

Abstract
The feasibility of using a liposome drug delivery system to formulate octylglycerol (OG) as a vaginal microbicide product was explored. A liposome formulation was developed containing 1% OG and phosphatidyl choline in a ratio that demonstrated in vitro activity against Neisseria gonorrhoeae, HSV-1, HSV-2 and HIV-1 while sparing the innate vaginal flora, Lactobacillus. Two conventional gel formulations were prepared for comparison. The OG liposome formulation with the appropriate OG/lipid ratio and dosing level had greater efficacy than either conventional gel formulation and maintained this efficacy for at least 2 months. No toxicity was observed for the liposome formulation in ex vivo testing in a human ectocervical tissue model or in vivo testing in the macaque safety model. Furthermore, minimal toxicity was observed to lactobacilli in vitro or in vivo safety testing. The OG liposome formulation offers a promising microbicide product with efficacy against HSV, HIV and N. gonorrhoeae.
Keywords: Gel, drug delivery, antiviral, HSV, lactobacilli, efficacy, safety
Introduction
With 33.3 million people living with AIDS and 2.6 million newly infected in 2009, sexually transmitted diseases (STDs), including HIV, continue to be a significant health problem worldwide1, more severe and frequent in women than in men. Currently, behavioral changes and condom use are the only mechanisms available for prevention of STDs; but unfortunately, have not been successful judging by the increased number of infections every year. Efforts have been made to develop vaccines against STDs with varying success2, but there is no evidence suggesting that any of the vaccines being studied will prevent HIV-1. Before women can protect themselves, a strategy is required that will allow them to control the prevention modality; vaginal microbicides offer such a platform. Microbicides offer prophylactic protection from STDs, including HIV, by inactivating pathogens deposited into the vagina during sexual intercourse. Ideally, this protection would occur without significant permeation of the active microbicide through vaginal or cervical tissues into the systemic circulation3–11.
Previous studies have shown that milk lipids could inactivate a number of bacteria and enveloped virus, including HIV12. Some of these antimicrobial lipids have been modified to increase their stability and aqueous solubility while maintaining their antimicrobial activity13 such as glycerol monolaurate (GML). Haase’s14group recently showed that GML formulated in a K-Y warming gel prevented mucosal transmission of simian immuno-deficiency virus (SIV) in the SIV-rhesus macaque model, the monkey version of HIV. More recently, Skinner, et al.15 showed that GML in a gel formulation inhibited Candida and Gardnerella vaginalis both in vitro and in vivo, but did not affect Lactobacillus.
Octylglycerol (OG), a synthetic lipid derived from lipids in human breast milk developed by Dr. Charles Isaacs at the Institute for Basic Research in Developmental Disabilities (Staten Island, NY), has been shown to have potential as a vaginal and rectal microbicide12,13,16,17. This compound destabilized viral envelopes and the membranes of pathogenic bacteria and fungi and inactivated HIV, HSV, gonorrhoeae, and Trichomonas without harming Lactobacillus. OG is a low molecular weight compound (M.W. 204.3) with excellent stability to heat and oxidation. Due to the ether linkage between the octyl group and glycerol, OG is resistant to degradation by mammalian and microbial lipases, heat and oxidation. Given that a topical microbicide should optimally inactivate a wide spectrum of microorganisms, OG provides an excellent drug candidate.
To date, the majority of microbicide drug candidates tested in the clinic have been formulated in aqueous hydrogel formulations. Although these hydrogel formulations provide a familiar inexpensive dosage form for vaginal delivery of microbicide drug candidates, one drawback for these formulations is that they can be associated with leaking and messiness. Liposomal formulations may provide an alternative to traditional gel products. OG is poorly soluble in water. Its hydrophobic nature makes it difficult to formulate in an aqueous hydrogel. The lipid phase of the liposome bilayer provides a non-polar environment that is ideal for hydrophobic drugs such as OG. Liposomes can be easily dispersed in aqueous environments, making them an effective tool for “solubilizing” and distributing poorly water-soluble drugs. Liposomes are composed of natural phospholipids, that are biodegradable and have therefore inherently very little toxicity. As a drug delivery system, liposome encapsulation has been investigated for therapeuticapplicationswidely,suchasanti-inflammatory drugs18–20, antitumor drugs21–23, treatments for Hepatitis B and C viruses24, and HIV treatments25,26. Thus, formulating OG into liposomes could enhance its bioactivity by providing a sustained and targeted release of OG.
In this study, the feasibility of using a liposome drug delivery system to formulate OG as a vaginal microbicide was investigated. A series of liposome formulations containing OG were developed which varied in preservative choice, OG to lipid ratio, viscosity agent and OG dosing levels. The impact of these liposome formulation modifications on HSV-1 and HSV-2 in vitro efficacy and Lactobacillus toxicity as compared to two conventional gel formulations comprised of OG in the base of either Carbopol® or poloxamer was studied. In these studies, it was seen that liposome formulation composition impacted both the observed product efficacy and toxicity. The results of this study demonstrate that a topical microbicide product of OG encapsulated in a liposome system may offer better in vitro efficacy than a conventional aqueous-based gel formulation against HSV-1, HSV-2, and HIV-1. OG in vitro release studies showed that the liposome formulation resulted in a sustained release of OG from the formulation as compared to the gel formulation. Furthermore, the liposome formulation was found to be non toxic to excised human tissues and Lactobacillus. In addition, the OG liposome formulation was found to be safe in a standard macaque safety model. Such an OG liposomal topical microbicide product could be applied intra-vaginally prior to coitus and would be a potentially effective way to prevent HIV.
Materials and methods
Materials
Octylglycerol was provided by the Institute of Basic Research in Developmental Disabilities (Staten Island, NY), citric acid, benzoic acid, sodium citrate, vitamin E, polysorbate 80, lactic acid, benzyl alcohol, propylene glycol, phosphatidylcholine, methylparaben, propylparaben, and propylene glycol were obtained from Spectrum Chemical (New Brunswick, NJ); benzethonium chloride (BZT) was obtained from ACROS Organics through Fisher Scientific (Fair Lawn, NJ); all chemicals used were either USP or NF grade. Carbopol® 934 and Carbopol® Ultrez 10 were obtained from Noveon (BF Goodrich, Cleveland, Ohio); poloxamer 407 (Lutrol® F127 NF) was provided by BASF Corporation (Mt. Olive, NJ); octanol and hexane were obtained from Fisher Scientific (Fair Lawn,NJ); ethanol was obtained from AAPER Alcohol and Chemical Company (Shelbyville, KY); N,O-bis(trimethylsiyl)trifluoroacetamide (BSTFA) was obtained from Pierce (Rockford, IL).
