;10(1):e1004000. doi: 10.1371/journal.pgen.1004000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.UK Collaborative Group on HIV Drug Resistance UK CHIC Study Group Long-term probability of detecting drug-resistant HIV in treatment-naive patients initiating combination antiretroviral therapy. Clin Infect Dis. 2010;50(9):1275–1285. doi: 10.1086/651684. [DOI] [PubMed] [Google Scholar]
- 56.Wright S. Isolation by distance. Genetics. 1943;28(2):114–138. doi: 10.1093/genetics/28.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uecker H, Otto SP, Hermisson J. Evolutionary rescue in structured populations. Am Nat. 2014;183(1):E17–E35. doi: 10.1086/673914. [DOI] [PubMed] [Google Scholar]
- 58.Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26(3):585–615. [Google Scholar]
- 59.Greco WR, Bravo G, Parsons JC. The search for synergy: A critical review from a response surface perspective. Pharmacol Rev. 1995;47(2):331–385. [PubMed] [Google Scholar]
- 60.Jilek BL, et al. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med. 2012;18(3):446–451. doi: 10.1038/nm.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ocampo PS, et al. Antagonism between bacteriostatic and bactericidal antibiotics is prevalent. Antimicrob Agents Chemother. 2014;58(8):4573–4582. doi: 10.1128/AAC.02463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michel J-B, Yeh PJ, Chait R, Moellering RC, Jr, Kishony R. Drug interactions modulate the potential for evolution of resistance. Proc Natl Acad Sci USA. 2008;105(39):14918–14923. doi: 10.1073/pnas.0800944105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chait R, Craney A, Kishony R. Antibiotic interactions that select against resistance. Nature. 2007;446(7136):668–671. doi: 10.1038/nature05685. [DOI] [PubMed] [Google Scholar]
- 64.Sanjuán R, Moya A, Elena SF. The contribution of epistasis to the architecture of fitness in an RNA virus. Proc Natl Acad Sci USA. 2004;101(43):15376–15379. doi: 10.1073/pnas.0404125101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bonhoeffer S, Chappey C, Parkin NT, Whitcomb JM, Petropoulos CJ. Evidence for positive epistasis in HIV-1. Science. 2004;306(5701):1547–1550. doi: 10.1126/science.1101786. [DOI] [PubMed] [Google Scholar]
- 66.Holmes EC. Evolution and Emergence of RNA Viruses. Oxford Univ Press; New York: 2009. [Google Scholar]
- 67.Bretscher MT, Althaus CL, Müller V, Bonhoeffer S. Recombination in HIV and the evolution of drug resistance: For better or for worse? BioEssays. 2004;26(2):180–188. doi: 10.1002/bies.10386. [DOI] [PubMed] [Google Scholar]
- 68.Carvajal-Rodríguez A, Crandall KA, Posada D. Recombination favors the evolution of drug resistance in HIV-1 during antiretroviral therapy. Infect Genet Evol. 2007;7(4):476–483. doi: 10.1016/j.meegid.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Althaus CL, Bonhoeffer S. Stochastic interplay between mutation and recombination during the acquisition of drug resistance mutations in human immunodeficiency virus type 1. J Virol. 2005;79(21):13572–13578. doi: 10.1128/JVI.79.21.13572-13578.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kearney BP, Aweeka FT. The penetration of anti-infectives into the central nervous system. Neurol Clin. 1999;17(4):883–900. doi: 10.1016/s0733-8619(05)70171-7. [DOI] [PubMed] [Google Scholar]
- 71.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6(8):583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 72.Fu F, Nowak MA, Bonhoeffer S. Spatial heterogeneity in drug concentrations can facilitate the emergence of resistance to cancer therapy. PLoS Comput Biol. 2015;11(3):e1004142. doi: 10.1371/journal.pcbi.1004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sarathy J, Dartois V, Dick T, Gengenbacher M. Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57(4):1648–1653. doi: 10.1128/AAC.02202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis. 2011;204(12):1951–1959. doi: 10.1093/infdis/jir658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.