

Abstract
Background
The bacterium Corynebacterium pseudotuberculosis (Cp) causes caseous lymphadenitis (CLA), mastitis, ulcerative lymphangitis, and oedema in a number of hosts, comprising ruminants, thereby intimidating economic and dairy industries worldwide. So far there is no effective drug or vaccine available against Cp. Previously, a pan-genomic analysis was performed for both biovar equi and biovar ovis and a Pathogenicity Islands (PAIS) analysis within the strains highlighted a large set of proteins that could be relevant therapeutic targets for controlling the onset of CLA. In the present work, a structural druggability analysis pipeline was accomplished along 15 previously sequenced Cp strains from both biovar equi and biovar ovis.
Methods and results
We computed the whole modelome of a reference strain Cp1002 (NCBI Accession: NC_017300.1) and then the homology models of proteins, of 14 different Cp strains, with high identity (≥ 85%) to the reference strain were also done. Druggability score of all proteins pockets was calculated and only those targets that have a highly druggable (HD) pocket in all strains were kept, a set of 58 proteins. Finally, this information was merged with the previous PAIS analysis giving two possible highly relevant targets to conduct drug discovery projects. Also, off-targeting information against host organisms, including Homo sapiens and a further analysis for protein essentiality provided a final set of 31 druggable, essential and non-host homologous targets, tabulated in table S4, additional file 1. Out of 31 globally druggable targets, 9 targets have already been reported in other pathogenic microorganisms, 3 of them (3-isopropylmalate dehydratase small subunit, 50S ribosomal protein L30, Chromosomal replication initiator protein DnaA) in C. pseudotuberculosis.
Conclusion
Overall we provide valuable information of possible targets against C. pseudotuberculosis where some of these targets have already been reported in other microorganisms for drug discovery projects, also discarding targets that might be physiologically relevant but are not amenable for drug binding. We propose that the constructed in silico dataset might serve as a guidance for the scientific community to have a better understanding while selecting putative therapeutic protein candidates as druggable ones as effective measures against C. pseudotuberculosis.
Keywords: Corynebacterium pseudotuberculosis (Cp), Druggable genome, Structural proteomics approach, Putative globally/conserved druggable/bindable targets, Caseous lymphadenitis
Background
Efforts to find new bacterial drug and/or vaccine targets are becoming indispensable due to the antimicrobial resistance, rapid loss of effectiveness in antibiotic treatment and the quantitative emergence of multi-resistant microbial strains that pose a global challenge and threat. Corynebacterium pseudotuberculosis (Cp) is a pathogen of great veterinary and economic importance, since it affects a broad spectrum of animal livestock worldwide, mainly sheep and goats, as well as mammals in numerous Asiatic, Arabic and African countries, North and Latin America and Australia [1]. C. pseudotuberculosis is a Gram-positive, facultative intracellular and pleomorphic organism; it possesses fimbriae but is non-motile in nature [2]. The rpoB gene analysis for CMNR group of bacteria (Corynebacterium, Mycobacterium, Nocardia and Rhodococcus), which has a great medical, veterinary and biotechnological importance, has shown a close phylogenetic relationship [3]. A number of pathogenic strains from a wide range of hosts have already been sequenced, demonstrating the importance of this microorganism [3]. The pathogen infects goat and sheep populations (biovar ovis), causing caseous lymphadenitis (CLA), a chronic contagious disease with abscess formation in superficial lymph nodes and subcutaneous tissues. Biovar equi, on the other hand, infects lung, kidney, liver and spleen in higher mammals like cow, camel, buffalo etc. thereby, threatening the life of herd animals [2,4]. There are few reports in humans of symptoms similar to lymphadenitis abscesses, caused by an occupational exposure to the infected animals [5-7]. Bearing in mind the medical importance of C. pseudotuberculosis due to a lack of efficient medicines, here, we have made an effort and applied a computational strategy to search for new therapeutic molecular targets from this bacterium.
Homology modelling is a widely used technique that has proved good results to expand structural space of pathogens [8,9]. We have designed and implemented a protein structure prediction pipeline using homology modelling based on Martí-Renom methodologies [10]. The pipeline was applied to the randomly selected Cp1002 genome as a reference strain; the genomes of other strains were modelled using a mutation procedure, the sequences that present homology in both, i.e., the core genome, were already modelled in the reference strain Cp1002.