Analytical
Gas chromatograph (GC) assay for OG
A GC method with a split injector and flame ionization detector (FID) was developed for the quantitation of OG. OG was extracted from an aqueous medium using a mixture of chloroform and aqueous medium at a 2:1 ratio. The chloroform layer was then evaporated to dryness and 100 μL of BSTFA was added to convert the OG to a trimethyl sialo derivative. The split ratio was set to 1:30. The injection volume was 1 μL. The GC system used was a Hewlett Packard (Model #5890A), and the GC column used was DB17 J & W Scientific capillary column, 30 m × 0.25 mm i.d. with 0.25 μm stationary film thickness. The carrier gas was helium and make-up gas was nitrogen. The carrier gas and make-up gas flow rates were approximately 30 and 400 mL/min, respectively. The injector was maintained at 250°C. The temperature program was set at 100°C, increased at a rate of 10°C/min up to a temperature of 250°C, then kept at 250°C for 7 min. The FID was operated at 250°C; hydrogen and air flows were set at 40 and 400 mL/min, respectively. The peak area of the chromatogram was analyzed by Series II Chem Station software.
HPLC (high pressure liquid chromatography) assay for OG
Given the complexity of the GC method, a HPLC method was also developed to determine the concentration of OG. A modular Waters (Waters Corp, Milford, MA) system equipped with Waters 600E pump, Waters W717 autosampler, evaporative light scattering detector (ELSD) PL-ELS 2100 (Polymer Laboratories Inc. Amherst, MA), and Milennium 3.2 software (Waters) were used in this assay with a Luna 5 μm NH2 100 Å 4.6 × 150 mm (Phenomenex Corp., Torrance, CA) column. Separations were done in an isocratic system using a mixture of acetonitrile (ACN): Milli Q water (97.5:2.5) at a flow rate of 1.0 mL/min, with an injection volume of 25 μL.
Formulation preparation
Liposome formulation
OG liposome formulations were manufactured at Optime Therapeutics Inc, (Petaluma, CA). Formulations were prepared by mixing two phases through an OptiMix Mixer (technology currently owned by Lippomix Incorporated (Novato, CA)). The OptiMix Mixer is an in-line static mixing device equipped with two storage compartments and a mixing chamber. The two storage compartments, one for the lipid phase and one for the aqueous phase, are vertical and provide continuous flow of two phases which is metered precisely by pumps. The two storage compartments for the two phases are maintained under controllable temperature conditions27. The compositions for the different liposomal formulations tested are shown in Table 1. Briefly, the aqueous phase consisted of deionized water (DI) or a citrate buffer at pH 5.0. The lipid phase consisted of phosphatidylcholine (1 to 10%), propylene glycol, vitamin E acetate, preservative (BZT or a combination of methylparaben and propylparaben), and 0 to 1% OG. The two phases were loaded into the two storage compartments separately and heated to 65°C. In the OptiMix system, the fluid flow rates are set at a range of 5–30 cm3/s and a range of 20–80 cm3/s for the lipid phase and aqueous phase, respectively, depending on the ratio of lipid phase to aqueous phase in the formulation. The lipid phase was continuously dispersed into the aqueous phase in the mixing chamber. A placebo liposome formulation was prepared which consisted of all ingredients except OG. For evaluation of the incorporation of viscosity agents, Carbopol® 934 or Carbopol® Ultrez 10 was hydrated in the aqueous phase by stirring prior to mixing with the lipid phase.
Table 1.
Percent composition (%w/w) for OG liposome formulations developed and tested in these studies.
| Ingredients | Prototype | OG/Lipid ratio optimization |
Effect of viscosity agent | Dosing study | Final |
|---|---|---|---|---|---|
| OG | 0.25/0.5/0.75/1 | 0.5 | 0.5 | 0.1/0.3/0.5/,1.0/3.0/6.0 | 1.0 |
| Phosphatidylcholline | 5 | 1/2.5/5/10 | 0.5/1/5/10 | 10 | 10 |
| Propylene glycol | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 |
| Ethanol | 5.00 | 5.00 | 5.00 | 5.00 | 5.00 |
| Vitamin E acetate | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Methylparaben | 0/0.15 | 0.04 | 0.04 | ||
| Propylparaben | 0/0.05 | 0.02 | 0.02 | ||
| BZT | 0 or 0.1 | ||||
| Carbopol® 934 or Ultrez 10 |
0 or 0.4 | ||||
| Citric acid anhydrous | 0.12 | 0.12 | |||
| Sodium citrate | 0.3 | 0.3 | |||
| DI water | qs to 100 | qs to 100 | qs to 100 | qs to 100 | qs to 100 |
qs, quantity sufficient.
Carbopol® gel
OG gel formulations were prepared with either of the two polymers, Carbopol® or poloxamer, as the gelling agent (see Table 2). Briefly, the Carbopol® was added to the water using an overhead propeller mixer (Model RW20, IKA Works, Inc. Wilmington, NC) at room temperature and mixed until dissolved. OG in 1/5 of the glycerin was added and mixed until uniform followed by the methylparaben, propylparaben and methylcellulose dispersed in the remainder of the glycerin. The sodium hydroxide solution, 1N, was added to adjust the pH to 4.5 ± 0.2.
Table 2.
Formulation of OG conventional gels.
| Carbopol®-based |
Poloxamer-based |
||
|---|---|---|---|
| Ingredients | (%) | Ingredients | (%) |
| OG | 1 | OG | 1 |
| Carbopol® 934 | 0.25/0.43/0.61 | Poloxamer 407 | 17 |
| Methylcellulose | 1 | Lactic acid | 0.8 |
| Glycerin | 5 | Citric acid anhydrous | 1 |
| Methylparaben | 0.15 | Sodium citrate | 0.3 |
| Propylparaben | 0.05 | Benzyl alcohol | 0.5 |
| Milli Q water | qs | Propylene glycol | 1.5 |
| Milli Q water | qs | ||
| Total | 100 | 100 | |
Poloxamer gel
Poloxamer was added slowly to 50% of the amount of water needed at 4 to 8°C using an overhead propeller mixer (Model RW20, IKA Works, Inc. Wilmington, NC). After storing overnight in a refrigerator, the citric acid, sodium citrate and lactic acid (dissolved in 10% of the water) were added sequentially and mixed until uniform. OG and benzyl alcohol dissolved the propylene glycol was added, mixed until uniform and the pH adjusted to 4.5 ± 0.2 with lactic acid.
Formulation properties
Viscosity
Rheological profiles were determined using a cone and plate viscometer (Brookfield HADV III+ and LVDV III ultra) recording shear stress over a range of shear rates at both increasing and decreasing shear rates, i.e. up curve and down curve, respectively. All viscosities reported are “apparent” viscosities calculated as the ratio of shear stress to shear rate. For comparison purposes, viscosities were calculated at a fixed shear rate corresponding to a cone rotational speed of 10 rpm.
pH
The pH was determined using an Accumet pH meter (Fisher Scientific) with an AccuFet solid-state probe calibrated with pH 4.0, 7.0 and 10.0 standards.
