

Abstract
Disorders of vascular function contribute importantly to cardiovascular disease which represents a substantial cause of morbidity and mortality worldwide. An emerging paradigm in the study of cardiovascular diseases is that protein ubiquitination and turnover represent key pathological mechanisms. Our understanding of these processes in the vasculature is growing but remains incomplete. Since protein ubiquitination and turnover can represent a terminal event in the life of a given protein, entry into these pathways must be highly regulated. However, at present understanding of these regulatory mechanisms, particularly in the vasculature, is fragmentary. The COP9 (constitutive photomorphogenic mutant 9) signalosome (CSN) is a heteromeric protein complex implicated in the control of protein degradation. The CSN participates critically in the control of Cullin Ring Ligases (CRLs), at least in part via the detachment of a small protein, Nedd8 (deneddylation). CRLs are one of the largest groups of ubiquitin ligases, which represent the most selective control point for protein ubiquitination. Thus, the CSN by virtue of its ability to control the CRLs ubiquitin ligase activity is ideally positioned to effect selective modulation of protein turnover. This review surveys currently available data regarding the potential role of the CSN in control of vascular function. Data potentially linking the CSN to control of regulatory proteins involved in vascular smooth muscle proliferation and to vascular smooth muscle contraction are presented with the intent of providing potentially intriguing possibilities for future investigation.
Keywords: The COP9 signalosome, cullin-RING ligases, ubiquitination, vascular smooth muscle proliferation, vascular smooth muscle contraction
Introduction
Cardiovascular diseases such as atherosclerosis, hypertension, heart failure, ischemic heart disease, myocardial infarction, stroke, and peripheral artery disease represent a leading cause of morbidity and mortality globally[1]. Of the approximately 53 million deaths recorded worldwide in 2010, fully one third are attributed to cardiovascular diseases [1]. The economic impact of this disease burden is staggering. It is estimated that the economic costs of these diseases amounted to $320 billion in 2011 for the United States alone [2]. Moreover, while the incidence of some adverse cardiovascular outcomes is decreasing others are increasing. Analyses of National Health and Nutrition Examination Survey (NHANES) data suggest that hypertension prevalence is increasing [3,4]. Recent American Heart Association predictions suggest that hypertension prevalence will increase by 10% by 2030 [2,5] by which time ~40% of the US population will suffer from this disease. Given the high prevalence of cardiovascular diseases increased understanding of the pathological mechanisms underlying these diseases is critical to revealing new targets for therapy. New approaches to treat cardiovascular disease could have substantial impact on worldwide health.
Ultimately, cell function and dysfunction hinge on cellular proteins. The amount and function of cellular proteins depend on a dynamic balance between protein synthesis and degradation. It is well recognized that degradation of cellular proteins by the ubiquitin proteasome system (UPS) or macroautophagy is critical to cell signaling and cell function [6,7]. Recent work from our laboratories showed that these processes are involved in both cardiac and vascular disease [8,9]. Since UPS protein processing can be a terminal event in the life of a given protein, entry into these pathways must be highly regulated. However, at present, understanding of these regulatory mechanisms is fragmentary. Ubiquitin E3 ligases, the substrate recognition components of the UPS, represent a major mechanism conferring specificity to proteasomal protein clearance. As such their regulation is an attractive target for therapeutic manipulation. The COP9 signalosome (CSN) represents a key mechanism for regulating a major subset of E3 ligases, the cullin-RING ligases (CRLs). The role of the CSN in regulating cardiac function is discussed in a companion paper in this issue [10]. This paper presents a brief overview of the potential for the CSN to participate in vascular function. Vascular dysregulation leading to the major cardiovascular diseases is represented largely by either VSM cell proliferation (e.g. neointimal growth in restenosis) or VSM contraction (e.g. elevated vascular resistance in hypertension). Accordingly, this review will focus on the potential role of the CSN in VSM cell proliferation and contraction.
Overview of the ubiquitin proteasome system, cullin Ring ligases and the COP9 signalosome
Since the UPS, CRLs and CSN systems are described in detail in a companion paper in this issue, this section will present only a brief overview. Readers interested in greater detail are directed to the companion paper [10]. Recent proteomic studies have revealed that ubiquitination is a major form of posttranslational modification in the cell [11], similar to or perhaps even more common than phosphorylation [12], which underscores the significance of ubiquitination. Ubiquitination, especially polyubiquitination with lys48-linkage, often targets the substrate for degradation by the 26S proteasome. Lys-48 ubiquitination targeting for proteasomal degradation represents the most studied function of ubiquitination, and is, in fact, responsible for the degradation of most cellular proteins [13]. Beyond targeting proteins for proteasomal degradation, ubiquitination can also serve as a signal to regulate a variety of other cellular processes, depending on the number of ubiquitins involved and the nature of ubiquitin chain linkage [14-17]. In all cases, ubiquitination attaches a small protein, ubiquitin, via an isopeptide bond to a lysine residue of the target protein or the preceding ubiquitin. This is accomplished by a series of stepwise reactions catalyzed by the ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3). The E3 ligase is responsible for substrate recognition and facilitates the transfer of an activated ubiquitin from E2 to the substrate target; hence, the E3 ligase determines which substrate protein to be ubiquitinated, conferring specificity of ubiquitination [18]. Consequently, ubiquitin E3 ligases are rightfully one of the most critical points in the UPS to be targeted for regulating the degradation of a target protein, which represents an emerging and potentially powerful strategy for therapeutic development [19].
The ubiquitin E3 ligases can be grouped based on certain common structural features. A ubiquitin E3 ligase usually carries a HECT (Homologous to E6AP Carboxyl Terminus) [20], RING (Really Interesting New Gene) [21], or U-box [22] domain. The RING domain containing group encompasses the largest number of ligases [22]. A large number of RING type ubiquitin ligases function in the form of protein complexes using a cullin (CUL) protein as the backbone, known as cullin-RING ligases (CRLs) [23,24]. The prototype of CRLs is the SCF (SKP1-CUL1-F-box) complex, in which CUL1 serves as the scaffold. CUL1 uses its carboxyl terminal to bind to a RING protein (RBX1) which recruits ubiquitin-charged E2 and uses its amino-terminal to bind a substrate receptor module consisting of SKP1 and variable F-box proteins. The F-box protein serves as the substrate receptor to directly recruit its specific substrate targets [25] (Figure 1). There are at least 7 cullins found in mammalian cells (CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5, and CUL7); each of them can form a subfamily of CRLs. Thus, based on the specific cullin used, CRLs are further classified into CRL1 (SCF), CRL2, CRL3, CRL4A, CRL4B, CRL5, and CRL7 subfamilies [24]. All subfamilies of CRLs share the same basic architecture but use different substrate receptor modules (for a detailed description of CRL subfamilies, please refer to the companion article by Wang and Martin in this Issue of Am J Cardiovasc Dis [10] and references therein).
Figure 1.
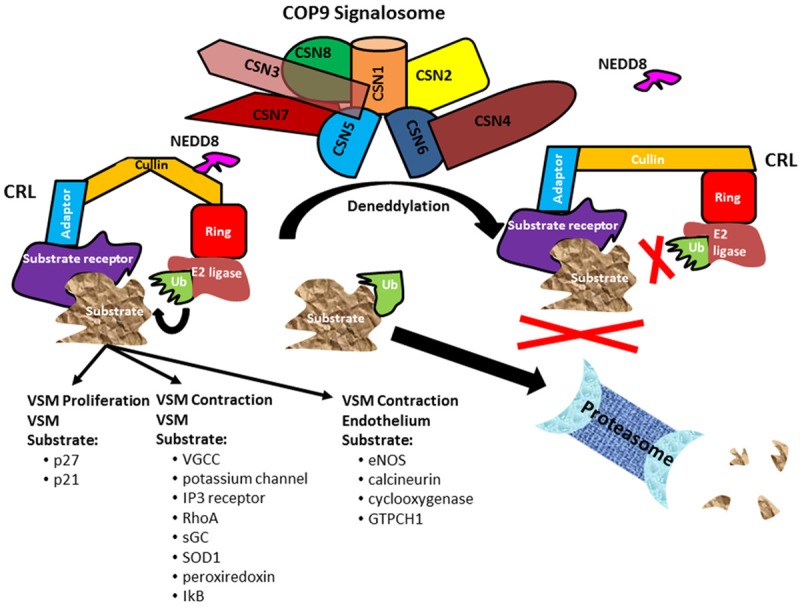
A summary of potential CSN targets in vasculature. The CSN is a heteromeric complex consisting of 8 subunits, CSN1~CSN8. The CSN is responsible for cullin deneddylation, with the deneddylase activity residing in CSN5. Neddylation/deneddylation is a key mechanism for controlling the activity of cullin RING ligases (CRLs). Attachment of NEDD8 (neddylation) renders the CRL able to ubiquitinate substrates to direct them to the proteasome for degradation. One of the major actions of the CSN is to remove NEDD8 (deneddylate) and inactivate CRLs. The deneddylated form of CRLs prevents the ubiquitination and proteasomal degradation of targeted substrates. The substrate recognition receptor of the CRL allows selective targeting of substrates. Thus by controlling CRLs the CSN may provide selective control of protein turnover in vascular smooth muscle (VSM). Key target substrates that may be under CSN control and participate in VSM proliferation include the cyclin dependent kinase inhibitors, p27 and p21. The CSN may also be involved in the control of vascular contraction by modulating the ubiquitination of substrates such as the voltage gated calcium channel (VGCC), potassium channel, IP3 receptor, RhoA, soluble guanylate cyclase (sGC), super oxide dismutase (SOD1), peroxiredoxin, or IkB. In the endothelium the CSN may control ubiquitination of endothelial nitric oxide synthase (eNOS), calcineurin, cyclooxygenase or GTP cyclohydrolase 1 (GTPCH1) to modulate the production and release of endothelium dependent vasodilators or constrictors.
A major mechanism controlling CRL function is the process of neddylation. For CRLs to perform ubiquitin ligation, their cullin backbone must be covalently modified with a ubiquitin-like protein NEDD8 (Neural precursor cell Expressed Developmentally Downregulated protein 8) via a ubiquitination-like process known as neddylation (for a detailed description of the biochemistry of neddylation and enzymes involved, please refer to another article by Kandala et al. in this Issue of Am J Cardiovasc Dis [26] and references therein) [27,28]. Conversely, removal of NEDD8 from the neddylated cullin via a process termed deneddylation inactivates the CRL. Cullin deneddylation not only deactivates the E3 ligase function but also allows the CRL to be partially disassembled. The components are then recycled to form a new CRL with a different substrate receptor module to target other substrates in the cell [24]. Thus cullin deneddylation is a mechanism regulating the activity of CRLs and also provides a degree of molecular flexibility and efficiency to this system.