The main purpose of this study is to offer information based on recently reported structure-based prediction of protein druggability that might be valuable for target selection in drug design projects. Druggability is a concept used to describe the ability of a given protein to bind a drug-like molecule, which in turn modulates its function in a desired way. Purely, from a structural point of view, it is the likelihood that small drug-like molecules bind a given target protein with high affinity (< 1µM), a concept also referred to "bind ability". Early attempts to determine the druggable genome of an organism were based on counting the number of targets belonging to known druggable domains that have yielded values in range of 10-14% for the human genome. According to our knowledge, a structural druggability assessment for the C. pseudotuberculosis multi-strain proteome was never performed before.
Recently, we developed a fast method for druggability prediction based on the open source pocket detection code "fpocket", which combines several physicochemical descriptors to estimate the pocket druggability and that can be used on a genomic scale [11]. Druggable proteins should have a pocket with suitable features that enable binding of a drug-like compound. After computing proteins that remains druggable along the 15 Cp genomes, 58 target candidates were selected.
The Cp genome has been reported to include seven putative pathogenicity islands (PAIS) [12], which contain several classical virulence factors, including genes for fimbrial subunits, adhesion factors, iron uptake and secreted toxins. Additionally, all of the virulence factors in the islands have characteristics that indicate the phenomenon of horizontal gene transfer. The importance of our dataset is enhanced with the emerged information from the literature regarding the PAIS, pan-genome and also with the pan-modelome strategies for target selection [38].
Methods
General concept
The druggability analysis pipeline consisted of the following steps (Figure 1). The Open Reading Frame (ORFs) sequences of C. pseudotuberculosis were obtained from the GenBank database [13,14]. All ORFs were then analysed with the HMMer software and the structural domains were assigned. Then, each ORF was used to perform a BLAST search against the Protein Data Bank to determine which structure(s) will be used as template(s) to perform homology modelling of the ORFs or computed domains. For all the 3D modelled structures, a set of structural properties were computed, including: i) the Druggability Score (DS) for each pocket, ii) the active site residues (if available) according to the template structures, iii) the conserved or family relevant residues. This information was later combined with the essentiality criteria and the previously related pathogenicity information present in the literature. A detailed description of the programs and databases used to perform each of the above mentioned pipeline step is given below in detail (Figure 1).
Figure 1.
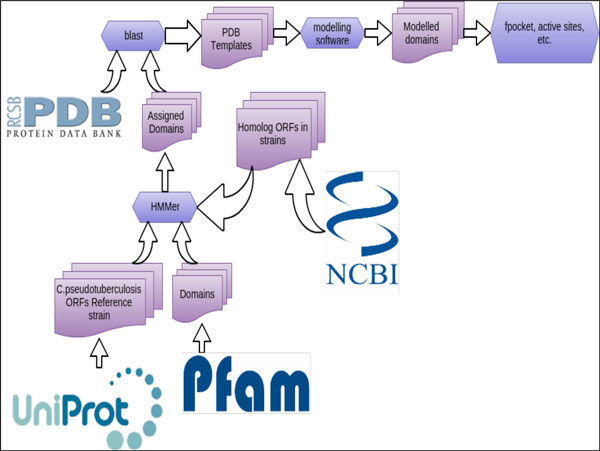
A general sketch of the pipeline. All outputs, steps and summaries are available for download purpose and later analyses; links are available in supplementary material.
Initial dataset construction
All ORFs or possible proteins for all the strains of Cp were obtained by downloading the information available at the NCBI database (ftp://ftp.ncbi.nih.gov/genomes/Bacteria). The randomly selected strain used as a reference genome for further calculations is Cp1002, according to recent work by Hassan et al., 2014 [38]. The Cp1002 genome has 2097 reported ORFs.
Pfam domain assignment
All the ORFs in the reference proteome were analysed with the HMMer program and were later assigned the Pfam families or domains, leading to a total of 2455 domains assignments from Pfam-A entries and 509 ORFs with no domain assigned. However, as expected, more than one ORF can be assigned to the same domain. Thus, considering this information, a total of 1327 unique (i.e. different) domains were assigned to a whole Cp reference genome. On average, the Cp reference genome has 1.87 domains per ORF and 1.58 unique domains per ORF.