In vitro release
An in vitro release test was utilized for formulated OG as a quality control measure to establish product function and reproducibility and was not intended to simulate biologically relevant conditions. The test was conducted as follows: a Franz-cell system with a 15-mm-diameter opening (1.77 cm2 cross sectional area) was used with a 33-mm regenerated cellulose dialysis flat membrane (Spectra/Por1, 6000–8000 molecular weight cut-off (MWCT), Spectrum Laboratories Inc.) sandwiched between the donor and receptor compartments. A circulating water-bath maintained the system at 37°C. The formulated product, 0.2 g, was placed in the donor compartment and 12 mL of pH 7.4 phosphate buffered saline (PBS) was used as the receptor medium. Precautions were taken not to entrap air bubbles between the PBS and the membrane. Samples, 200 μL (replacement with fresh PBS), were taken from the receptor compartment at 0, 0.5, 1, 2, 3, 4, 5 and 6 h, and analyzed by HPLC and GC for OG content.
Lipid stability
Since phosphatidylcholine can be hydrolyzed into stearic acid and lyso-phosphatidylcholine, the integrity of the phosphatidylcholine in the liposome OG formulation was evaluated at a 1-month-time point using a normal phase analytical HPLC method. The HPLC system (Waters Corp, Milford, MA) equipped with a Waters 2487 UV detector and an Alltech evaporative light scattering detector (ELSD 2000) was used. The stearic acid control eluted at 5.2 min, lyso-phosphatidylcholine at 18.5 min, and phosphatidylcholine at approximately 12 min (see Figure 1). All other standards, including vitamin E acetate, OG, and cholesterol were observed to elute in the void volume (1.6 min); propylene glycol was not detected using the ELSD.
Figure 1.

Typical HPLC chromatograms of distearylphosphatidylcholine (b) and its hydrolysis products: stearic acid (a) and lysophosphatidylcholine (c) using an ELSD detector.
Stability studies
In order to study the efficacy of OG formulations upon storage, the optimized OG liposome formulation containing 1% OG described in Table 1 along with its placebo formulation were placed on stability at three conditions: 5 ± 2, 25 ± 2, and 35 ± 2°C. In a separate study, the stability of two polymer-based gel formulations containing 1% OG in either a 0.43% Carbopol® or a 17% poloxamer base was monitored at two conditions: 25 ± 2°C with relative humidity of 60 ± 5% and 40 ± 2°C with relative humidity of 75 ± 5%. The liposome and gel products were monitored for efficacy against HSV-1, HSV-2, and HIV for a period of 2 months. Physicochemical stability including appearance, pH, osmolality, and viscosity at 25°C were monitored up to 6 months for liposome formulations and 3 months for gel formulations.
Safety studies
Human ectocervical tissue
Freshly excised ectocervical tissue was obtained from the Tissue Procurement Facility at the Magee Women’s Hospital according to IRB protocol number MWH-98-065 from premenopausal women undergoing hysterectomy for benign conditions. The age range of subjects was 35–49 years old. No patient identifiers were provided and all tissues collected were anonymized, deidentified, unlinking any patient ID to the investigators. All tissue specimens were obtained within 1 h of surgical excision. After the surgery, the tissue was held at 5°C in either PBS or Dulbecco’s modified Eagle Medium (Mediatech Inc, Herndon, VA) media until used. Excess stroma from cervical tissue was removed using a Thomas-Stadie-Riggs tissue slicer (Thomas Scientific, Swedesboro, NJ).
Exposure of human ectocervical tissue to OG formulations
The same Franz-cell system as described in the permeability study was used to determine toxicity to a short-term exposure of OG formulations to human ectocervical tissue. Formulated product was placed in the donor compartment; the tissue removed after 2-h exposure, fixed in Clark’s solution (ethanol/acetic acid 75:25) for 24 h, transferred to ethanol for 24 h, subsequently embedded in paraffin, and stained with Hematoxylin and Eosin to identify any morphological changes.
Permeability of OG through human ectocervical tissue
Permeability studies were conducted in a water-jacketed Franz-cell system (maintained at 37°C via a circulating water-bath) with a 7-mm diameter opening (0.385 cm2 cross sectional area) and a receptor compartment volume of 4.9 mL. PBS (pH 7.4) solution was used in the receptor compartment. Human ectocervical tissue was sandwiched between the two compartments with the epithelial side facing the donor compartment and equilibrated with 400 μL of PBS (pH 7.4) in the donor compartment for 5 min. After the equilibration period, the PBS (pH 7.4) solution was removed from the donor compartment and replaced with 400 μL of OG solutions in PBS at 0.25, 0.5 and 1.0% concentration. Samples, 200 μL, were taken from the receiver compartment (replacement with fresh PBS) at the following time intervals: 0, 0.5, 1, 2, 4, 6, 8, and 20 h and analyzed by GC for OG content. Duplicate experiments in four tissues were conducted.
Safety evaluation in macaque monkeys
Product safety was assessed in the rectal exposure pigtailed macaque safety model28,29. Rectal product application was utilized since the rectal tissues are more susceptible to perturbation than vaginal tissues. In these studies, six breeding-aged female Macaca nemestrina were obtained from colony animals at the Washington National Research Center (University of Washington, Seattle). The animals were housed and cared for under conditions that meet NIH standards as stated in the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, 1996), ILAR recommendations, and AAALAC accreditation standards for animals of this species. All six animals were treated identically, in that each underwent the same experimental protocol twice. Each experiment lasted 4 days: with daily applications over 3 days of either 2.5 mL active optimized final liposome formulation (Table 1) or no product administered intrarectally. Animals were under general sedation with telazol (3–6 mg/kg body weight, IM) during application of the products. Study endpoints evaluated included microbiological assessment, rectal pH, and evidence of blood or epithelial sloughing in rectal lavage samples.
Efficacy studies
HSV-1 and HSV-2
HSV-1 and HSV-2 (American Type Culture Collection ATCC, Manassas, VA) were grown in Vero and CV-1 cells, respectively and assayed as described previously30. Briefly, between 105 and 106 tissue culture infectious doses of HSV-1 or HSV-2 were mixed with the formulated product and incubated for 1 h at 37°C in the presence of 1% heat inactivated fetal bovine serum (Atlanta Biologicals, Norcross, GA). Following incubation, viral infectivity was determined by serial dilution end point method as described by Reed et al.31. Dilutions (10-fold) were inoculated into mono-layers of Vero cells. The plates were examined for 3 to 5 days and examined daily for cytopathic effect.