Cullin deneddylation is catalyzed by the COP9 (COnstitutively Photomorphogenic mutant 9) signalosome [29-31] (Figure 1). The COP9 signalosome (CSN) was initially discovered in a model plant Arabidopsis [32,33]. The CSN is a protein complex conserved from yeast to humans and apparently evolved in parallel to the UPS. The CSN is composed of 8 unique proteins termed CSN1 through CSN8, considered a paralogue of the lid complex of the 19S proteasome as both display a similar electron microscopic morphology and the same domain structure of their subunits [34-36]. The catalytic activity of CSN deneddylase resides in subunit CSN5 but cullin deneddylation by the CSN requires all 8 subunits to form a holocomplex [37]; hence, ablation of any of the CSN subunits leads to loss of cullin deneddylation. Therefore, the CSN is central to regulation of CRL-dependent ubiquitination. The (patho) physiological significance of the CSN in mammalian system has just begun to be deciphered.
Vascular structure
The vascular wall is comprised of the tunica intima made up of endothelial cells, the tunica media which consists primarily of vascular smooth muscle (VSM) cells and the tunica adventitia which contains a mixture of cells including fibroblasts and immune cells. The major function of these structures is to provide contractile ability and compliance to blood vessels to modulate vascular resistance, pressure waves and blood pressure. However, the vascular wall is extremely plastic and responds to both physiological (e.g. pressure) or pathophysiological (e.g. oxidized lipoproteins) stimuli with changes in vascular proliferation, vascular function (contraction/relaxation), or both. Direct investigation of the CSN on blood vessel structure and function is sparse. Nevertheless, indirect evidence suggests that the CSN modulates each of these aspects of blood vessel biology.
Vascular smooth muscle proliferation
Under physiological conditions, VSM cells are more or less quiescent exhibiting relatively low rates of proliferation. However, unlike cardiac cells, VSM cells are not terminally differentiated and under the appropriate stimuli can dedifferentiate to alter their phenotype in subtle or extreme fashion [38]. At its most profound, this process termed “phenotypic switching” results in transformation of VSM cells from a predominantly contractile phenotype to a predominantly proliferative/secretory phenotype that has been implicated in the pathophysiology of atherosclerosis, neointima formation in stent restenosis, pulmonary hypertension, systemic hypertension and cancer [38,39].
Phenotypic switching of VSM cells requires reentry into the cell cycle. Normally, VSM cells exist in the G0 phase of the cycle [40,41], however under the appropriate stimuli VSM cells can enter the G1 phase and progress through the cell cycle. An important mechanism regulating this process is inhibition of cyclin dependent kinase(s) by cyclin dependent kinase inhibitors (CKDI) [40-42]. An important group of CDKI is the KIP/CIP family consisting of p21, p27 and p57. While these proteins share a similar N terminal structure, the remainder of their structure is sufficiently different that each may serve different functions [42]. Moreover, different mechanisms may control these CDKIs at different stages of the cell cycle. For example, p27 is elevated during the G0 phase to effectively inhibit the cell cycle and maintain cells in a quiescent state. At the G0-G1 transition, current evidence suggests that p27 degradation is promoted via a ubiquitination complex termed the Kip1 ubiquitination-promoting complex (KPC) [43]. However, at the S/G2 interface, p27 levels are regulated by an E3 ligase complex, SCFSkp2 [44]. p57 is regulated post-transcriptionally in a manner similar to p27 [45]. Conversely, p21 is maintained at low levels during G0 but is increased transcriptionally later in G1 as a counter-regulatory mechanism [46,47].
These regulatory mechanisms have been shown to have important effects on vascular pathophysiology. Vascular injury was associated with decreased p27 expression and increased proliferative markers in VSM cells in dogs and mice [48,49]. Moreover, VSM cell proliferation in response to vascular injury was increased in p27 null mice [50]. On the other hand, overexpression of p27 in blood vessels attenuated balloon injury-induced VSM cell proliferation in rats, rabbits and dogs [47,51-53]. Similarly, drug treatments that increased p21 expression were found to attenuate VSM cell proliferation in response to injury in the carotid artery of rats [54,55] while overexpression of p21 reduced VSM cell proliferation in response to balloon injury in the pig [56]. Thus, the role of CDKI in VSM cell proliferation for arterial repair following mechanical injury is relatively well established. p27 is also implicated as a regulatory factor in the vascular proliferative response in disease. Closely related to the increased VSM cell proliferation observed with mechanical injury is the increase in VSM cell proliferation associated with the atherosclerotic process. p27 was identified as a critical regulatory factor in this setting. Double transgenic mice lacking apoE and p27 showed increased atherosclerotic burden compared to apoE knock out alone, suggesting that loss of p27 accelerated the atherosclerotic process [57]. Qualitatively similar results were obtained in p21 null mice [58]. Direct comparison of the effects of p27 or p21 knockout suggested that the magnitude was substantially less with p21 knockout than that seen in p27 null mice [57]. Conversely, treatment that reduced plaque burden in apoE null mice was associated with induction of p27 expression and reduced intraplaque VSM cell proliferation [59]. Likewise, adenovirus mediated overexpression of p21 inhibited atheroma formation in apoE null mice [60]. Thus, CKDI appear to play a critical role in the control of focal VSM proliferation in response to injury or atherosclerosis.
These control mechanisms have also been implicated in more diffuse vascular disease models. Pulmonary hypertension is a debilitating disease characterized by excessive proliferation of VSM cells in the pulmonary arteries to the point of vascular occlusion [61]. Chronic exposure to hypoxia and endothelin 1 are potent triggers of pulmonary hypertension. Exposure to hypoxia or endothelin 1 was associated with increased proliferation of VSM cells and a decrease in p27 expression [62,63]. On the other hand, treatments that upregulated p27 expression were found to reduce pulmonary artery VSM cell proliferation [63,64]. VSM cell proliferation associated with systemic hypertension may also involve changes in p27. Angiotensin II infusion to produce hypertension in rats increased proliferation indices in mesenteric arterial VSM cells and caused a reduction in p27 [65]. Similarly, wild type mice made hypertensive with angiotensin exhibited increased aortic medial thickness and aortic VSM cell proliferation (PCNA staining), effects that were exaggerated in p27 null mice [66]. In the spontaneously hypertensive rat, a genetic model of hypertension, agonist-induced VSM cell proliferation was exaggerated compared to normotensive Wistar Kyoto rats [67,68]. The enhanced VSM cell proliferative response was associated with a reduction in p27 expression in spontaneously hypertensive compared to Wistar Kyoto rats [67]. In addition drug treatment that reduced the VSM cell proliferative response triggered an increase in p21 expression [68]. Thus CKDI appear to play a central role in the VSM cell proliferation associated with pulmonary or systemic hypertension. Collectively these data suggest that CDKIs represent an important control mechanism for modulating VSM cell proliferation in a number of cardiovascular diseases.
The COP9 signalosome and vascular smooth muscle proliferation
Direct study of the role of the CSN in control of VSM cell proliferation is limited. In non-vascular cells there are data showing that the CSN is a critical regulator of cell proliferation. Perhaps the best established link between the CSN and cell cycle regulation is the recognition that p27 degradation is regulated by the CSN. Initial evidence to support this possibility was provided by studies showing that CSN5 (formerly Jab1) bound p27 and that overexpression of CSN5 resulted in accelerated degradation of p27 and increased cell proliferation [69]. Subsequent work suggested that CSN5 contributed to p27 degradation by also enhancing nuclear to cytoplasmic export thus enhancing delivery to the UPS [70]. Moreover, ectopic expression of CSN5 could rescue NIH3T3 cells from p27-induced cell cycle arrest [69] showing that these interactions were important functionally. Similarly, overexpression of CSN6 was found to release NIH3T3 cell from p57 induced G1 arrest [71]. Conversely, knockdown of CSN4 or CSN5 reduced S phase entry in HeLa or HEK293T cells [72]. Similar findings were obtained in thymocytes where selective CSN5 knockdown arrested the cell cycle in the S/G2/M phase [73]. These data provided a picture wherein CSN function was a positive regulator of the cell cycle. Indeed impairment of CSN function by deleting various CSN subunits (CSN2, CSN3) proved to be embryonically lethal in mice at least in part due to defective cell proliferation [74,75].
However, further study has shown that the picture is not nearly so straightforward. Complicating interpretation is the fact that CSN5, the most frequently targeted CSN subunit (presumably because of its catalytic activity), has functions independent of the CSN complex [76,77]. In addition, in contrast to some earlier work, it has been reported that siRNA induced reduction in CSN3 increased proliferation in mouse fibroblasts [78]. Subsequently, it was shown that conditional silencing of CSN5 or CSN8 in the same cell system resulted in opposite effects on proliferation of HeLa cells. Reduction in CSN5 expression caused a marked attenuation of cell proliferation consistent with earlier studies, whereas CSN8 reduction was associated with accelerated growth [79]. A similar increase in proliferation was observed in mouse embryonic fibroblasts harvested from CSN8 hypomorphic mice which exhibit a reduction in CSN8 expression and CSN function [79]. On the other hand, conditional knockdown of CNS8 in postnatal hepatocytes resulted in impaired hepatocyte proliferation [80]. Thus, perhaps fittingly for a complex as intricate as the CSN, more recent work suggests that the CSN may both promote and inhibit the cell cycle. The net effect of CSN disruption may depend on the CSN subunit affected and the tissue/cell type in which the deficit occurs. Nevertheless in aggregate, these findings indicate that the CSN participates critically in the control of the cell cycle. Accordingly, the CSN has the potential to be involved in conditions which exhibit alterations in VSM cell proliferation.
Given the strong data tying CSN function to regulation of the cell cycle, it is curious that direct study (knockdown, overexpression) of CSN in VSM proliferation is lacking. Nevertheless, indirect evidence ties the CSN to the control of VSM proliferation via regulation of the CKDIs. One of the important mechanisms regulating the CKDIs is UPS-mediated degradation [81]. Substantial evidence supports the view that CKDI degradation during the cell cycle is controlled at least in part by SCF complexes by targeting of the CKDIs for ubiquitination and degradation [82]. SCF complexes represent a subfamily of CRLs that contain four primary components: Skp1 (an adaptor protein), Cullin1, Roc1/Rbx1 (a RING finger protein), and an F-box protein which represents the substrate recognition site [83]. SCF complexes containing the F-box protein Skp2 (SCFSkp2) have the CKDIs as one class of its targets. Convincing data show that SCFSkp2 contributes to regulation of VSM proliferation by controlling the turnover of CKDIs. In fact, Skp2 was shown to be required for p27 ubiquitination and degradation [84]. While p27 is constitutively expressed in quiescent VSM, Skp2 expression is quite low [85,86]. Overexpression of Skp2 in rat aortic smooth muscle cells triggered a reduction in p27 and an increase in proliferative indices in isolated rat aortic VSM cells (thymidine incorporation) or rat aorta organ culture (BrDU incorporation) [87]. Moreover, physiologic stimuli such as balloon injury that promote VSM proliferation trigger an increase in Skp2 and a concomitant reduction p27 [86,88]. A similar finding was reported for pulmonary artery VSM cells that were stimulated to proliferate with endothelin [62]. Conversely, siRNA suppression of Skp2 or expression of a dominant negative form of Skp2 in rat thoracic aorta VSM cells resulted in an increase in p27 expression and reduced proliferation [86,89,90]. In vivo relevance of these findings was further strengthened by the demonstration that Skp2 null mice exhibited reduced neointima formation in response to balloon injury [88]. On the other hand, AMPK null mice which exhibited an increase in Skp2 expression developed more severe neointimal VSM growth in response to wire injury of the carotid artery [90]. AMPK activation was shown to inhibit pulmonary artery VSM cell proliferation by preventing endothelin mediated increases in Skp2 expression [62]. In the same vein, treatment of rats with hypoxic pulmonary hypertension that reduced pulmonary artery thickness was reported to prevent hypoxic induction of Skp2 expression [91]. While the preponderance of data has addressed the role of SCFSkp2 control of VSM proliferation via interactions with p27, there are data showing that other CDKIs may also be involved. Overexpression of Skp2 triggered p21 degradation in rat [92] and in mouse [93] aortic VSM cells.