Generation of structural homology-based models
The strain Cp1002 was used a basis for the structural study. For each sequence in this strain, several models were built using the following procedures: First, a PSI-Blast [15] search was performed against UniRef50, using 3 iterations and an e.value threshold of 0.0001, in order to compute a checkpoint that will be used as a profile for the target sequence. Second, PSI-Blast search is restarted using the aforementioned checkpoint against a template library. The template library consisted of all sequences from every individual protein chain in the Protein Data Bank (PDB), grouped at 95% sequence identity threshold using CD-HIT [16]. Afterward, the target-template alignments were computed in the last step, with an e-value of ≤ 0.00001 to build the model structures using the MODELLER software [17,18]. For each target-template alignment, ten different target models were built, and their quality measures were assessed using the GA341 [19,20] and QMEAN methods [21]. Only the models with GA341 score ≥ 0.7 (score for the reliability of a model derived from statistical potentials) [22], were retained. A reliable model has a probability of correct fold larger than 95% and coverage of over 50%. Every sequence belonging to the other 14 strains were compared to strain Cp1002 using BLASTp. For each sequence that gives a hit with sequence identity above 85%, a mutation methodology was applied on each amino acid substitution using MODELLER between this sequence and the sequence belonging to Cp1002.
Structural assessment of druggability
Structural druggability of each modelled and potential target was assessed by determining and characterizing the ability of putative pockets to bind a drug-like molecule by using the fpocket program and the recently developed Drug Score (DS) [23]. Briefly, the method is based upon Voronoi Tessellation algorithm to identify pockets and computes suitable physicochemical descriptors (polar and apolar surface area, hydrophobic density, hydrophobic and polarity score) that are combined to yield the DS, which ranges between 0 (non-druggable, ND) to 1 (highly druggable, HD). In this work we separated the druggability score in four sets: non-druggable proteins (ND; DS ≤ 0.2), poorly druggable (PD; 0.2 < DS ≤ 0.5), druggable (D; 0.5 < DS ≤ 0.7), and highly druggable (HD; DS ≥ 0.7). This distribution based on our previous study where we computed the druggability score for all pockets present in all unique proteins in the PDB, which were crystallized in complex with a drug-like compound [11]. In Figure 2, we compare the druggability distribution of all structures of Cp genome built in this work. Although the distribution has a small shift to higher values; we use the same bounds to define the sets of druggable proteins (Figure 2).
Figure 2.
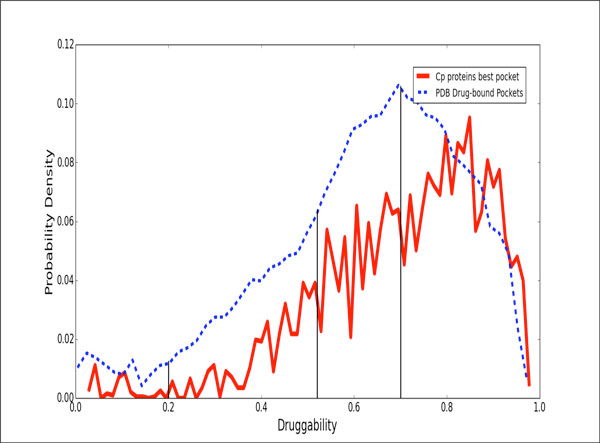
Histogram of druggability score (DS). All ligand-bound structures in the PDB (blue pointed line) and all the modeled structures of the C. pseudotuberculosis genome (red line) are represented in the histogram. The scores were computed using the fpocket program for all pockets present in all unique proteins in the PDB, which were crystallized in complex with a drug-like compound. A Gaussian fit of the data made to define these sets was performed in Radusky et al., 2014 [11]. The sets are: non-druggable proteins (ND; DS ≤ 0.2), poorly druggable (PD; 0.2 < DS ≤ 0.5), druggable (D; 0.5 < DS ≤ 0.7), and highly druggable (HD; DS ≥ 0.7). The red line is the distribution of DS over all the models built in this work.
Extending this information along the different strains, those structures labelled as HD in the reference strain were analysed in the other ones. A protein target, which remained druggable in all the strains, was classified as Globally Druggable (GD), also, a protein druggable in all the virulent strains is classified as Virulent Druggable (VD). The GD, obviously, remained a subset of VD.
Active site identification
In order to identify the active site pocket, our pipeline implements two different analyses that rely on; i) the information from the CSA, and ii) a Pfam position site, as the importance criteria.
The data from CSA was downloaded from http://www.ebi.ac.uk/thornton-srv/databases/CSA/ that consisted of a list of PDB_IDs linked to a number of residues, which comprised the corresponding protein active site. To map the active site residues to as many Cp protein domains as possible, each PDB_ID used as a template for the homology modelling in CSA was assigned to the modelled ORFs.