HIV-1
Experiments were conducted using the method described previously32. PHA-stimulated Peripheral bood mononuclear cells (PBMC), 2 × 105, were plated per well in a 96-well-plate and centrifuged at 800 rpm for 5 min to pellet the cells at the bottom of the well. Plates were held in the incubator until ready to be inoculated with the virus. Hundred microliter of HIV (50,000 pg/mL p24 equivalent) were incubated for 30 min at 25°C with equal volume of test material. As a control, HIV was incubated with medium for the same period of time. After incubation, samples were centrifuged at 10,000 rpm for 15 min, filtered through a 0.45-μm-filter, and re-centrifuged at 22,000 rpm for 1 h to pellet the virus. Pelleted viruses were resuspended in 200 μL of the RPMI medium containing 20% fetal calf serum (FCS) and 5% natural IL2 and inoculated onto PBMC in the 96-well-plate. After 5 days of incubation at 37°C, production of HIV was monitored by measuring HIV p24 in the culture supernatant using the antigen capture assay (Dupont/Perkin Elmer, Boston, MA). At the end of incubation, cell viability was also measured by the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. HIV suppression was calculated using the following equation:
N. gonorrhoea and normal vaginal microflora
The minimum cidal concentration (MCC) assay was used to determine the minimum concentration of OG required to kill 99.99% of the test organisms in 30 min17,33. Reference strains were obtained from the American Type Culture Collection, ATCC (Manassas, VA). Field isolates were obtained from human vaginal samples and identified to the species level as described previously33. Organisms were stored frozen at −70°C in litmus milk until needed. Stock cultures were revived by plating onto Chocolate agar with supplements (PML Microbiologicals, or prepared in house) for Neisseria gonorrhoeae or blood agar plates (Columbia blood agar base) for Lactobacillus. Cultures were incubated at 35°C in air enriched to 6% CO2 overnight or until good growth was observed. The OG liposome and gel formulations were used directly. Killing was determined by suspending the test organisms in saline to a density of a 0.5 McFarland standard, diluting 1 to 10 in sterile saline, 10 μL of bacterial suspensions were added to 90 μL of test solution. After incubation at 35°C for 30 min, 25 μL samples were taken and plated on to the appropriate medium, allowed to absorb and dry for 10–15 min then spread over the surface of the agar plate. Plates were incubated as described above for 24 h and evaluated for killing of the test microorganisms by examination. Samples yielding 10 or fewer colony forming units were considered sensitive to killing (99.99% of organisms killed). ACES buffer was used at a pH of 7.0 and lactate buffer was used at pH 5.0. The isotonic strength of each lot of buffer was determined in a vapor pressure osmometer (Vapro 5520, Wescor, Inc.) and adjusted to 200–300 mmol/kg prior to use.
Results
Formulation development
Prototype formulation development
Initial development of the OG liposome formulation utilized benzethonium chloride (BZT) as a preservative. Several formulations were prepared with and without preservative, and varying concentration of OG (0.25 to 1%) (Table 1). This first generation of liposome formulation presented significant efficacy against HIV-1 (above 90% suppression). However, toxicity was observed with the formulations containing BZT in the tissue exposure assay. Formulations without BZT did not show toxicity while maintaining significant anti-HIV activity. Consequently, BZT was eliminated as a preservative in subsequent formulations. Methyl and propyl parabens replaced BZT as non toxic preservatives.
Optimization of OG/lipid ratio
After elimination of BZT, formulation development continued by varying the OG/lipid ratio with the intent of optimizing the lipid concentration (Tables 1 and 3). The concentration of OG was maintained at 0.5% whereas the phospholipid, phosphatidylcholine, was varied from 1 to 10%. Efficacy of the formulations against N. gonorrhoeae, (ATCC 19425, GC 131, DOD 633, UPS 1170, ATCC 49226), HSV-1, HSV-2 and HIV-1 showed that the OG liposome formulations effectively killed all strains of gonococcus tested as well as HSV-1 and HSV-2. PHA-simulated PBMC infection by HIV appeared to be suppressed by all active OG liposome formulations tested, but was not suppressed by the placebo controls. The 1% phospholipid formulation suppressed about 91% of the HIV infection as compared to 80% suppression for the 10% phospholipid formulation suggesting an inverse correlation between the concentration of lipid and the activity of the formulation. Without being bound by any theory, it was believed that this reduced activity at higher lipid concentration was due to less active agent being present per liposome. In addition, tests to check preservation of normal vaginal flora were conducted with Lactobacillus crispatus (ATCC 6099, and 1211). Formulations with an OG/lipid ratio less than 0.2 were not toxic to Lactobacillus present in the normal vaginal flora as no killing was observed. However, killing of lactobacilli was observed with the formulation containing an OG/lipid ratio of 0.5.
Table 3.
Efficacy of OG liposome formulations as a function of OG/lipid ratio. Efficacy test against Gonococcus (ATCC 19425, GC 131, DOD 633, UPS 1170, ATCC 49226), Lactobacillus crispatus (ATCC 6099, and 1211), HSV-1 and HSV-2, and HIV-1.
| Ratio OG/Lipid | % Lipid | % OG | GC | Lactobacillus | HSV-1 | HSV-2 | HIV-1 (% Suppression) |
|---|---|---|---|---|---|---|---|
| 0.5 | 1.0 | 0.5 | × | × | + | + | 91 |
| 0.2 | 2.5 | 0.5 | × | / | + | + | 87 |
| 0.1 | 5 | 0.5 | × | / | + | + | 87 |
| 0.05 | 10 | 0.5 | × | / | + | + | 80 |
× = Killing by showing greater than a 4-log drop compared to control after exposure to the formulation mixture for 30 min.
/ = No killing by showing less than a 4-log drop compared to control after exposure to the formulation mixture for 30 min.
+ = Killing by showing greater than a 3-log drop compared to non-treated control.
Evaluation of viscosity increasing agents and preservative
A series of formulations incorporating Carbopol® 934 or Ultrez 10 with fixed 0.5% OG, variable 0–10% phospholipid, with and without preservatives were prepared (Table 1) and tested for HSV efficacy. The viscosity ranged from 2500 to 7200 cps and the in vitro release rate ranged from 1.05 to 2.32 μg/cm2/min1/2. However, all the formulations showed reduced efficacy against both of HSV-1 and HSV-2 by showing less than a 2-log drop compared to non-treated control group. This suggested interference in efficacy by the viscosity increasing agents. Consequently, viscosity increasing agents were not used in the final formulation.