Collectively these findings suggest that SCFSkp2 mediated degradation of CDKI plays a critical role in the control of VSM cell proliferation. Given that SCFSkp2 E3 ligase complex is cullin based it is logical to predict that the CSN is involved in controlling VSM cell proliferation under various conditions by modulating the degradation of CDKI such as p27 and p21 (Figure 1). Nevertheless direct further investigation is needed to directly link the CSN to VSM cell proliferation, to establish the precise role of CSN subunits, and to decipher the molecular mechanisms by which the CSN regulates SCFSkp2 complex function in VSM.
Vascular smooth muscle contraction
Vascular tone
Vascular smooth muscle is responsible for the dynamic maintenance of blood vessel diameter, a primary determinant of vascular resistance. Vascular tone plays a major role in maintaining peripheral vascular resistance and systemic blood pressure to provide a constant pressure for tissues to regulate blood flow in proportion to their metabolic demand. In addition, VSM in regional vascular beds (organs) is also subject to a myriad of local influences that help determine regional vascular resistance and blood flow distribution, again mostly in proportion to the local tissue demands. Unlike skeletal and cardiac muscle whose contractions are predominantly episodic, VSM exists in a state of perpetual contraction, so called vascular tone that is highly modulated by a number of control mechanisms.
Vascular contraction mechanisms
As in other muscle, an increase in intracellular calcium is the primary stimulus for VSM contraction. Increases in intracellular calcium are mediated via calcium entry from the extracellular space or from intracellular stores in the sarcoplasmic reticulum [94-96]. One important control point for VSM contraction is calcium entry via the L type voltage gated calcium channel which is regulated in large part via membrane voltage. Plasmalemmal potassium channels are a major determinant of VSM membrane voltage [97,98]. Four types of potassium channel have been implicated in control of VSM tone: the voltage dependent potassium channel (Kv), the inward rectifying potassium channel (Kir), the ATP dependent potassium channel (KATP) and the calcium activated potassium channel (KCa) [97-99]. The KCa is further classified into small (SKCa), intermediate (IKCa) and large conductance (BKCa), with the BKCa recognized as a major contributor to vascular tone [100,101]. Calcium release from the sarcoplasmic reticulum is mediated by inositol 1,4,5-trisphosphate (IP3) released from the plasma membrane acting on IP3 receptors present in the sarcoplasmic reticulum [102]. Changes in IP3 receptor expression were recently shown to play a critical role in modulating vascular tone [103].
Subsequent to an elevation in intracellular calcium, the contractile mechanisms differ substantially from the heart and skeletal muscle. Calcium binds to calmodulin and the calcium calmodulin complex stimulates myosin light chain kinase to phosphorylate myosin light chain which then stimulates force generation via the canonical actin myosin interaction [94,104]. Thus, myosin light chain phosphorylation is the proximal event triggering VSM contraction. A key regulatory feature of VSM is that myosin light chain is also dephosphorylated by myosin light phosphatase [95,96,104]. Therefore, the magnitude of the VSM contractile response to calcium is determined by the balance of myosin light chain phosphorylation and dephosphorylation. Accordingly, myosin light chain kinase and myosin light chain phosphatase represent key control points for vascular tone.
Signaling mechanisms that impact these core processes provide further fine tuning of vascular tone. One important such mechanism is provided by the phosphorylation status of myosin light chain phosphatase. Myosin light chain phosphatase consists of a catalytic subunit (PP1c), a regulatory subunit (MYPT1) and a 20 kDa subunit of unknown function [95,96,104]. Phosphorylation of MYPT1 by Rho kinase is an inhibitory signal that reduces myosin light chain phosphatase activity [104,105]. A second mode of control is through activation of protein kinase C which also inhibits myosin light chain phosphatase. This inhibitory input is mediated by a small protein CPI17 that inhibits the PP1c subunit to reduce myosin light chain phosphatase activity [95,104]. Both these pathways ultimately result in an increase in myosin light chain phosphorylation and vascular tone for any given level of intracellular calcium rise, a process called “calcium sensitization”. Moreover both pathways are modulated by factors that affect the increased vascular tone in hypertension [105-108]. The reverse process of calcium desensitization can also occur. In this situation myosin light chain phosphatase is targeted by vasodilator mechanisms that activate protein kinase G (PKG) or protein kinase A (PKA) signaling. PKA or PKG phosphorylate MYPT1 at sites that impair the ability of Rho kinase or PKC to inhibit myosin light chain phosphatase [109]. Thus, there are several control points at which the CSN may modulate VSM contraction.
The COP9 signalosome and vascular contraction
The role of the CSN in modulating vascular contraction has received very little direct research attention. Nevertheless, there are indirect clues that the CSN is well positioned to interact with several VSM control points and thus impact control of vascular tone and blood pressure regulation. An increase in intracellular calcium is clearly a requisite event for VSM contraction. While not studied directly in VSM, there is evidence to suggest that the CSN is involved in the control of L type voltage gated calcium channels. The L type calcium channel is a multimeric transmembrane protein of which the α1C (Cav1.2) subunit confers most of the primary functional properties [110]. The α1C subunit is a predominant form of the pore forming unit expressed in cardiac and VSM cells [111]. Studies in the rat showed that CSN5 co-immunoprecipitates with α1C subunit consistent with an interaction between these two proteins [112]. Moreover, this interaction appeared to be functionally relevant since siRNA knockdown of CSN5 increased calcium currents significantly [112]. This increase in calcium current occurred without significant changes in calcium channel kinetics suggesting an effect on voltage gated calcium channel density [112]. Thus, CNS5 may be involved in controlling voltage gated calcium channel turnover, at least in cardiac cells. The similarities in voltage gated calcium channel structure between cardiac and vascular muscle suggests a potentially similar role in the vasculature. However, this remains to be tested experimentally.
Other data, albeit circumstantial, may implicate the CSN in the control of VSM potassium channels. Expression of the β1 subunit of the BKCa potassium channel was reduced in aortic VSM harvested from diabetic rats and mice or human coronary artery VSM cells exposed to high glucose [113,114]. Since there were no changes in BKCa transcript levels and BKCa channel protein expression was increased by MG-132 [114], this change in expression was linked to proteasomal degradation. Under diabetic conditions, increased expression of the F-box proteins, atrogin-1 (FBXO32) and FBXO9 were associated with concomitant reductions in BKCa channel expression [113]. Moreover, siRNA reduction of atrogin-1 was paralleled by an increase in BKCa β1 subunit expression [114]. Furthermore, drug treatment that reduced atrogin-1 expression was associated with an increase in BKCa expression and vasodilation in response to NS-1619, a specific activator of BKCa [113]. Since atrogin-1 and FBXO9 are substrate recognition subunits for SCF ligase complexes, it seems plausible that BKCa channel proteins may be regulated by the CSN. This possibility requires experimental validation. It is interesting to note that in the case of voltage gated calcium channels CSN activity may inhibit the channel thus producing a vasodilator response. In contrast, at least in the diabetic state, CSN activity would be predicted to enhance the degradation of BKCa potassium channels, which would result in vasoconstriction. Thus, while there are clues that CSN may be involved in controlling VSM ion channel function, there is a need for direct experimentation to unravel the precise effects.
The CSN may also be implicated in the control of calcium release from intracellular stores. It has been recognized for some time that stimuli which increase the production of IP3 trigger a reduction in IP3 receptor protein in the endoplasmic reticulum. Since this effect was blocked by proteasome inhibitors, IP3 receptor downregulation appears to be mediated via the UPS [115]. More recently a similar phenomenon has been reported for VSM [116]. Rat aortic VSM cells treated with either hydrogen peroxide or angiotensin II exhibited a sustained increase in IP3 that was associated with an increase in IP3 receptor ubiquitination and a concomitant reduction in IP3 receptor protein content. Cotreatment with a proteasome inhibitor (MG132) prevented the decrease IP3 receptor content. Importantly, this study also demonstrated that these effects were functionally relevant since the reduction in IP3 receptor expression was associated with reduced IP3 mediated calcium release and decreased agonist-induced contraction in isolated VSM [116]. The mechanisms regulating IP3 receptor degradation have not been fully elucidated. RING ligases were implicated in IP3 receptor turnover [117]. However, it is not known at this point whether CRL E3 ligases are responsible for targeting IP3 receptors for proteasomal degradation. If this proves to be the case, the CSN may be involved in modulating vascular contraction by influencing agonist stimulated intracellular calcium release.
The CSN may also control cellular pathways that modulate myosin light chain phosphorylation via myosin light chain phosphatase. A variety of agonists that induce VSM contraction are known to do so, at least in part, via activation of the RhoA-RhoKinase (ROCK) pathway [118,119], which as outlined above potentiates vascular contraction. The application of MLN4924, an inhibitor of neddylation, triggers an increase in Rho A protein levels in Hela cells [120]. Since the CSN is involved in controlling CRLs via deneddylation, this finding suggests that the CSN may be involved in controlling RhoA-ROCK function via modulation of CRL activity. Indeed, siRNA deletion of cullin-3 also increased Rho A protein levels. The increase in Rho A protein was concomitant with decreased Rho A polyubiquitination and an extended the half life of Rho A [121], consistent with the possibility that a cullin-3 based ubiquitin ligase is involved in the cellular clearance of Rho A. Cullin-3 knockdown was also reported to increase the active form of Rho A, Rho A-GTP [121]. An important step in activating Rho A is the exchange of GDP for GTP which is mediated by the Rho guanine exchange factors (Rho GEFs) [122]. siRNA knockdown of KLHL20, a BTB protein that functions as the substrate recognition unit for a cullin-3 based E3 ligase, increased PDZ-RhoGEF and Rho A activation [123]. Thus, CRL3 ligases appear to play a critical role modulating activation of the Rho A pathway and, by inference, the CSN is implicated as well. While these data were not obtained in VSM cells, recent work suggests that these findings have relevance for control of VSM contraction. Consistent with work described above, siRNA knockdown of cullin-3 in rat aortic VSM cells increased Rho A protein [124]. Treatment of rat aortic rings with MLN4924 to inhibit cullin activation also increased Rho A protein levels and amplified the vasoconstrictor response to G protein coupled receptor agonists such as phenylephrine and endothelin [124]. Perhaps most importantly, this work also showed that subcutaneous administration of MLN4924 over the course of 3 days lead to a significant increase in mean arterial blood pressure (~20 mmHg) in conscious mice [124]. Collectively these data are consistent with the idea that one mechanism by which the CSN may control vascular tone is via regulating the activity of cullin based E3 ligases that are responsible for modulating agonist activated vasoconstrictor pathways such as RhoA-ROCK. Validation of this idea will require studies that directly manipulate CSN function while monitoring cardiovascular parameters such as blood pressure, cardiac output and total peripheral resistance.