As an alternative approach to determine the relevance of a given pocket (or residues), we looked for residues of a given Pfam family/domain that were located in an important position and are well conserved. Important positions were defined as those positions in the corresponding HMMer model whose information content was larger than a defined importance cut-off value (icov). The nature of the conserved amino acids in the corresponding position was determined by comparing each residue type emission probability (ep) with icov. If the ratio between ep and icov was larger than a conserved type cut-off value (ctcov), the corresponding residue type was assumed as conserved. Optimal values of icov and ctcov [11] were 0.27 and 0.24, respectively. These values were calculated as the ones that allowed labelling all the important residues of the CSA database in their respective protein's domains. Briefly, this approach is a strategy to extend the definition of catalytic site capturing all the sites described in CSA. CSA is a curated database with limited amount of data. Our strategy gives us a clue about new candidates of being catalytic sites, with some false positives.
By using these analyses, for each Pfam domain, the pipeline provided a list of position-residue type relevant residues, which could thus be mapped to all Cp ORFs with assigned Pfam domain.
Pathogenicity islands and pan-modelomics information
Information collected from other software pipelines is used in the present work to enhance the target selection process.
All the ORFs that belonged to a pathogenicity island (PAIS) were properly labelled with this indicator. The PAIS were previously computed and identified using the PIPS software [24] that has predicted 16 pathogenicity islands in C. pseudotuberculosis. PIPS predicts pathogenicity islands by taking into account some important features, i.e., flanking tRNA, codon usage bias, GC content deviation; transposases, virulence factors and their absence in non-pathogenic organism of the same genus or related species. Pathogenicity islands are large regions that were acquired through horizontal gene transfer that represent the genome plasticity of a species and possess a high concentration of virulence factors. Virulence factors are proteins whose function is related to bacterial virulence and pathogenicity. They help the pathogenic bacterium in adhesion mechanisms, invasion, and surviving through colonization, and replication inside the host as well as in immune system evasion [24].
A novel integrative approach has been adapted in a recent work for the identification of new therapeutic targets in C. pseudotuberculosis [38] where a final set of 10 proteins has been selected that was essential for the bacterial survival. Here, too, a focus was made only on the previously selected homologous target proteins in the reference Cp1002 genome, whose detailed analysis with our pipeline is reported.
Bacterial essential and non-host homologous proteins analysis
The pool of global highly druggable (GD) 58 target proteins was subjected to NCBI-BLASTp (e-value lesser than 1e-07, bit score ≥100, identity ≥ 50%) against human and ovine proteomes to identify non-host homologs targets, and coverage of all the sequences of the proteins greater than 80% [25]. The exhaustive list of GD and Virulent highly druggable (VD) ORFs is available in supplementary material table S1, additional file 1.
Furthermore, from the filtered list of 41 highly druggable non-host homologous target proteins, an approach based on a subtractive genomics was made for the conserved GD targets that were essential to bacteria [26]. Briefly, the set of proteins from C. pseudotuberculosis were submitted to the Database of Essential Genes (DEG, which contains experimentally validated essential genes from 20 bacteria) for homology analyses [27]. The BLASTp cut-off values used were: e-value = 1e-05, bit score ≥100, identity ≥ 35% [26]. The final list of targets based on previously described criteria contained 31 essential and non-host homologous target proteins. The list of putative targets was subjected to biochemical pathway analysis to KEGG (Kyoto Encyclopaedia of Genes and Genomes) [28], virulence using PIPS (Pathogenicity Island Prediction Software) [29], functionality using UniProt (Universal Protein Resource) [30], and cellular localization using CELLO (subCELlular LOcalization predictor) [31].
Results and discussion
Models in the reference Cp1002 strain
The following subsections describe the database data summary after running the HMMer software and the modelling pipeline.
A total of 2598 models were built from the ORFs of the reference genome and then a part of the ORFs were assigned to the Pfam families. Here, 1206 unique ORFs were involved. The models from the Pfam family were 1546 in total, where only 879 unique Pfam families were used. The other 1051 models correspond to full ORF models (table 1).
Table 1.