Final formulation development
A series of liposome formulations were prepared containing 10% phospholipid while varying the concentrations of OG from 0.1 to 6% with a citrate buffer to maintain the pH of the formulation around 4.5–5.0 (Table 1). Efficacy of the formulations against N. gonorrhoeae, (UPS 1155, UPS 2015, UPS 1170, DOD 633, ATCC 19425, UPS 1137), HSV-1, HSV-2 and HIV-1 appeared to be concentration-related (Table 4). Concentrations below 0.3% OG were not able to kill STD pathogens while significant killing efficacy was observed at ≥0.5% OG. Lactobacillus crispatus (ATCC 6013, 6090, 6027, 2047ATCC 6099, and 1211) toxicity at pH 5.0 (10 mM ACES buffer) and pH 7.0 (10 mM Citrate buffer) was observed at ≥3% OG. In this stage of the development, toxicity to human ectocervical tissue was also evaluated. Figure 2 shows the comparison of histology, of human ectocervical tissue before and after 2-h exposure to 1% OG liposome formulation. No gross morphological changes in the tissue were found post exposure.
Table 4.
Dose response of OG in liposome formulation. Efficacy test against Gonococcus (GC, 5 strains: UPS 1155, UPS 2015, UPS 1170, DOD 633, ATCC 19425, UPS 1137), HSV-1 and HSV-2, HIV-1, and Lactobacillus crispatus (ATCC 6013, 6090, 6027, 2047).
| Ratio OG/Lipid | % Lipid | % OG | GC | Lactobacillus | HSV-1 | HSV-2 | HIV-1 (% Suppression) |
|---|---|---|---|---|---|---|---|
| 0 | 10 | 0 | / | / | − | − | NA |
| 0.01 | 10 | 0.1 | / | / | − | − | 52 |
| 0.03 | 10 | 0.3 | / | / | − | − | 27 |
| 0.05 | 10 | 0.5 | × | / | + | + | 80 |
| 0.1 | 10 | 1.0 | × | / | + | + | 91 |
| 0.3 | 10 | 3.0 | × | × | + | + | 95 |
| 0.6 | 10 | 6.0 | × | × | + | + | 99 |
× = Killing by showing greater than a 4-log drop compared to control after exposure to the formulation mixture for 30 min.
/ = No killing by showing less than a 4-log drop compared to control after exposure to the formulation mixture for 30 min.
+ = Killing by showing greater than a 3-log drop compared to non-treated control.
− = No killing by showing less than a 3-log drop compared to non-treated control.
Figure 2.

Comparison of histology, 20× magnification, of human ectocervical tissue before (a) and after (b) 2-h exposure to 1% OG liposome formulation.
The final liposome formulation developed as shown in Table 1 consists of 1% OG, citrate buffer, 10% phosphatidylcholine, 5% propylene glycol, 5% ethanol, 1% vitamin E acetate, 0.04% methyl paraben and 0.02% propyl paraben.
In vitro release
OG in vitro release rate from liposome formulations were compared with that from the conventional gels in Table 5. The release rate was obtained by linear regression of the slope of amount of OG released per area against square root of time34. OG exhibited greater release rates in Carbopol® gels than in liposome formulations and the release rate appeared to be viscosity dependent. Increasing the concentration of Carbopol® from 0.25 to 0.61% decreased the release rate by 30%. The release rate from the poloxamer-based gel was similar to that of the liposome formulation even though the viscosity of the poloxamer gel was 10-fold greater than that of the OG liposome formulation.
Table 5.
Characterization of 1%OG liposome and gel formulations.
| Product desciption |
Release Rate*
μg/cm2/min1/2 Medium: PBS |
Viscosity cps,10 rpm |
|||
|---|---|---|---|---|---|
| 25°C | 37°C | pH | |||
| Conventional gels | |||||
| Polymer base | Conc. of polymer (%) | ||||
| Carbopol | 0.25 | 8.92 | 1208 | NA | 4.5 |
| 0.43 | 8.00 | 4086 | NA | 4.5 | |
| 0.61 | 6.24 | 7117 | NA | 4.5 | |
| Poloxamer | 17 | 0.91 | 5970 | 7055 | 4.5 |
| Liposome | |||||
| Placebo | 0 | 0 | 573.4 | 579.2 | 5.3 |
| Active | 0 | 1.65 | 501.8 | 513 | 5.3 |
Data obtained from linear portion of in vitro release profile plotting from 30 to 240 min.
pH and viscosity stability
No significant difference was observed in pH during stability testing over a period of 6 months for both the OG and placebo liposome formulation (see Figure 3). The rheological profile of the final liposome formulation was non-Newtonian, pseudoplastic (shear thinning). This is a desired profile for microbicide products in that it indicates a formulation which has retention at the applied site, but can be easily spread during intercourse35. Viscosity was also monitored to ensure physical product stability (see Figure 4). An initial decrease in viscosity was observed for the active liposome formulation stored at 5°C attributed to an insufficient holding time for the formulation to achieve equilibrium. Once equilibrium was obtained, no further change in viscosity was observed.
Figure 3.
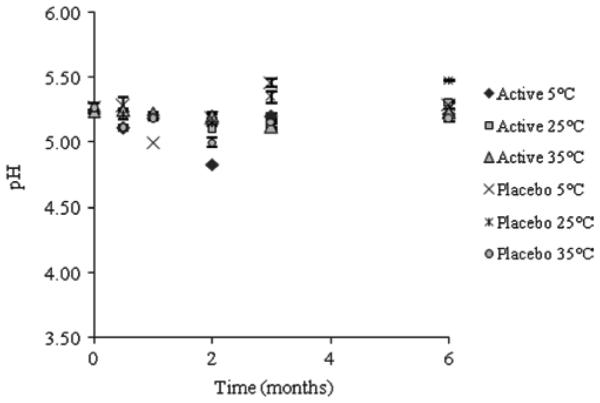
pH stability of OG liposome formulations during stability studies at 5°C, 25°C and 35°C for a period of 6 months.
Figure 4.
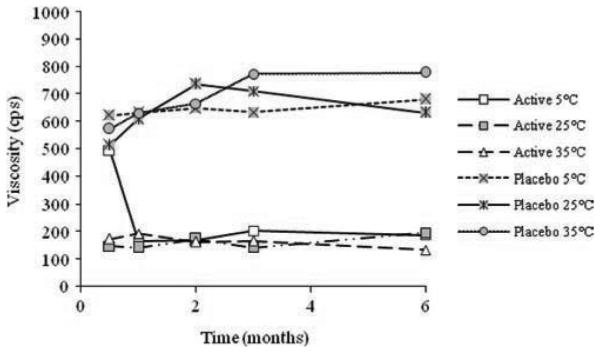
Viscosity stability of OG liposome formulations during stability studies at 5°C, 25°C and 35°C for a period of 6 months.
The conventional OG gel formulations using two different polymers (Table 2) were stable over the 6-month-stability testing period at both of 25 and 40°C; the pH ranged from 4.2 to 4.8, the osmolality was less than 1000 mmol/kg and the viscosity ranged from 4086 to 7000 cps.
Lipid stability evaluation
Analysis of the OG liposome formulation, by monitoring phosphatidylcholine hydrolysis, showed no presence of fatty acid or lyso-phosphatidylcholine over a 30-day testing period. This suggests that phosphatidylcholine present in the liposome formulation remained stable with no evidence of hydrolysis.