In summary, there may be multiple points of interaction between the CSN and mechanisms controlling vascular smooth muscle contraction (Figure 1). The CSN may control the trigger for contraction, calcium flux, via effects on voltage gated calcium channels, potassium channels or IP3 receptors. Additionally, the CSN might also interact with intracellular signaling pathways such as RhoA-ROCK that impact sensitivity of the contractile machinery to calcium or to vasoactive agents released from the endothelium (described below). The current findings linking the CSN to control of vascular smooth muscle contraction are limited but sufficiently intriguing to spur more investigation in the area.
Endothelial control of vascular function
The endothelium makes up only a small proportion of the blood vessel wall, a single cell lining the vascular lumen. Nevertheless, the vascular endothelium plays a pivotal role in cardiovascular homeostasis as a pleiotropic endocrine/paracrine organ involved in functions ranging from control of vascular tone to secretion of factors involved in remodeling the extracellular matrix [125]. One major function of the endothelium is control of VSM tone via the release of endothelium derived relaxing factors. The best characterized of these factors is nitric oxide (NO). As a diffusible gas, NO is not stored but is synthesized on demand by the actions of nitric oxide synthase (NOS), of which three isoforms exist: endothelial nitric oxide synthase (eNOS; NOS-3), neuronal nitric oxide synthase (nNOS; NOS-1) or inducible nitric oxide synthase (iNOS; NOS-2). The canonical pathway for NO medicated dilation is activation of soluble guanylate cyclase and protein kinase G [125,126]. NO may also activate BKCa [126]. In addition, the endothelium releases prostacyclin, a vasodilator arachidonic acid metabolite that generates cAMP by activating adenylate cyclase and ultimately stimulates protein kinase A [127]. A third group of endothelial dilators, the endothelial hyperpolarizing factors (EDHF) is less well characterized but triggers VSM hyperpolarization and thus relaxation via the opening of SKCa, and IKCa potassium channels [125,127].
The endothelium is also a source for endothelial derived contracting substances. Endothelin and arachidonic acid metabolites such as thromboxane or 20-hydroxyeicosatetraeonic (20-HETE) represent the best studied of these factors [128-130]. Endothelin has been characterized as the most potent vasoconstrictor agent known [128,131]. Endothelin acts on ETA, ETB and ETC receptors, the balance of which determines the ultimate effect of this peptide paracrine factor [128,129]. Thromboxane is a prostanoid vasoconstrictor agent generated by cyclooxygenase metabolism of arachidonic acid to endoperoxides which are subsequently acted upon by thromboxane synthase [130]. 20-HETE is also generated by cyclooxygenase metabolism of arachidonic acid, however in this pathway the resultant endoperoxides are processed by specific cytochrome P450 isoforms [129,130]. Thus, cyclooxygenase metabolism of arachidonic acid is a major source of endothelial derived contracting substances. Lastly, reactive oxygen species (ROS) can be an important endothelial derived factor that impacts VSM function. Important sources for ROS include NADPHoxidase, xanthine oxidase, mitochondria and NOS in its “uncoupled” form [132-135]. Various conditions can lead to NOS uncoupling including persistent oxidative stress, lack of the substrate L-arginine or deficiency in essential cofactors such as tetrahydrobyopterin (BH4) [133,135]. In addition a number of systems are in place to counterbalance ROS production by scavenging these reactive molecules. These systems include superoxide dismutase (SOD), catalase and the peroxiredoxins [133,136]. Ultimately, the equilibrium amongst these factors determines the prevailing level of ROS, which in turn can trigger VSM contraction or proliferation [133-136]. In summary, the endothelium is a source for a number of factors that can influence VSM function. While a great deal of research effort and information is now available characterizing the endothelial control of VSM at various levels, the role of the CSN in this aspect of vascular function remains largely unexplored.
The COP9 signalosome and endothelial control of vascular function
As a key mechanism for endothelial modulation of vascular function, NOS is modulated by a variety of mechanisms including posttranslational mechanisms such as phosphorylation and protein-protein interactions [125,126]. Increasing evidence suggests that ubiquitination and proteasomal processing may represent yet another mechanism for posttranslational control of NOS function. A number of studies have shown that iNOS is regulated by ubiquitination and proteasomal degradation [137,138]. Comparatively fewer studies have studied the relationship between eNOS and the ubiquitin proteasome pathway. Nevertheless it has been reported that treatment with the proteasome inhibitor lactacystin increased eNOS protein levels in COS-7 cells transfected to express the protein [139]. Lactacystin was similarly found to increase eNOS in bovine aortic endothelial cells in culture [140]. Graded inhibition of proteasomal function with MG-132 resulted in graded increases in eNOS expression in human umbilical vein endothelial cells [141]. A similar effect was reported in bovine pulmonary artery endothelial cells [142]. In some cases eNOS mRNA was increased after proteasome inhibition [142] whereas in others there were no changes in eNOS mRNA [141]. Thus, whether the effects of proteasome inhibition on eNOS expression represent a transcriptional or posttranslational effect remains an open question. Nevertheless, these data are at least consistent with the possibility that eNOS is regulated by proteasomal degradation. The E3 ligases involved in mediating eNOS degradation remain to be determined. While carboxyl terminus of Hsp70-interacting protein (CHIP) was found to interact with eNOS, it reportedly does not direct eNOS to the proteasome for degradation [139]. The cullin based E3 ligase Elongin B/C-Cullin-5-SPRY domain- and SOCS box-containing protein (ECS (SPSB)) was reported to interact with iNOS but not with eNOS [143]. On the other hand, resveratrol, an activator of the sirtuin pathway, was found to increase eNOS expression in rat pulmonary vessels [144]. Resveratrol is reported to increase cullin 5 expression in cancer cells [145], suggesting the possibility the eNOS expression may be susceptible to CSN based regulation.
Aside from direct control by eNOS protein turnover, eNOS function is also regulated via a number of other posttranslational mechanisms. Several observations are consistent with the possibility that the CSN could modulate eNOS activity at this level. It is relatively well established that eNOS activity is controlled in part via phosphorylation/dephosphorylation at either excitatory (e.g. serine 1177) or inhibitory (e.g. threonine 495) sites [146]. It was reported that calcineurin (protein phosphatase 3) mediated dephosphorylation of serine 116 leads to an increase in agonist stimulated eNOS activity [147,148]. Interestingly, an SCF complex, SCFatrogin-1 promoted ubiquitination of calcineurin, and atrogin-1 overexpression reduced calcineurin levels in cardiac cells [149]. Similarly, BH4 is a necessary co-factor for nitric oxide synthase activity [146]. BH4 synthesis was decreased in hypertensive or diabetic mice due to increased proteasomal degradation of GTP cyclohydrolase 1 (GTPCH1), the enzyme responsible for BH4 synthesis [150-152]. Treatment with a PPAR gamma agonist, GW501516, increased BH4, eNOS activity and nitric oxide availability in BH4 deficient mice, at least in part due to an increase in GTPCH1 activity [153]. Since transgenic mice expressing a dominant negative form of PPAR gamma exhibited reduced neddylation of cullin 3, collectively these data are consistent with the possibility that GTPCH1 degradation is mediated through a cullin based E3 ligase. Lastly, soluble guanylate cyclase (sGC) is a major downstream target of nitric oxide and is thought to mediate many of the vascular actions of nitric oxide. Studies in HEK cells showed that sGC subunits undergo polyubiquitination and proteasomal degradation [154]. Recent data in cancer cells suggest that the CSN is involved in modulating proteasomal degradation of sGC. siRNA knockdown of CSN4 or CSN5 resulted in decreased sGC protein, an effect that was reversed by treatment with a proteasome inhibitor [155]. As a whole these findings suggest that in addition to controlling eNOS protein, the CSN may regulate endothelial function by modulating the turnover of regulatory factors that control eNOS activity or downstream targets that mediate its effects.
Cyclooxygenase metabolites of arachidonic acid represent another pathway by which the endothelium regulates vascular smooth muscle. Studies in human megakaryocytic cells showed that both cyclooxygenase 1 and prostaglandin synthase D were ubiquitinated and degraded by the proteasome [156]. Similarly cyclooxygenase 2 was found to be a substrate for proteasomal degradation [157,158]. In a study using CaCo cells, which constitutively express COX2, it was found that immunoprecipitation of CSN7 pulled down COX2. Moreover, in Hela cells the reverse immunoprecipitation of COX2 pulled down CSN5 [157]. Importantly, this study also revealed that COX2 along with the CSN subunits formed complexes with cullin 1 and cullin 4 as well as with the RING protein ROC1 [157]. These data suggest that COX2 physically interacts with a CSN/CRL complex. Collectively these findings are consistent with the possibility that the CSN is involved in modulating proteasomal degradation of cyclooxygenase. Since the activity of this enzyme is critical for the formation of prostacyclin, a vasodilator, and of thromboxane, a vasoconstrictor, the net effect of CSN on endothelial vasoactive pathways involving cyclooxygenase is unclear at this time, but does warrant further study.