Summary of modelling pipeline over non-reference strains.
| NCBI accession | C. pseudotuberculosis strains | ORFs | Homolog ORFs to Cp1002 | Modelled ORFs |
|---|---|---|---|---|
| NC_017308.1 | Cp1/06-A | 1963 | 1654 | 493 |
| NC_017730.1 | Cp31 | 2088 | 1663 | 441 |
| NC_017945.1 | Cp258 | 2106 | 1661 | 503 |
| NC_016932.1 | Cp316 | 2057 | 1696 | 502 |
| NC_017307.1 | CpCIP52.97 | 2057 | 1704 | 489 |
| NC_018019.1 | Cp162 | 2002 | 1652 | 444 |
| NC_017031.1 | CpP54B96 | 2084 | 1808 | 543 |
| NC_017462.1 | Cp267 | 2148 | 1826 | 561 |
| NC_017306.1 | Cp42/02-A | 2097 | 1783 | 544 |
| NC_017301.1 | CpC231 | 2051 | 1821 | 552 |
| NC_017303.1 | CpI19 | 2095 | 1826 | 552 |
| NC_016781.1 | Cp3/99-5 | 2099 | 1759 | 530 |
| NC_017305.1 | CpPAT10 | 2142 | 1814 | 542 |
| NC_014329.1 | CpFRC41 | 2089 | 1827 | 559 |
Models in non-reference strains
The details of these constructed models are presented here in a tabulated form.
Druggability summary of 15 C. pseudotuberculosis strains
A summary of the calculated structural druggability scores is presented in table 2. In parentheses are the pockets containing residues from CSA database or at least one important residue from the Pfam family, assigned to the corresponding ORF in the Cp1002 genome. It should be noted that the pocket calculations over the non-reference strains were done only for the homologous models (proteins with identity > 85% to the reference genome). ORFs with high druggability over all the strains are labelled as Global druggable and those druggable in the pathogenic islands as Virulent druggable. For Cp1002 strain, only those ORFs in the list appear having at least one homolog ORF in another strain. In other strains, only the ORFs homologous to a Cp1002 ORF are considered for this table.
Table 2.
Summary of druggability for all C. pseudotuberculosis strains, Global highly druggable (GD) ORFs and Virulent highly druggable (VD) ORFs.
| NCBI accession | C. pseudotuberculosis Strain | ND or PD | D | HD |
|---|---|---|---|---|
| NC_017300.1 | Cp1002 | 20 | 185 | 377 |
| NC_017308.1 | Cp1/06-A | 42 | 224 | 227 |
| NC_017730.1 | Cp31 | 36 | 202 | 203 |
| NC_017945.1 | Cp258 | 39 | 226 | 238 |
| NC_016932.1 | Cp316 | 44 | 232 | 226 |
| NC_017307.1 | CpCIP52.97 | 39 | 227 | 223 |
| NC_018019.1 | Cp162 | 32 | 196 | 216 |
| NC_017031.1 | CpP54B96 | 43 | 241 | 259 |
| NC_017462.1 | Cp267 | 44 | 255 | 262 |
| NC_017306.1 | Cp42/02-A | 45 | 249 | 250 |
| NC_017301.1 | CpC231 | 48 | 248 | 256 |
| NC_017303.1 | CpI19 | 44 | 251 | 257 |
| NC_016781.1 | Cp3/99-5 | 44 | 246 | 240 |
| NC_017305.1 | CpPAT10 | 42 | 246 | 254 |
| NC_014329.1 | CpFRC41 | 46 | 251 | 262 |
| Global highly druggable (GD) ORFs | 58 | |||
| Virulent highly druggable (VD) ORFs | 2 | |||
Bacterial essential and non-host homologous proteins analyses
An exhaustive literature review of candidate druggable targets
As aforementioned, the list of 58 most druggable proteins (table S1, additional file 1) was compared to the corresponding host proteomes, leading to the identification of 41 non-host homologous proteins (table S2, additional file 1) and 17 host homologous proteins (table S3, additional file 1). A final list of 31 essential and non-host homologous targets from C. pseudotuberculosis is given in table S4, additional file 1 after computing the 41 non-host homologous proteins to the database of essential genes (DEG), using the earlier mentioned default parameters.