Safety studies
Permeability and exposure − human ectocervical tissue
OG permeability, assessed using the Franz-cell system, showed no detectable level of OG in the receptor compartment over 20 h. This indicates a lack of tissue permeability by OG in this model, but does not rule out tissue absorption of OG.
Histological evaluation of the tissues showed no significant morphological alterations after 2-h exposure to 1% OG liposome formulation as compared to pre exposure tissue (Figure 2).
Macaque safety studies
The 1% liposome formulation or no product was administered rectally at a dose of 2.5 mL daily for 3 days. pH, epithelial sloughing, and microbiological assessments were examined 15 min and 24 h after each application. Microbiological assessments indicated that the presence of lactobacilli remained equally constant after active liposome formulation or no product use. Endogenous hydrogen peroxide production by viridans streptococci was likewise unaffected. In general, the rectal microbiology remained unaffected by test product used, and compared favorably to rectal microbiology after no product use.
Both the no product and active liposome formulation treatments had minimal effect on rectal pH (Table 6). Although fluctuations in rectal pH measurements were noted throughout the study in active liposome and no product groups, no trend of product-related effects were seen. Observations of rectal lavage viewed under 7× magnifications, indicated no association between active OG product administration and increased epithelial desquamation (evidence of epithelial sheets (one side ≥3 mm)). Shedding of epithelial sheets in rectal lavage samples (Table 7) was noted sporadically in both active liposome and no product arms of the study.
Table 6.
Safety study in monkeys. Rectal pH evaluations after exposure to no productand liposome formulation after stability of 8 weeks (n = 6).
| Day 1 |
Day 2 |
Day 3 |
Day 4 |
||||
|---|---|---|---|---|---|---|---|
| Formulation | Time 0 | 15 min | Time 0 | 15 min | Time 0 | 15 min | Final |
| No product | 6.5 ± 0.8 | 7.0 ± 0.9 | 6.6 ± 0.8 | 6.8 ± 0.4 | 7.6 ± 0.7 | 7.5 ± 0.6 | 7.4 ± 0.7 |
| Liposome | 6.8 ± 0.9 | 7.3 ± 0.8 | 6.9 ± 0.7 | 6.8 ± 0.6 | 7.1 ± 0.7 | 6.8 ± 0.7 | 7.3 ± 0.6 |
Table 7.
Rectal lavage evaluationsafter exposure to no productand liposome formulation. Stroma and blood associated with measurable epithelial sheets (Evidence of epithelial sheets (one side ≥3mm)).
| Day 1 |
Day 2 |
Day 3 |
Day 4 Final |
|||||
|---|---|---|---|---|---|---|---|---|
| Monkey # | Time 0 | 15 min | Time 0 | 15 min | Time 0 | 15 min | ||
| Liposome | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 1a | 0 | |
| 3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |
| 4 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 5 | 0 | 1a | 1b | 0 | 0 | 0 | 2 | |
| 6 | 0 | 0 | 2c | 0 | 2 | 0 | 2 | |
| No product | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 4 | 0 | 0 | 0 | 0 | 5 | 4 | 0 | |
| 5 | 0 | 0 | 0 | 0 | 1a | 8e | 2 | |
| 6 | 0 | 0 | 0 | 1d | 0 | 0 | 1 | |
Trace blood noted on sheet.
Trace blood and stroma noted on sheet.
Trace blood noted on 1 sheet.
Trace blood and stroma noted on sheet.
Trace blood noted on 2 of 8 sheets.
Efficacy studies
HSV-1 and HSV-2
OG and placebo liposome formulations were tested against HSV-1 and HSV-2 at time 0, 1, 1.25 and 2 months stability at 5, 25 and 35°C (see Table 8). OG liposome formulations were fully active against HSV-1 over a period of at least 2 months by consistently decreasing the HSV-1 titer by greater than 6 logs. HSV-2 titers were decreased by greater than 5 logs at time 0, but only by approximately 2 logs at time periods of 1 month or more. The liposome placebo formulation had no activity.
Table 8.
Efficacy of OG liposome formulation vs. gel formulations against HSV-1 and HSV-2 for a period of 2 months storage.
| HSV-1 |
HSV-2 |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Formulations | Time (months) | 0 | 1 | 1.25 | 2 | 0 | 1 | 1.25 | 2 |
| Liposome | No Treatment Control | 6.38 | 6.25 | 6.38 | 6.50 | 6.25 | 5.63 | 6.00 | 5.63 |
| Placebo | |||||||||
| 5°C | 6.75 | 6.25 | 5.50 | 5.75 | 6.25 | 5.63 | 5.50 | 5.75 | |
| 25°C | 6.75 | 6.00 | 5.75 | 6.00 | 6.25 | 6.25 | 6.00 | 5.38 | |
| 35°C | 6.75 | 6.25 | 5.63 | 6.00 | 6.25 | 5.75 | 5.50 | 5.38 | |
| Active | |||||||||
| 5°C | <0 | <0 | <0 | <0 | >1 | 3.75 | 2.75 | 3.25 | |
| 25°C | <0 | <0 | <0 | <0 | >1 | 3.50 | 3.00 | 3.75 | |
| 35°C | <0 | <0 | <0 | <0 | >1 | 3.75 | 2.75 | 3.63 | |
| Gel | Carbopol-based | ||||||||
| Placebo | NA | 5.13 | NA | 4.60 | NA | 4.50 | NA | 4.50 | |
| Active | NA | 4.13 | NA | 5.00 | NA | 4.50 | NA | 3.50 | |
| Poloxamer-based | |||||||||
| Placebo | NA | 4.13 | NA | 5.00 | NA | 4.50 | NA | 3.50 | |
| Active | NA | 5.13 | NA | 5.50 | NA | 5.00 | NA | 4.50 | |
Data presented as logarithmic values of tissue cultrue infectious doses.
OG showed less activity against HSV-1 and HSV-2 in the two gel formulations by showing only a 1 to 2-log drop in viability as compared to non-treated control. This level of activity was maintained throughout the stability testing period. Furthermore, HSV in vitro bioactivity studies showed that both gel placebos exhibited some degree of activity revealing that gel placebo formulations themselves had baseline activity.