ROS play an important role in VSM regulation both as a signaling molecule generated by a variety of agonists and by virtue of their ability to scavenge NO. Relatively little data are currently available regarding the impact of the CSN on ROS formation or scavenging. Direct evidence of CSN8/CSN involvement in redox regulation was recently provided when Su and colleagues reported that an increased amount of oxidized proteins was detected in mouse hearts with adult-onset cardiomyocyte-restricted CSN8 knockout [159]. The greatest amount of evidence to link the CSN to control of ROS is less direct and comes from study of the NF-Kappa B (NFκB) pathway. NFκB becomes activated when phosphorylation of its cognate inhibitor IκB is triggered by agonist stimulation. IκB is then acted upon by SCFβ-TrCP, a cullin based E3 ligase that targets IκB for proteasomal degradation [160-162]. The CSN is implicated in this control mechanism. Immunoprecipitation experiments demonstrated that CSN2 associated with IkBα in TNFα stimulated Hela cells [161]. Other work revealed that the CSN interacts with other components of the NFκB activation pathway. Co-immunoprecipitation experiments showed that CSN3 physically interacts with IKK8, a component of the IKKα-IKKβ-IKK8-complex responsible for phosphorylating IκBα [163]. A subsequent study showed that IKKβ was complexed with CSN3 and SCFβ-TrCP, the E3 ligase that ubiquitinates IκBα, suggesting that an assembled CSN/CRL complex associated with the NFκB control machinery [160]. The functional impact of this association has also been demonstrated. siRNA knockdown of CSN2 was associated with decreased IκBα protein and increased NFκB translocation into the nucleus [161]. Similarly, siRNA knockdown of CSN5 was reported to increase basal NFκB activity in 293T cells [160,164]. CSN3 oligo antisense treatment also resulted in an increase, albeit slight, in basal NFκB activity [163]. Some work has shown that CSN knockdown also increased TNFα stimulated NFκB activation [163], whereas other work failed to detect such an effect [160,161]. Nevertheless, on the whole these data suggest that the CSN acts as a negative modulator of NFκB activation. The precise mechanism by which the CSN attenuates NFκB activation remains an open question since some authors suggest a primary role for the deubiquitinating activity associated with the CSN [161], while other data suggest that the deneddylating activity is critical [160]. At least in cancer cells, inhibition of neddylation with ML4924 was associated with an increase in NFκB activation and downstream target protein expression [165].
A complex and as yet not fully understood interaction occurs between ROS and NFkB. ROS are a stimulus for activation of NFκB. Amongst the many downstream effects of NFκB activation this pathway can, under the appropriate conditions, trigger an increase in the expression of antioxidant enzymes such as super oxide dismutase or catalase [166] to act as a negative feedback loop. Yet other evidence suggests that activation of NFκB results in increased expression of ROS generating enzymes such as NADPH oxidase [166,167], a positive feedback loop implicated in inflammatory processes. The ultimate outcome of NFκB activation may depend on the composition of the NFκB dimer formed or the cell type [166]. Accordingly, it is difficult to predict the precise effect that CSN regulation of NFκB activation will have on ROS generation and scavenging. Further study is required to identify the precise nature of this control under various conditions and in specific cell types.
In addition to affecting transcription of pro-oxidant or antioxidant proteins, indirect evidence suggests that the CSN may also be linked to clearance of oxidant-redox proteins themselves. Proteomic analysis of control and Cullin 4B null HEK293 cells revealed upregulation of protein for superoxide dismutase 1 and for peroxiredoxin III [168] both of which are involved in scavenging ROS. Peroxiredoxin III was chosen for follow up. Since there were no changes in peroxiredoxin III mRNA expression, these changes were ascribed to reduced degradation of the protein. Consistent with this possibility, ubiquitination of peroxiredoxin III was reduced and cycloheximide chase experiments revealed a markedly increased half-life for peroxiredoxin III in the Cullin 4B null cells. The effects were functionally significant since ROS indices were markedly reduced in the Cullin 4B knockdown cells [168].
As a whole, these findings suggest that the CSN may modulate endothelial function by interacting with multiple control points (Figure 1). The CSN may control eNOS function either by regulating turnover of eNOS itself or of proteins that regulate its activity such as calcineurin or GTPCH1. The CSN may also affect endothelial vasoactive factors by regulating COX degradation. Lastly, the CSN may be involved in controlling ROS generation or scavenging which will greatly impact endothelial control of VSM. While suggestive, currently available data are less than conclusive. Accordingly, much more work is necessary in this area to establish the precise role of the CSN in endothelial function.
Concluding remarks and perspectives
Cardiovascular diseases represent a leading cause of morbidity and mortality worldwide. An emerging paradigm in understanding cardiovascular regulation and dysregulation is the recognition that protein modification via ubiquitination can play a critical role in modifying cellular function. A bottleneck in moving this concept forward to therapeutic advantage has been deciphering ways to selectively manipulate protein ubiquitination. The CSN represents a control mechanism for a large group of E3 ligases, the CRLs, which represent the substrate recognition component of the ubiquitination system and are thus the most selective control point. The role of the CSN in cardiovascular control is just beginning to be explored. Perhaps the best characterized interaction is with VSM proliferation. There is sound evidence linking SCF complexes (CRL1s) to degradation of CKDI proteins and control of proliferation in VSM. Current data do not paint an entirely clear picture however, since the effects of CSN manipulation on VSM cell proliferation appear to be dependent on the specific cell type or CSN subunit manipulated. Nevertheless, further exploration of these mechanisms may provide a new avenue to target diseases characterized by excessive VSM proliferation such as neointimal hyperplasia after balloon angioplasty or vascular wall hypertrophy in hypertensive vascular remodeling.
The data linking the CSN to VSM contractile function are less well developed. However, current findings suggest that the CSN may be involved in controlling the turnover of voltage gated calcium and potassium channels and the IP3 receptor. These proteins represent mechanisms controlling calcium flux in VSM. In addition, available data also imply that the CSN may regulate intracellular signaling mechanisms such as the RhoA-Rho kinase pathway which modulate calcium sensitivity in VSM. Lastly, the CSN may interact with endothelial mechanisms modulating vascular tone. This may occur through turnover of eNOS protein itself. Alternatively, eNOS regulatory mechanisms such phosphorylation or downstream targets such as sGC may be subject to CSN control. Collectively these data are consistent with the possibility that the CSN regulates vascular tone at several levels (Figure 1). Accordingly, further understanding of these interactions could reveal targets for development of vasoactive agents for the treatment conditions such as hypertension, Raynaud’s disease or coronary artery spasm.
Admittedly, current evidence linking the CSN to vascular control remains largely circumstantial. However, the available data provide intriguing clues for further investigation. The CSN is an intricate control mechanism that is itself not as yet fully understood. However, within that complexity lays the promise of the requisite specificity of protein ubiquitination regulation that will be necessary for development of novel therapeutics to treat vascular disease.
References
- 1.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker-Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez-Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O’Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez-Ruiz F, Perico N, Phillips D, Pierce K, Pope CA 3rd, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui-Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres J, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. Heart Disease and Stroke Statistics-2015 Update: A Report From the American Heart Association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 3.Cutler JA, Sorlie PD, Wolz M, Thom T, Fields LE, Roccella EJ. Trends in hypertension prevalence, awareness, treatment, and control rates in United States adults between 1988-1994 and 1999-2004. Hypertension. 2008;52:818–827. doi: 10.1161/HYPERTENSIONAHA.108.113357. [DOI] [PubMed] [Google Scholar]
- 4.Hajjar I, Kotchen JM, Kotchen TA. Hypertension: trends in prevalence, incidence, and control. Annu Rev Public Health. 2006;27:465–490. doi: 10.1146/annurev.publhealth.27.021405.102132. [DOI] [PubMed] [Google Scholar]
- 5.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27. doi: 10.1016/j.yjmcc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 8.Li S, Wang X, Li Y, Kost CK Jr, Martin DS. Bortezomib, a proteasome inhibitor, attenuates angiotensin II-induced hypertension and aortic remodeling in rats. PLoS One. 2013;8:e78564. doi: 10.1371/journal.pone.0078564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su H, Li J, Menon S, Liu J, Kumarapeli AR, Wei N, Wang X. Perturbation of cullin deneddylation via conditional Csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res. 2011;108:40–50. doi: 10.1161/CIRCRESAHA.110.230607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Martin DS. The COP9 signalosome and cullin-RING ubiquitin ligases in the heart. Am J Cardiovasc Dis. 2015;5:1–18. [PMC free article] [PubMed] [Google Scholar]
- 11.Kim E, Yoon SJ, Kim EY, Kim Y, Lee HS, Kim KH, Lee KA. Function of COP9 signalosome in regulation of mouse oocytes meiosis by regulating MPF activity and securing degradation. PLoS One. 2011;6:e25870. doi: 10.1371/journal.pone.0025870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Terpstra EJ. Ubiquitin receptors and protein quality control. J Mol Cell Cardiol. 2013;55:73–84. doi: 10.1016/j.yjmcc.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chernorudskiy AL, Gainullin MR. Ubiquitin system: direct effects join the signaling. Sci Signal. 2013;6:pe22. doi: 10.1126/scisignal.2004251. [DOI] [PubMed] [Google Scholar]