Based on this filter, the number of selected targets was drastically reduced to a final set of 31 targets. This list was considered as druggable, essential and non-host homologous target proteins. We have further extrapolated our search to find out whether some of these putative targets have already been reported in the literature or not. This way 9 such putative targets were found where 3 of them including 3-isopropylmalate dehydratase small subunit, 50S ribosomal protein L30 and Chromosomal replication initiator protein DnaA have previously been reported in C. pseudotuberculosis [25,26], while the other 6 druggable target proteins have been reported in other pathogenic microorganisms, both in bacteria and parasites (table 3). The remaining 22 druggable targets that are not yet reported as putative targets were searched for molecular functions, biological processes, cellular compartmentalisations and metabolic pathway roles (table S5, additional file 1).
Table 3.
Essential and non-host homologous bacterial protein already reported as drug target in other pathogenic microorganisms.
| Sr. No | Reference Protein Loci | UniProt Accession | Protein Name | PMID/Reference |
|---|---|---|---|---|
| 1 | Cp1002_0854 | D9Q7U7 | 1,4-alpha-glucan branching enzyme GlgB | [32] |
| 2 | Cp1002_0907 | D9Q7Z8 | 3-isopropylmalate dehydratase small subunit | [25] |
| 3 | Cp1002_0948 | D9Q838 | Acetylglutamate kinase | [33] |
| 4 | Cp1002_0412 | D9Q6L7 | Alanine racemase | [34] |
| 5 | Cp1002_0374 | D9Q6I0 | 50S ribosomal protein L30 | [26] |
| 6 | Cp1002_1592 | D9Q409 | Bifunctional protein folC | [35] |
| 7 | Cp1002_1239 | D9Q8X6 | Diaminopimelate epimerase | [36] |
| 8 | Cp1002_1270 | D9Q906 | Riboflavin biosynthesis protein ribF | [37] |
| 9 | Cp1002_0001 | D9Q5G4 | Chromosomal replication initiator protein DnaA | [25] |
Conclusions and perspectives
In the present work we have attempted to show a comprehensive study of the druggability scores along the known completely sequenced strains of C. pseudotuberculosis species to complement further the research work performed by our colleagues and collaborators. After our pipeline was executed, a list of cross-strain highly druggable 58 ORFs was obtained. These ORFs had the information about their membership in the set of virulent strains. We too have provided the information if the most druggable pockets of the ORF have highly conserved residues in the Pfam families they belong to, and if a catalytic site is reported in the template structure or structures that were used to build the homology model for each ORF. We expect that the constructed dataset might serve as a guide for the scientific community to have a better understanding while selecting protein candidates as therapeutic and druggable ones. All the data is available via web at http://www.inquimae.fcen.uba.ar/turjanski_adrian.
The information obtained here was also used to exhaustively analyse the target proteins actually considered as druggable targets in previous works. Our analyses have proposed a set of putative druggable targets in the veterinary pathogen C. pseudotuberculosis on one hand, while on the other hand it has also demonstrated the efficiency and the high-throughput nature of our pipeline used in this study. All this work is expandable and could be applied to the emergence of new strains of the same organism species as well as to the new organisms with the same characteristics.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
Coordinated entire work: LGR SSH EL AGT. Performed all in silico analyses: LGR SSH EL ST SBJ. Cross-analysed genome contents, conserved pan-modelome, subtractive/genome modelome approach, residue level structural comparison: SSH LGR EL DB AGT. Provided timely consultation and reviewed the manuscript: VA DB AA JA RSF AS. Read and approved the final manuscript: RSF SSH JA DB AGT AS VA. Conceived and designed the work: SSH LGR EL. Analysed the data: SSH RSF ST SBJ DB AGT AM AS VA. Wrote the paper: LGR SSH AGT.
Supplementary Material
Supplementary tables.
Contributor Information
Leandro G Radusky, Email: leandro.radusky@gmail.com.
Syed Shah Hassan, Email: hassan_chemist@yahoo.com.
Esteban Lanzarotti, Email: estiben@gmail.com.
Sandeep Tiwari, Email: sandip_sbtbi@yahoo.com.
Syed Babar Jamal, Email: syedbabar.jamal@gmail.com.
Javed Ali, Email: javedgm@yahoo.com.
Amjad Ali, Email: amjad_uni@yahoo.com.
Rafaela Salgado Ferreira, Email: rafaelasf@gmail.com.
Debmalya Barh, Email: dr.barh@gmail.com.
Artur Silva, Email: asilva@ufpa.br.
Adrián G Turjanski, Email: vasco@icb.ufmg.br.
Vasco AC Azevedo, Email: adrian@qi.fcen.uba.ar.