HIV-1
OG and placebo liposome formulations were tested against HIV-1 in vitro at time 0 and 2 months. Results are shown in Figure 5. No significant variations were observed. More than 80% HIV-1 was suppressed by the OG liposome formulations, which indicated that anti-HIV-1 activity of OG liposome formulation was maintained after stored at 5, 25 and 35°C for a period of 2 months. The liposome placebo formulation did not show activity against HIV-1 at any conditions of storage.Gel formulations were evaluated for anti HIV-1 activity after storage as well. The data, summarized in Figure 6 showed that all gel formulations including placebos presented significant suppression of HIV infection at the initial time point. Both the Carbopol® and liposome formulations containing OG maintained suppression of HIV infection at greater than 80% over the 2-month period. However, the poloxamer formulation appeared to decrease in efficacy with increased storage time. Efficacy of the OG containing poloxamer formulation was decreased by 26 and 28% after 2 months storage at 25 and 40°C, respectively. This observed decrease was potentially due to formulation destabilization.
Figure 5.
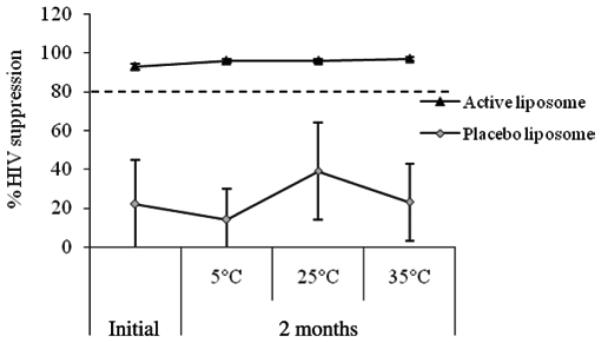
HIV-1 suppression of OG liposomal formulations after 2 months stability.
Figure 6.
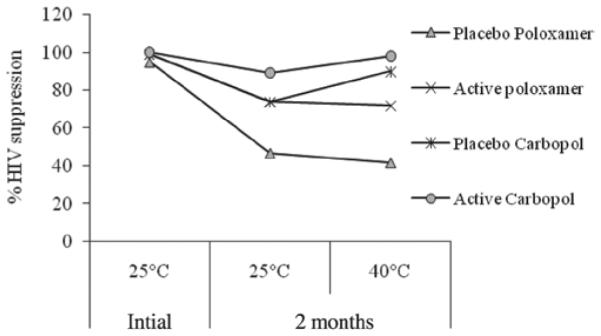
HIV-1 suppression of OG gel formulations after 2 months stability.
N. gonorrhoeae
Efficacy of the formulations was tested against N. gonorrhoeae as shown in Table 9. Formulations were exposed to the microorganism (ATCC 49226 and ATCC 19425) and activities were measured by killing or not killing17,33. OG Liposome formulation showed efficacy against N. gonorrohoeae even after 2-month storage. The placebo formulation showed no activity.
Table 9.
Efficacy of OG liposome formulation against Neisseria gonorrhoeae during stability studies.
| Time (month) |
|||||
|---|---|---|---|---|---|
| Microorganism | Formulation | 0 | 0.5 | 1 | 2 |
| N. gonorrhoeae | Active | × | × | × | × |
| (ATCC 49226) | Placebo | / | / | / | / |
| N. gonorrhoeae | Active | × | × | × | × |
| (ATCC 19425) | Placebo | / | / | / | / |
× = Killing by showing greater than a 4-log drop as compared to control group.
/ = No killing by showing less than a 4-log drop as compared to control group.
Discussion
AIDS continues to take a devastating toll on lives across the world. Several earlier clinical trials of vaginal topical microbicides failed to show efficacy when evaluated in large clinical trials36–39. However, recently it was shown in the CAPRISA trial that a vaginal gel product containing the nucleotide reverse transcriptase inhibitor tenofovir resulted in a 39% reduction in new HIV infections40. This study demonstrates the feasibility of such products for use in prevention of HIV infection, but also demonstrate the need for the design of better product which can more efficiently deliver active microbicide drug candidates resulting in increased product efficacy. A number of drug candidatesbeingevaluatedaspotentialmicrobicidesshow potency against HIV, but are hydrophobic in nature such as OG. Liposomal vaginal gel products can be an effective means to deliver such hydrophobic drug candidates to the vagina. Although liposome encapsulation as a drug delivery system has been investigated in numerous therapeutic applications as indicated previously, little work with the application of liposomal creams for the delivery of microbicide drug candidates has been published to date. A liposomal microbicide product containing a non-nucleoside reverse transcriptase inhibitor (MC1220) was reported to protect Rhesus Macaques from infection following intravaginal challenge with RT-SHIV41.
One critical specification for topical microbicide products is that the product should be non toxic to either vaginal or rectal tissues. These studies assessed toxicity using two models: ex vivo excised human tissue exposure and in vivo macaque safety rectal exposure. We used exposure studies to human ectocervical tissue to evaluate the vaginal safety of formulation products. Both of the OG liposome formulation and gel formulations did not show evidence of toxicity to freshly excised ectocervical tissue. A further safety study in the pigtailed macaque model demonstrated that the OG liposome formulation and no product had no effect on rectal pH, no impact on microbiological flora, and caused no epithelial desquamation.
Another challenge is to preserve the normal microflora, which plays a significant role in natural defense mechanisms, while maintaining activity against STD pathogens; lactobacilli are a major component of the vaginal microflora in premenopausal women. The in vitro biocompatibility study with Lactobacillus, demonstrated that Lactobacillus toxicity could be avoided by controlling the OG/lipid ratio. An OG/lipid ratio of less than 0.5 at a fixed concentration of OG (0.5%) and a ratio less than 0.3 at fixed lipid concentration (10%) spared four strains of Lactobacillus tested (ATCC 6013, 6090,6027, and 2047). However, it should be noted the accepted MCC for OG is 0.05 and 0.1% for most Lactobacillus. Thus, the MCC for OG drug substance is less than it is for the OG containing formulated liposome product. Other studies have demonstrated that the binding of the OG renders the molecule ineffective at killing the bacteria until the binding molecule is saturated with OG, or until the equilibrium dynamics reaches the point where the MCC is met. The binding may occur with albumin or fatty acids17.
Product efficacy is a second critical specification for microbicide products. A dose response study established that 1% OG with 10% lipid resulted in sufficient in vitro efficacy against HSV-1, HSV-2, HIV, and GC while sparing Lactobacillus and showing no toxicity in the excised tissue model.
Our studies demonstrate that both the OG liposome and gel formulations exhibit activity against HSV-1 and HSV-2 in the Vero and CV-1 cell model. The OG liposome formulation showed better activity against HSV-1 and HSV-2 as compared to the conventional OG gel formulations at the 0 time point with the liposome formulation having greater than a 6-log drop for HSV-1 and greater than a 5-log drop for HSV-2. Our results correlate with previous studies showing HSV-2 to be inherently slightly less sensitive to OG than HSV-142. Liposome product efficacy as measured by those in vitro evaluations was maintained throughout the 2 months of stability monitored for HSV-1. However, efficacy of the liposome product against HSV-2 decreased over time. The placebo liposome formulations did not show activity against HSV-1 and HSV-2. The two gel formulations, Carbopol®-based and poloxamer-based, showed approximately a 1.5–2 logs drop for HSV-1 and HSV-2 following 2 months of storage. The baseline activity observed in the gel placebo formulation is probably due to the preservative in these formulations.