- 15.Ye Y, Blaser G, Horrocks MH, Ruedas-Rama MJ, Ibrahim S, Zhukov AA, Orte A, Klenerman D, Jackson SE, Komander D. Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature. 2012;492:266–270. doi: 10.1038/nature11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13:508–523. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 17.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 18.Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- 19.Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 20.Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci U S A. 1995;92:5249. doi: 10.1073/pnas.92.11.5249-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 22.Yang Y, Lorick KL, Jensen JP, Weissman AM. Expression and evaluation of RING finger proteins. Methods Enzymol. 2005;398:103–112. doi: 10.1016/S0076-6879(05)98010-5. [DOI] [PubMed] [Google Scholar]
- 23.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 24.Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–1061. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–381. doi: 10.1038/nrm3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kandala S, Kim IM, Su H. Neddylation and deneddylation in cardiac biology. Am J Cardiovasc Dis. 2014;4 [Epub ahead of print] [PMC free article] [PubMed] [Google Scholar]
- 27.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 28.Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- 29.Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114:663–671. doi: 10.1016/s0092-8674(03)00722-0. [DOI] [PubMed] [Google Scholar]
- 30.Schwechheimer C, Deng XW. COP9 signalosome revisited: a novel mediator of protein degradation. Trends Cell Biol. 2001;11:420–426. doi: 10.1016/s0962-8924(01)02091-8. [DOI] [PubMed] [Google Scholar]
- 31.Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Shevchenko A, Deshaies RJ. Promotion of NEDD-CUL1 conjugate cleavage by COP9 signalosome. Science. 2001;292:1382–1385. doi: 10.1126/science.1059780. [DOI] [PubMed] [Google Scholar]
- 32.Wei N, Chamovitz DA, Deng XW. Arabidopsis COP9 is a component of a novel signaling complex mediating light control of development. Cell. 1994;78:117–124. doi: 10.1016/0092-8674(94)90578-9. [DOI] [PubMed] [Google Scholar]
- 33.Wei N, Deng XW. COP9: a new genetic locus involved in light-regulated development and gene expression in arabidopsis. Plant Cell. 1992;4:1507–1518. doi: 10.1105/tpc.4.12.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei N, Deng XW. Characterization and purification of the mammalian COP9 complex, a conserved nuclear regulator initially identified as a repressor of photomorphogenesis in higher plants. Photochem Photobiol. 1998;68:237–241. doi: 10.1562/0031-8655(1998)068<0237:capotm>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 35.Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell. 1998;94:615–623. doi: 10.1016/s0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Deng XW. The COP9 signalosome: an alternative lid for the 26S proteasome? Trends Cell Biol. 2003;13:507–509. doi: 10.1016/j.tcb.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Wei N, Deng XW. The COP9 signalosome. Annu Rev Cell Dev Biol. 2003;19:261–286. doi: 10.1146/annurev.cellbio.19.111301.112449. [DOI] [PubMed] [Google Scholar]
- 38.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 39.Rzucidlo EM. Signaling pathways regulating vascular smooth muscle cell differentiation. Vascular. 2009;17(Suppl 1):S15–20. doi: 10.2310/6670.2008.00089. [DOI] [PubMed] [Google Scholar]
- 40.Charron T, Nili N, Strauss BH. The cell cycle: a critical therapeutic target to prevent vascular proliferative disease. Can J Cardiol. 2006;22(Suppl B):41B–55B. doi: 10.1016/s0828-282x(06)70986-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4:104–111. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 43.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6:1229–1235. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 44.Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 2006;20:47–64. doi: 10.1101/gad.1384406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakano N, Urasawa K, Takagi Y, Saito T, Kaneta S, Ishikawa S, Higashi H, Tsutsui H, Hatakeyama M, Kitabatake A. Downregulation of cyclin-dependent kinase inhibitor; p57(kip2), is involved in the cell cycle progression of vascular smooth muscle cells. Biochem Biophys Res Commun. 2005;338:1661–1667. doi: 10.1016/j.bbrc.2005.10.093. [DOI] [PubMed] [Google Scholar]
- 46.Braun-Dullaeus RC, Mann MJ, Dzau VJ. Cell cycle progression: new therapeutic target for vascular proliferative disease. Circulation. 1998;98:82–89. doi: 10.1161/01.cir.98.1.82. [DOI] [PubMed] [Google Scholar]
- 47.Tanner FC, Boehm M, Akyurek LM, San H, Yang ZY, Tashiro J, Nabel GJ, Nabel EG. Differential effects of the cyclin-dependent kinase inhibitors p27(Kip1), p21(Cip1), and p16(Ink4) on vascular smooth muscle cell proliferation. Circulation. 2000;101:2022–2025. doi: 10.1161/01.cir.101.17.2022. [DOI] [PubMed] [Google Scholar]
- 48.Ii M, Hoshiga M, Fukui R, Negoro N, Nakakoji T, Nishiguchi F, Kohbayashi E, Ishihara T, Hanafusa T. Beraprost sodium regulates cell cycle in vascular smooth muscle cells through cAMP signaling by preventing down-regulation of p27(Kip1) Cardiovasc Res. 2001;52:500–508. doi: 10.1016/s0008-6363(01)00411-4. [DOI] [PubMed] [Google Scholar]
- 49.Reis ED, Roque M, Cordon-Cardo C, Drobnjak M, Fuster V, Badimon JJ. Apoptosis, proliferation, and p27 expression during vessel wall healing: time course study in a mouse model of transluminal femoral artery injury. J Vasc Surg. 2000;32:1022–1029. doi: 10.1067/mva.2000.109763. [DOI] [PubMed] [Google Scholar]
- 50.Boehm M, Olive M, True AL, Crook MF, San H, Qu X, Nabel EG. Bone marrow-derived immune cells regulate vascular disease through a p27(Kip1)-dependent mechanism. J Clin Invest. 2004;114:419–426. doi: 10.1172/JCI20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen D, Krasinski K, Sylvester A, Chen J, Nisen PD, Andres V. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J Clin Invest. 1997;99:2334–2341. doi: 10.1172/JCI119414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McArthur JG, Qian H, Citron D, Banik GG, Lamphere L, Gyuris J, Tsui L, George SE. p27-p16 Chimera: a superior antiproliferative for the prevention of neointimal hyperplasia. Mol Ther. 2001;3:8–13. doi: 10.1006/mthe.2000.0239. [DOI] [PubMed] [Google Scholar]
- 53.Tsui LV, Camrud A, Mondesire J, Carlson P, Zayek N, Camrud L, Donahue B, Bauer S, Lin A, Frey D, Rivkin M, Subramanian A, Falotico R, Gyuris J, Schwartz R, McArthur JG. p27-p16 fusion gene inhibits angioplasty-induced neointimal hyperplasia and coronary artery occlusion. Circ Res. 2001;89:323–328. doi: 10.1161/hh1601.094482. [DOI] [PubMed] [Google Scholar]
- 54.Lim Y, Kwon JS, Kim DW, Lee SH, Park RK, Lee JJ, Hong JT, Yoo HS, Kwon BM, Yun YP. Obovatol from Magnolia obovata inhibits vascular smooth muscle cell proliferation and intimal hyperplasia by inducing p21Cip1. Atherosclerosis. 2010;210:372–380. doi: 10.1016/j.atherosclerosis.2009.11.037. [DOI] [PubMed] [Google Scholar]
- 55.Yoo SH, Lim Y, Kim SJ, Yoo KD, Yoo HS, Hong JT, Lee MY, Yun YP. Sulforaphane inhibits PDGF-induced proliferation of rat aortic vascular smooth muscle cell by up-regulation of p53 leading to G1/S cell cycle arrest. Vascul Pharmacol. 2013;59:44–51. doi: 10.1016/j.vph.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Yang ZY, Simari RD, Perkins ND, San H, Gordon D, Nabel GJ, Nabel EG. Role of the p21 cyclin-dependent kinase inhibitor in limiting intimal cell proliferation in response to arterial injury. Proc Natl Acad Sci U S A. 1996;93:7905–7910. doi: 10.1073/pnas.93.15.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akyurek LM, Boehm M, Olive M, Zhou AX, San H, Nabel EG. Deficiency of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1 accelerates atherogenesis in apolipoprotein E-deficient mice. Biochem Biophys Res Commun. 2010;396:359–363. doi: 10.1016/j.bbrc.2010.04.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khanna AK. Enhanced susceptibility of cyclin kinase inhibitor p21 knockout mice to high fat diet induced atherosclerosis. J Biomed Sci. 2009;16:66. doi: 10.1186/1423-0127-16-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shahzad K, Thati M, Wang H, Kashif M, Wolter J, Ranjan S, He T, Zhou Q, Blessing E, Bierhaus A, Nawroth PP, Isermann B. Minocycline reduces plaque size in diet induced atherosclerosis via p27(Kip1) Atherosclerosis. 2011;219:74–83. doi: 10.1016/j.atherosclerosis.2011.05.041. [DOI] [PubMed] [Google Scholar]
- 60.Condorelli G, Aycock JK, Frati G, Napoli C. Mutated p21/WAF/CIP transgene overexpression reduces smooth muscle cell proliferation, macrophage deposition, oxidation-sensitive mechanisms, and restenosis in hypercholesterolemic apolipoprotein E knockout mice. FASEB J. 2001;15:2162–2170. doi: 10.1096/fj.01-0032com. [DOI] [PubMed] [Google Scholar]
- 61.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115:115–130. doi: 10.1161/CIRCRESAHA.115.301146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu Y, Liu L, Zhang Y, Wang G, Han D, Ke R, Li S, Feng W, Li M. Activation of AMPK inhibits pulmonary arterial smooth muscle cells proliferation. Exp Lung Res. 2014;40:251–258. doi: 10.3109/01902148.2014.913092. [DOI] [PubMed] [Google Scholar]
- 63.Zhang L, Pu Z, Wang J, Zhang Z, Hu D, Wang J. Baicalin inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via the AKT/HIF-1alpha/p27-associated pathway. Int J Mol Sci. 2014;15:8153–8168. doi: 10.3390/ijms15058153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu DQ, Luo Y, Liu Y, Wang J, Zhang B, Xu M, Wang YX, Dong HY, Dong MQ, Zhao PT, Niu W, Liu ML, Gao YQ, Li ZC. Beta-estradiol attenuates hypoxic pulmonary hypertension by stabilizing the expression of p27kip1 in rats. Respir Res. 2010;11:182. doi: 10.1186/1465-9921-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diep QN, El Mabrouk M, Touyz RM, Schiffrin EL. Expression of cell cycle proteins in blood vessels of angiotensin II-infused rats: role of AT(1) receptors. Hypertension. 2001;37:604–608. doi: 10.1161/01.hyp.37.2.604. [DOI] [PubMed] [Google Scholar]
- 66.Kanda T, Hayashi K, Wakino S, Homma K, Yoshioka K, Hasegawa K, Sugano N, Tatematsu S, Takamatsu I, Mitsuhashi T, Saruta T. Role of Rho-kinase and p27 in angiotensin II-induced vascular injury. Hypertension. 2005;45:724–729. doi: 10.1161/01.HYP.0000153316.59262.79. [DOI] [PubMed] [Google Scholar]
- 67.Seasholtz TM, Zhang T, Morissette MR, Howes AL, Yang AH, Brown JH. Increased expression and activity of RhoA are associated with increased DNA synthesis and reduced p27 (Kip1) expression in the vasculature of hypertensive rats. Circ Res. 2001;89:488–495. doi: 10.1161/hh1801.096337. [DOI] [PubMed] [Google Scholar]
- 68.Sung JY, Choi HC. Aspirin-induced AMP-activated protein kinase activation regulates the proliferation of vascular smooth muscle cells from spontaneously hypertensive rats. Biochem Biophys Res Commun. 2011;408:312–317. doi: 10.1016/j.bbrc.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 69.Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27 Kip1 is instigated by Jab1. Nature. 1999;398:160–165. doi: 10.1038/18230. [DOI] [PubMed] [Google Scholar]