Acknowledgements
This work resulted from a mutual collaboration among various, especially the two leading research and academic institutions of Argentine and Brazil, the Structural Bioinformatics Group, Institute of Physical Chemistry of Materials, Environment and Energy, University of Buenos Aires, Argentine and the Laboratory of Cellular and Molecular Genetics, PG Program in Bioinformatics, Institute of Biological Sciences, Federal University of Minas Gerais, Belo Horizonte, Minas Gerais, Brazil. We acknowledge an immense support from all the co-authors who have worked hard in accomplishing this task. We are grateful to our Brazilian collaborators, especially Prof. Dr. Vasco A.C Azevedo, Dr. Syed Shah Hassan (postdoctoral fellow) and other team members for their collaboration, timely consultation in theoretical and biological knowledge regarding the genetics background of the microorganism, C. pseudotuberculosis.
Declarations
Publication costs for this article were funded by grant from the Argentinian National Agency PICT-2010-2805 and VA from the Programa Nacional de Pós-doutorado da CAPES-PNPD/2011: Concessão Institucional, Brazil.
This article has been published as part of BMC Genomics Volume 16 Supplement 5, 2015: Proceedings of the 10th International Conference of the Brazilian Association for Bioinformatics and Computational Biology (X-Meeting 2014). The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S5.
References
- Hassan SS, Schneider MP, Ramos RT, Carneiro AR, Ranieri A, Guimaraes LC. et al. Whole-genome sequence of Corynebacterium pseudotuberculosis strain Cp162, isolated from camel. J Bacteriol. 2012;194(20):5718–5719. doi: 10.1128/JB.01373-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorella FA, Pacheco LG, Oliveira SC, Miyoshi A, Azevedo V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet Res. 2006;37(2):201–218. doi: 10.1051/vetres:2005056. [DOI] [PubMed] [Google Scholar]
- Soares SC, Trost E, Ramos RT, Carneiro AR, Santos AR, Pinto AC. et al. Genome sequence of Corynebacterium pseudotuberculosis biovar equi strain 258 and prediction of antigenic targets to improve biotechnological vaccine production. J Biotechnol. 2013;167(2):135–141. doi: 10.1016/j.jbiotec.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Khamis A, Raoult D, La Scola B. Comparison between rpoB and 16S rRNA gene sequencing for molecular identification of 168 clinical isolates of Corynebacterium. J Clin Microbiol. 2005;43(4):1934–1936. doi: 10.1128/JCM.43.4.1934-1936.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis MA, Lunetta AC. [Alcohol and drugs: preliminary survey of Brazilian nursing research] Rev Lat Am Enfermagem. 2005. pp. 1219–1230. [DOI] [PubMed]
- Peel MM, Palmer GG, Stacpoole AM, Kerr TG. Human lymphadenitis due to Corynebacterium pseudotuberculosis: report of ten cases from Australia and review. Clin Infect Dis. 1997;24(2):185–191. doi: 10.1093/clinids/24.2.185. [DOI] [PubMed] [Google Scholar]
- Williamson LH. Caseous lymphadenitis in small ruminants. Vet Clin North Am Food Anim Pract. 2001;17(2):359–371. doi: 10.1016/s0749-0720(15)30033-5. vii. [DOI] [PubMed] [Google Scholar]
- Kinnings SL, Xie L, Fung KH, Jackson RM, Xie L, Bourne PE. The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLoS Comput Biol. 2010;6(11):e1000976. doi: 10.1371/journal.pcbi.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Sankaran S, Mukherjee S, Yeturu K, Laskowski R, Bhardwaj A. et al. Structural annotation of Mycobacterium tuberculosis proteome. PLoS One. 2011;6(10):e27044. doi: 10.1371/journal.pone.0027044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Radusky L, Defelipe LA, Lanzarotti E, Luque J, Barril X, Marti MA, Turjanski AG. TuberQ: a Mycobacterium tuberculosis protein druggability database. Database (Oxford) 2014;2014(0):bau035. doi: 10.1093/database/bau035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz JC, D'Afonseca V, Silva A, Ali A, Pinto AC, Santos AR. et al. Evidence for reductive genome evolution and lateral acquisition of virulence functions in two Corynebacterium pseudotuberculosis strains. PLoS One. 2011;6(4):e18551. doi: 10.1371/journal.pone.0018551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilofsky HS, Burks C. The GenBank genetic sequence data bank. Nucleic Acids Res. 1988;16(5):1861–1863. doi: 10.1093/nar/16.5.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39(Database issue):D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Jaroszewski L, Godzik A. Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics. 2001;17(3):282–283. doi: 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. In: Curr Protoc Protein Sci. Unit 2.9. John E Coligan [et al], editor. Chapter 2. 2007. Comparative protein structure modeling using MODELLER. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277(5):1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- Melo F, Sali A. Fold assessment for comparative protein structure modeling. Protein Sci. 2007;16(11):2412–2426. doi: 10.1110/ps.072895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert P, Kunzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37(Web Server issue):W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo F, Sanchez R, Sali A. Statistical potentials for fold assessment. Protein Sci. 2002;11(2):430–448. doi: 10.1002/pro.110430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velec HF, Gohlke H, Klebe G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. Journal of medicinal chemistry. 2005;48(20):6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- Soares SC, Abreu VA, Ramos RT, Cerdeira L, Silva A, Baumbach J. et al. PIPS: pathogenicity island prediction software. PLoS One. 2012;7(2):e30848. doi: 10.1371/journal.pone.0030848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barh D, Gupta K, Jain N, Khatri G, Leon-Sicairos N, Canizalez-Roman A. et al. Conserved host-pathogen PPIs. Globally conserved inter-species bacterial PPIs based conserved host-pathogen interactome derived novel target in C. pseudotuberculosis, C. diphtheriae, M. tuberculosis, C. ulcerans, Y. pestis, and E. coli targeted by Piper betel compounds. Integr Biol (Camb) 2013;5(3):495–509. doi: 10.1039/c2ib20206a. [DOI] [PubMed] [Google Scholar]
- Barh D, Jain N, Tiwari S, Parida BP, D'Afonseca V, Li L. et al. A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chem Biol Drug Des. 2011;78(1):73–84. doi: 10.1111/j.1747-0285.2011.01118.x. [DOI] [PubMed] [Google Scholar]
- Zhang R, Ou HY, Zhang CT. DEG: a database of essential genes. Nucleic Acids Res. 2004;32(Database issue):D271–D272. doi: 10.1093/nar/gkh024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SH, Park YK, Lee S, Choi D, Oh TK, Hur CG, Kim JF. Towards pathogenomics: a web-based resource for pathogenicity islands. Nucleic Acids Res. 2007;35(Database issue):D395–D400. doi: 10.1093/nar/gkl790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CS, Lin CJ, Hwang JK. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004;13(5):1402–1406. doi: 10.1110/ps.03479604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg SK, Alam MS, Kishan KV, Agrawal P. Expression and characterization of alpha-(1,4)-glucan branching enzyme Rv1326c of Mycobacterium tuberculosis H37Rv. Protein Expr Purif. 2007;51(2):198–208. doi: 10.1016/j.pep.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martinez M, Marcos E, Tauler R, Field M, Crehuet R. Conformational compression and barrier height heterogeneity in the N-acetylglutamate kinase. J Phys Chem B. 2013;117(46):14261–14272. doi: 10.1021/jp407016v. [DOI] [PubMed] [Google Scholar]
- Anishetty S, Pulimi M, Pennathur G. Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput Biol Chem. 2005;29(5):368–378. doi: 10.1016/j.compbiolchem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Mathieu M, Debousker G, Vincent S, Viviani F, Bamas-Jacques N, Mikol V. Escherichia coli FolC structure reveals an unexpected dihydrofolate binding site providing an attractive target for anti-microbial therapy. J Biol Chem. 2005;280(19):18916–18922. doi: 10.1074/jbc.M413799200. [DOI] [PubMed] [Google Scholar]
- Pillai B, Cherney MM, Diaper CM, Sutherland A, Blanchard JS, Vederas JC, James MNG. Structural insights into stereochemical inversion by diaminopimelate epimerase: An antibacterial drug target. Proc Natl Acad Sci U S A. 2006;103(23):8668–8673. doi: 10.1073/pnas.0602537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia DF. [Novel targets for antibiotics discovery: riboswitches] Yao Xue Xue Bao. 2013;48(9):1361–1368. [PubMed] [Google Scholar]
- Hassan SS, Tiwari S, Guimarães LC, Jamal SB, Folador E, Sharma NB. et al. Proteome Scale Comparative Modeling for Conserved Drug and Vaccine Targets Identification in Corynebacterium pseudotuberculosis. BMC Genomics. 2014;15(Suppl 7):S3. doi: 10.1186/1471-2164-15-S7-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary tables.