- 70.Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY. The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem. 2002;277:2302–2310. doi: 10.1074/jbc.M104431200. [DOI] [PubMed] [Google Scholar]
- 71.Chen B, Zhao R, Su CH, Linan M, Tseng C, Phan L, Fang L, Yang HY, Yang H, Wang W, Xu X, Jiang N, Cai S, Jin F, Yeung SC, Lee MH. CDK inhibitor p57 (Kip2) is negatively regulated by COP9 signalosome subunit 6. Cell Cycle. 2012;11:4633–4641. doi: 10.4161/cc.22887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Denti S, Fernandez-Sanchez ME, Rogge L, Bianchi E. The COP9 signalosome regulates Skp2 levels and proliferation of human cells. J Biol Chem. 2006;281:32188–32196. doi: 10.1074/jbc.M604746200. [DOI] [PubMed] [Google Scholar]
- 73.Panattoni M, Sanvito F, Basso V, Doglioni C, Casorati G, Montini E, Bender JR, Mondino A, Pardi R. Targeted inactivation of the COP9 signalosome impairs multiple stages of T cell development. J Exp Med. 2008;205:465–477. doi: 10.1084/jem.20070725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lykke-Andersen K, Schaefer L, Menon S, Deng XW, Miller JB, Wei N. Disruption of the COP9 signalosome Csn2 subunit in mice causes deficient cell proliferation, accumulation of p53 and cyclin E, and early embryonic death. Mol Cell Biol. 2003;23:6790–6797. doi: 10.1128/MCB.23.19.6790-6797.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yan J, Walz K, Nakamura H, Carattini-Rivera S, Zhao Q, Vogel H, Wei N, Justice MJ, Bradley A, Lupski JR. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol Cell Biol. 2003;23:6798–6808. doi: 10.1128/MCB.23.19.6798-6808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chamovitz DA, Segal D. JAB1/CSN5 and the COP9 signalosome. A complex situation. EMBO Rep. 2001;2:96–101. doi: 10.1093/embo-reports/kve028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14:1209–1225. doi: 10.1111/j.1365-2443.2009.01349.x. [DOI] [PubMed] [Google Scholar]
- 78.Yoneda-Kato N, Tomoda K, Umehara M, Arata Y, Kato JY. Myeloid leukemia factor 1 regulates p53 by suppressing COP1 via COP9 signalosome subunit 3. EMBO J. 2005;24:1739–1749. doi: 10.1038/sj.emboj.7600656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu C, Guo LQ, Menon S, Jin D, Pick E, Wang X, Deng XW, Wei N. COP9 signalosome subunit Csn8 is involved in maintaining proper duration of the G1 phase. J Biol Chem. 2013;288:20443–20452. doi: 10.1074/jbc.M113.468959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lei D, Li F, Su H, Tian Z, Ye B, Wei N, Wang X. COP9 signalosome subunit 8 is required for postnatal hepatocyte survival and effective proliferation. Cell Death Differ. 2011;18:259–270. doi: 10.1038/cdd.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Teixeira LK, Reed SI. Ubiquitin ligases and cell cycle control. Annu Rev Biochem. 2013;82:387–414. doi: 10.1146/annurev-biochem-060410-105307. [DOI] [PubMed] [Google Scholar]
- 82.Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323–333. doi: 10.1016/j.semcdb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 83.Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 84.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 85.Tanner FC, Yang ZY, Duckers E, Gordon D, Nabel GJ, Nabel EG. Expression of cyclin-dependent kinase inhibitors in vascular disease. Circ Res. 1998;82:396–403. doi: 10.1161/01.res.82.3.396. [DOI] [PubMed] [Google Scholar]
- 86.Wu YJ, Bond M, Sala-Newby GB, Newby AC. Altered S-phase kinase-associated protein-2 levels are a major mediator of cyclic nucleotide-induced inhibition of vascular smooth muscle cell proliferation. Circ Res. 2006;98:1141–1150. doi: 10.1161/01.RES.0000219905.16312.28. [DOI] [PubMed] [Google Scholar]
- 87.Bond M, Sala-Newby GB, Newby AC. Focal adhesion kinase (FAK)-dependent regulation of S-phase kinase-associated protein-2 (Skp-2) stability. A novel mechanism regulating smooth muscle cell proliferation. J Biol Chem. 2004;279:37304–37310. doi: 10.1074/jbc.M404307200. [DOI] [PubMed] [Google Scholar]
- 88.Wu YJ, Sala-Newby GB, Shu KT, Yeh HI, Nakayama KI, Nakayama K, Newby AC, Bond M. S-phase kinase-associated protein-2 (Skp2) promotes vascular smooth muscle cell proliferation and neointima formation in vivo. J Vasc Surg. 2009;50:1135–1142. doi: 10.1016/j.jvs.2009.07.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sicari BM, Troxell R, Salim F, Tanwir M, Takane KK, Fiaschi-Taesch N. c-myc and skp2 coordinate p27 degradation, vascular smooth muscle proliferation, and neointima formation induced by the parathyroid hormone-related protein. Endocrinology. 2012;153:861–872. doi: 10.1210/en.2011-1590. [DOI] [PubMed] [Google Scholar]
- 90.Song P, Wang S, He C, Wang S, Liang B, Viollet B, Zou MH. AMPKalpha2 deletion exacerbates neointima formation by upregulating Skp2 in vascular smooth muscle cells. Circ Res. 2011;109:1230–1239. doi: 10.1161/CIRCRESAHA.111.250423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 91.Luo Y, Xu DQ, Dong HY, Zhang B, Liu Y, Niu W, Dong MQ, Li ZC. Tanshinone IIA inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via Akt/Skp2/p27-associated pathway. PLoS One. 2013;8:e56774. doi: 10.1371/journal.pone.0056774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bond M, Sala-Newby GB, Wu YJ, Newby AC. Biphasic effect of p21Cip1 on smooth muscle cell proliferation: role of PI 3-kinase and Skp2-mediated degradation. Cardiovasc Res. 2006;69:198–206. doi: 10.1016/j.cardiores.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 93.Song GJ, Barrick S, Leslie KL, Bauer PM, Alonso V, Friedman PA, Fiaschi-Taesch NM, Bisello A. The scaffolding protein EBP50 promotes vascular smooth muscle cell proliferation and neointima formation by regulating Skp2 and p21(cip1) Arterioscler Thromb Vasc Biol. 2012;32:33–41. doi: 10.1161/ATVBAHA.111.235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Akata T. Cellular and molecular mechanisms regulating vascular tone. Part 1: basic mechanisms controlling cytosolic Ca2+ concentration and the Ca2+-dependent regulation of vascular tone. J Anesth. 2007;21:220–231. doi: 10.1007/s00540-006-0487-5. [DOI] [PubMed] [Google Scholar]
- 95.Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12:2165–2180. doi: 10.1111/j.1582-4934.2008.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 97.Ko EA, Han J, Jung ID, Park WS. Physiological roles of K+ channels in vascular smooth muscle cells. J Smooth Muscle Res. 2008;44:65–81. doi: 10.1540/jsmr.44.65. [DOI] [PubMed] [Google Scholar]
- 98.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 99.Sorensen CM, Braunstein TH, Holstein-Rathlou NH, Salomonsson M. Role of vascular potassium channels in the regulation of renal hemodynamics. Am J Physiol Renal Physiol. 2012;302:F505–518. doi: 10.1152/ajprenal.00052.2011. [DOI] [PubMed] [Google Scholar]
- 100.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- 101.Yang Q, Underwood MJ, He GW. Calcium-activated potassium channels in vasculature in response to ischemia-reperfusion. J Cardiovasc Pharmacol. 2012;59:109–115. doi: 10.1097/FJC.0b013e318210fb4b. [DOI] [PubMed] [Google Scholar]
- 102.Dabertrand F, Nelson MT, Brayden JE. Ryanodine receptors, calcium signaling, and regulation of vascular tone in the cerebral parenchymal microcirculation. Microcirculation. 2013;20:307–316. doi: 10.1111/micc.12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Abou-Saleh H, Pathan AR, Daalis A, Hubrack S, Abou-Jassoum H, Al-Naeimi H, Rusch NJ, Machaca K. Inositol 1,4,5-trisphosphate (IP3) receptor up-regulation in hypertension is associated with sensitization of Ca2+ release and vascular smooth muscle contractility. J Biol Chem. 2013;288:32941–32951. doi: 10.1074/jbc.M113.496802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirano K, Hirano M, Kanaide H. Regulation of myosin phosphorylation and myofilament Ca2+ sensitivity in vascular smooth muscle. J Smooth Muscle Res. 2004;40:219–236. doi: 10.1540/jsmr.40.219. [DOI] [PubMed] [Google Scholar]
- 105.Goulopoulou S, Webb RC. Symphony of vascular contraction: how smooth muscle cells lose harmony to signal increased vascular resistance in hypertension. Hypertension. 2014;63:e33–39. doi: 10.1161/HYPERTENSIONAHA.113.02444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Salamanca DA, Khalil RA. Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol. 2005;70:1537–1547. doi: 10.1016/j.bcp.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Song J, Eyster KM, Kost CK Jr, Kjellsen B, Martin DS. Involvement of protein kinase C-CPI-17 in androgen modulation of angiotensin II-renal vasoconstriction. Cardiovasc Res. 2010;85:614–621. doi: 10.1093/cvr/cvp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song J, Martin DS. Rho kinase contributes to androgen amplification of renal vasoconstrictor responses in the spontaneously hypertensive rat. J Cardiovasc Pharmacol. 2006;48:103–109. doi: 10.1097/01.fjc.0000245403.45406.d8. [DOI] [PubMed] [Google Scholar]
- 109.Wooldridge AA, MacDonald JA, Erdodi F, Ma C, Borman MA, Hartshorne DJ, Haystead TA. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J Biol Chem. 2004;279:34496–34504. doi: 10.1074/jbc.M405957200. [DOI] [PubMed] [Google Scholar]
- 110.Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ. Vascular calcium channels and high blood pressure: pathophysiology and therapeutic implications. Vascul Pharmacol. 2006;44:131–142. doi: 10.1016/j.vph.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch. 2009;458:481–487. doi: 10.1007/s00424-009-0635-5. [DOI] [PubMed] [Google Scholar]
- 112.Kameda K, Fukao M, Kobayashi T, Tsutsuura M, Nagashima M, Yamada Y, Yamashita T, Tohse N. CSN5/Jab1 inhibits cardiac L-type Ca2+ channel activity through protein-protein interactions. J Mol Cell Cardiol. 2006;40:562–569. doi: 10.1016/j.yjmcc.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 113.Lu T, Chai Q, Yu L, d’Uscio LV, Katusic ZS, He T, Lee HC. Reactive oxygen species signaling facilitates FOXO-3a/FBXO-dependent vascular BK channel beta1 subunit degradation in diabetic mice. Diabetes. 2012;61:1860–1868. doi: 10.2337/db11-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang DM, He T, Katusic ZS, Lee HC, Lu T. Muscle-specific f-box only proteins facilitate bk channel beta(1) subunit downregulation in vascular smooth muscle cells of diabetes mellitus. Circ Res. 2010;107:1454–1459. doi: 10.1161/CIRCRESAHA.110.228361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Oberdorf J, Webster JM, Zhu CC, Luo SG, Wojcikiewicz RJ. Down-regulation of types I, II and III inositol 1,4,5-trisphosphate receptors is mediated by the ubiquitin/proteasome pathway. Biochem J. 1999;339:453–461. [PMC free article] [PubMed] [Google Scholar]
- 116.Martin-Garrido A, Boyano-Adanez MC, Alique M, Calleros L, Serrano I, Griera M, Rodriguez-Puyol D, Griendling KK, Rodriguez-Puyol M. Hydrogen peroxide down-regulates inositol 1,4,5-trisphosphate receptor content through proteasome activation. Free Radic Biol Med. 2009;47:1362–1370. doi: 10.1016/j.freeradbiomed.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 117.Lu JP, Wang Y, Sliter DA, Pearce MM, Wojcikiewicz RJ. RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J Biol Chem. 2011;286:24426–24433. doi: 10.1074/jbc.M111.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- 119.Shi J, Wei L. Rho kinases in cardiovascular physiology and pathophysiology: the effect of fasudil. J Cardiovasc Pharmacol. 2013;62:341–354. doi: 10.1097/FJC.0b013e3182a3718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Leck YC, Choo YY, Tan CY, Smith PG, Hagen T. Biochemical and cellular effects of inhibiting Nedd8 conjugation. Biochem Biophys Res Commun. 2010;398:588–593. doi: 10.1016/j.bbrc.2010.06.128. [DOI] [PubMed] [Google Scholar]
- 121.Chen Y, Yang Z, Meng M, Zhao Y, Dong N, Yan H, Liu L, Ding M, Peng HB, Shao F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell. 2009;35:841–855. doi: 10.1016/j.molcel.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 122.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 123.Lin MY, Lin YM, Kao TC, Chuang HH, Chen RH. PDZ-RhoGEF ubiquitination by Cullin3-KLHL20 controls neurotrophin-induced neurite outgrowth. J Cell Biol. 2011;193:985–994. doi: 10.1083/jcb.201103015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, Keen HL, Weatherford ET, Faraci FM, Sigmund CD. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARgamma and RhoA/Rho-kinase. Cell Metab. 2012;16:462–472. doi: 10.1016/j.cmet.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Triggle CR, Samuel SM, Ravishankar S, Marei I, Arunachalam G, Ding H. The endothelium: influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol. 2012;90:713–738. doi: 10.1139/y2012-073. [DOI] [PubMed] [Google Scholar]
- 126.Feletou M, Kohler R, Vanhoutte PM. Nitric oxide: orchestrator of endothelium-dependent responses. Ann Med. 2012;44:694–716. doi: 10.3109/07853890.2011.585658. [DOI] [PubMed] [Google Scholar]
- 127.Kang KT. Endothelium-derived Relaxing Factors of Small Resistance Arteries in Hypertension. Toxicol Res. 2014;30:141–148. doi: 10.5487/TR.2014.30.3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thorin E, Clozel M. The cardiovascular physiology and pharmacology of endothelin-1. Adv Pharmacol. 2010;60:1–26. doi: 10.1016/B978-0-12-385061-4.00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Virdis A, Ghiadoni L, Taddei S. Human endothelial dysfunction: EDCFs. Pflugers Arch. 2010;459:1015–1023. doi: 10.1007/s00424-009-0783-7. [DOI] [PubMed] [Google Scholar]
- 130.Wong MS, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin. 2010;31:1095–1102. doi: 10.1038/aps.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Feletou M, Kohler R, Vanhoutte PM. En-dothelium-derived vasoactive factors and hypertension: possible roles in pathogenesis and as treatment targets. Curr Hypertens Rep. 2010;12:267–275. doi: 10.1007/s11906-010-0118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li H, Horke S, Forstermann U. Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol Sci. 2013;34:313–319. doi: 10.1016/j.tips.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 134.Li H, Horke S, Forstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014;237:208–219. doi: 10.1016/j.atherosclerosis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 135.Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 136.Ebrahimian T, Touyz RM. Thioredoxin in vascular biology: role in hypertension. Antioxid Redox Signal. 2008;10:1127–1136. doi: 10.1089/ars.2007.1985. [DOI] [PubMed] [Google Scholar]
- 137.Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285:F178–190. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- 138.Osawa Y, Lowe ER, Everett AC, Dunbar AY, Billecke SS. Proteolytic degradation of nitric oxide synthase: effect of inhibitors and role of hsp90-based chaperones. J Pharmacol Exp Ther. 2003;304:493–497. doi: 10.1124/jpet.102.035055. [DOI] [PubMed] [Google Scholar]
- 139.Jiang J, Cyr D, Babbitt RW, Sessa WC, Patterson C. Chaperone-dependent regulation of endothelial nitric-oxide synthase intracellular trafficking by the co-chaperone/ubiquitin ligase CHIP. J Biol Chem. 2003;278:49332–49341. doi: 10.1074/jbc.M304738200. [DOI] [PubMed] [Google Scholar]
- 140.Thomas S, Kotamraju S, Zielonka J, Harder DR, Kalyanaraman B. Hydrogen peroxide induces nitric oxide and proteosome activity in endothelial cells: a bell-shaped signaling response. Free Radic Biol Med. 2007;42:1049–1061. doi: 10.1016/j.freeradbiomed.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cokic VP, Beleslin-Cokic BB, Noguchi CT, Schechter AN. Hydroxyurea increases eNOS protein levels through inhibition of proteasome activity. Nitric Oxide. 2007;16:371–378. doi: 10.1016/j.niox.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 142.Stangl V, Lorenz M, Meiners S, Ludwig A, Bartsch C, Moobed M, Vietzke A, Kinkel HT, Baumann G, Stangl K. Long-term up-regulation of eNOS and improvement of endothelial function by inhibition of the ubiquitin-proteasome pathway. FASEB J. 2004;18:272–279. doi: 10.1096/fj.03-0054com. [DOI] [PubMed] [Google Scholar]
- 143.Matsumoto K, Nishiya T, Maekawa S, Horinouchi T, Ogasawara K, Uehara T, Miwa S. The ECS (SPSB) E3 ubiquitin ligase is the master regulator of the lifetime of inducible nitric-oxide synthase. Biochem Biophys Res Commun. 2011;409:46–51. doi: 10.1016/j.bbrc.2011.04.103. [DOI] [PubMed] [Google Scholar]
- 144.Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM, Hu F, Ballabh P, Podlutsky A, Losonczy G, de Cabo R, Mathew R, Wolin MS, Ungvari Z. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension. 2009;54:668–675. doi: 10.1161/HYPERTENSIONAHA.109.133397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lubbers J, Lewis S, Harper E, Hledin MP, Marquez GA, Johnson AE, Graves DR, Burnatowska-Hledin MA. Resveratrol enhances anti-proliferative effect of VACM-1/cul5 in T47D cancer cells. Cell Biol Toxicol. 2011;27:95–105. doi: 10.1007/s10565-010-9173-3. [DOI] [PubMed] [Google Scholar]
- 146.Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010;459:793–806. doi: 10.1007/s00424-009-0767-7. [DOI] [PubMed] [Google Scholar]
- 147.Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. J Biol Chem. 2002;277:29669–29673. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- 148.Ruan L, Torres CM, Buffett RJ, Kennard S, Fulton D, Venema RC. Calcineurin-mediated dephosphorylation of eNOS at serine 116 affects eNOS enzymatic activity indirectly by facilitating c-Src binding and tyrosine 83 phosphorylation. Vascul Pharmacol. 2013;59:27–35. doi: 10.1016/j.vph.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang S, Xu J, Song P, Viollet B, Zou MH. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes. 2009;58:1893–1901. doi: 10.2337/db09-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xu J, Wang S, Wu Y, Song P, Zou MH. Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension. 2009;54:625–632. doi: 10.1161/HYPERTENSIONAHA.109.133736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xu J, Wu Y, Song P, Zhang M, Wang S, Zou MH. Proteasome-dependent degradation of guanosine 5’-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation. 2007;116:944–953. doi: 10.1161/CIRCULATIONAHA.106.684795. [DOI] [PubMed] [Google Scholar]
- 153.Santhanam AV, d’Uscio LV, He T, Katusic ZS. PPARdelta agonist GW501516 prevents uncoupling of endothelial nitric oxide synthase in cerebral microvessels of hph-1 mice. Brain Res. 2012;1483:89–95. doi: 10.1016/j.brainres.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Meurer S, Pioch S, Pabst T, Opitz N, Schmidt PM, Beckhaus T, Wagner K, Matt S, Gegenbauer K, Geschka S, Karas M, Stasch JP, Schmidt HH, Muller-Esterl W. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ Res. 2009;105:33–41. doi: 10.1161/CIRCRESAHA.109.198234. [DOI] [PubMed] [Google Scholar]
- 155.Bhansali M, Shemshedini L. COP9 subunits 4 and 5 target soluble guanylyl cyclase alpha1 and p53 in prostate cancer cells. Mol Endocrinol. 2014;28:834–845. doi: 10.1210/me.2014-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yazaki M, Kashiwagi K, Aritake K, Urade Y, Fujimori K. Rapid degradation of cyclooxygenase-1 and hematopoietic prostaglandin D synthase through ubiquitin-proteasome system in response to intracellular calcium level. Mol Biol Cell. 2012;23:12–21. doi: 10.1091/mbc.E11-07-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Neuss H, Huang X, Hetfeld BK, Deva R, Henklein P, Nigam S, Mall JW, Schwenk W, Dubiel W. The ubiquitin- and proteasome-dependent degradation of COX-2 is regulated by the COP9 signalosome and differentially influenced by coxibs. J Mol Med (Berl) 2007;85:961–970. doi: 10.1007/s00109-007-0197-y. [DOI] [PubMed] [Google Scholar]
- 158.Singh M, Chaudhry P, Parent S, Asselin E. Ubiquitin-proteasomal degradation of COX-2 in TGF-beta stimulated human endometrial cells is mediated through endoplasmic reticulum mannosidase I. Endocrinology. 2012;153:426–437. doi: 10.1210/en.2011-1438. [DOI] [PubMed] [Google Scholar]
- 159.Su H, Li J, Osinska H, Li F, Robbins J, Liu J, Wei N, Wang X. The COP9 signalosome is required for autophagy, proteasome-mediated proteolysis, and cardiomyocyte survival in adult mice. Circ Heart Fail. 2013;6:1049–1057. doi: 10.1161/CIRCHEARTFAILURE.113.000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Orel L, Neumeier H, Hochrainer K, Binder BR, Schmid JA. Crosstalk between the NF-kappaB activating IKK-complex and the CSN signalosome. J Cell Mol Med. 2010;14:1555–1568. doi: 10.1111/j.1582-4934.2009.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schweitzer K, Bozko PM, Dubiel W, Naumann M. CSN controls NF-kappaB by deubiquitinylation of IkappaBalpha. EMBO J. 2007;26:1532–1541. doi: 10.1038/sj.emboj.7601600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schweitzer K, Naumann M. Control of NF-kappaB activation by the COP9 signalosome. Biochem Soc Trans. 2010;38:156–161. doi: 10.1042/BST0380156. [DOI] [PubMed] [Google Scholar]
- 163.Hong X, Xu L, Li X, Zhai Z, Shu H. CSN3 interacts with IKKgamma and inhibits TNF- but not IL-1-induced NF-kappaB activation. FEBS Lett. 2001;499:133–136. doi: 10.1016/s0014-5793(01)02535-2. [DOI] [PubMed] [Google Scholar]
- 164.Harari-Steinberg O, Cantera R, Denti S, Bianchi E, Oron E, Segal D, Chamovitz DA. COP9 signalosome subunit 5 (CSN5/Jab1) regulates the development of the Drosophila immune system: effects on Cactus, Dorsal and hematopoiesis. Genes Cells. 2007;12:183–195. doi: 10.1111/j.1365-2443.2007.01049.x. [DOI] [PubMed] [Google Scholar]
- 165.Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, Oberheu K, Padmanabhan S, O’Dwyer M, Nawrocki ST, Giles FJ, Carew JS. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115:3796–3800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- 166.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2010;396:901–907. doi: 10.1016/j.bbrc.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 168.Li X, Lu D, He F, Zhou H, Liu Q, Wang Y, Shao C, Gong Y. Cullin 4B protein ubiquitin ligase targets peroxiredoxin III for degradation. J Biol Chem. 2011;286:32344–32354. doi: 10.1074/jbc.M111.249003. [DOI] [PMC free article] [PubMed] [Google Scholar]