The effects of HIV-1 suppression were investigated using a PBMC cell-based model that was designed specifically to test potential microbicides in the form of a cream or gel32. OG liposome formulations showed efficacy against HIV in greater than 80% suppression and this efficacy was maintained throughout stability monitoring. The gel formulations studied including their placebos showed efficacy against HIV; however, the poloxamer gel formulation did not maintained this efficacy after 2-month storage.
In the process of liposome formulation development, we incorporated bioadhesive polymers, either Carbopol® 934 or Ultrez 10. These excipients were added to obtain a desirable viscosity of the formulation with the intent to achieve potentially greater retention in the vagina. The viscosities of liposome formulations were increased to a range of 3000–7000 cps which was about 6–14 times as compared to 502 cps for liposome formulations without bioadhesive polymers. Bioadhesive liposome gels have been studied for local therapy of vaginitis characterized and evaluated in vitro43. Other mucoadhesive liposomes were reported for prolonged and controlled release44. In our studies, it was observed that addition of polymers resulted in reduced activity against HSV. Additionally, activity against HIV was reduced. Although correlations could not be made between the increase in viscosity and reduction of bioactivity, the polymers were believed to inhibit the release of OG, further decreased the activity. Other formulation constituents can impact product efficacy. It should be noted that previously reported studies have also shown that differing the composition of lipid in the liposomal formulation can impact cytotoxicity as well as efficacy against HIV infection25,45.
Controlled release of liposome encapsulated OG was one of the defining characteristics observed in the developed liposome product. These observed controlled release properties agree with release profiles observed by other liposomal products described in the literature46–49. This attribute may provide more effective targeting, deposit and prolong the activity of active drug ingredient. In additiobn, controlled and targeted release may reduce product side effects. A liposome formulation containing nevirapine, a hydrophobic reverse transcriptase inhibitor, showed potential improvement for HIV treatment by targeted delivery of the anti-retroviral drug to select compartments and cells which resulted in reduction of systemic toxic side effects50. In our study, the release profile of OG from the liposome product was controlled and prolonged. The in vitro release rate of OG (1.65 μg/cm2/min1/2) from the liposome formulations was approximately 2.2–5.4 times less than that from Carbopol®-based gel formulations. The OG release rate from multiple batches of liposome formulations prepared during the formulation optimization process, were consistent in their demonstration of controlled and prolonged delivery of OG.
Unlike the Carbopol®-based gel formulation, OG poloxamer-based gel formulation showed similar release rate to the liposome formulations. This phenomenon may be due to the high viscosity nature of poloxamer gel which was 4.5- to 12-fold greater than the viscosity of the liposome formulations.
Our studies indicate that a liposome drug delivery system is more effective for the delivery of OG as a microbicide than typical gel formulations. Additional research is required to further optimize the liposome drug delivery system developed for OG. Further investigation is needed to define encapsulation efficiency and a more thorough characterization of the liposome system must be conducted. Additionally it will be essential to efficacy to fully elucidate the tissue targeting which is achieved with this developed OG containing liposome product.
Conclusions
Combined the described studies demonstrate the feasibility of formulating OG as a liposomal drug delivery system. Compared to conventional gel formulations, the liposome formulation appeared to offer better activity against HSV-1, HSV-2, and HIV. In addition, the formulation showed efficacy against N. gonorrhoeae. With regard to safety, exposure to the OG liposome formulation resulted in no harm to Lactobacillus, the predominant innate microflora organism. Additionally, safety was demonstrated in vitro, ex vivo, and in vivo. Notably no toxicity was observed when exposed to human ectocervial tissue for a period of 2 h and was shown in a macaque model. This formulation was stable and retained efficacy after 2 months of storage. The results indicate the developed OG liposomal formulation may provide a safe and effective microbicide product with advantage over traditional hydrogel dosage forms.
Acknowledgments
We are grateful for the significant contributions of Roger Schnaare toward editing of this manuscript. Additionally we would like to acknowledge Frank Sorgi (currently at DPT Laboratories, Ltd. San Antonio, TX) and Hans Hofland (currently at Stiefel Laboratories, Inc. North Carolina) who were previously at Optime for their contributions to liposome product manufacture and hydrolysis assessment. Finally, we would like to acknowledge the grant support provided by the National Institute of Allergy and Infectious Diseases (NIAID) at the National Institute of Health (5 P01 Transport and Activity of Microbicide Formulations, A139061, Principle Investigator: Hillier, Sharon, Magee Womens Research Institute) and the grant support by Public Health Services (WaNPRC RR00166). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- 1.UNAIDS UNAIDS Report on the global AIDS epidemic 2010. Available at: http://www.unaids.org/globalreport/documents/20101123_GlobalReport_full_en.pdf.Accessed on 18 October 2011.
- 2.Duerr A, Wasserheit JN, Corey L. HIV vaccines: New frontiers in vaccine development. Clin Infect Dis. 2006;43:500–511. doi: 10.1086/505979. [DOI] [PubMed] [Google Scholar]
- 3.Turpin JA. Considerations and development of topical microbicides to inhibit the sexual transmission of HIV. Expert Opin Investig Drugs. 2002;11:1077–1097. doi: 10.1517/13543784.11.8.1077. [DOI] [PubMed] [Google Scholar]
- 4.Moench TR, Chipato T, Padian NS. Preventing disease by protecting the cervix: The unexplored promise of internal vaginal barrier devices. AIDS. 2001;15:1595–1602. doi: 10.1097/00002030-200109070-00001. [DOI] [PubMed] [Google Scholar]
- 5.Sheryl L, Lard KLH, David Feigal. Considerations in the Development of Vaginal Products Intended to Prevent the Sexual Transmission of HIV. 1994 [Google Scholar]
- 6.Elias CJ, Coggins C. Female-controlled methods to prevent sexual transmission of HIV. AIDS. 1996;10(Suppl 3):S43–S51. [PubMed] [Google Scholar]
- 7.Darroch JE, Frost JJ. Women’s interest in vaginal microbicides. Fam Plann Perspect. 1999;31:16–23. [PubMed] [Google Scholar]
- 8.McGowan I. Microbicides: A new frontier in HIV prevention. Biologicals. 2006;34:241–255. doi: 10.1016/j.biologicals.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Maguire R. Microbicide product development. Curr Opin HIV AIDS. 2008;3:554–557. doi: 10.1097/COH.0b013e32830891e2. [DOI] [PubMed] [Google Scholar]
- 10.Rohan LC, Sassi AB. Vaginal drug delivery systems for HIV prevention. AAPS J. 2009;11:78–87. doi: 10.1208/s12248-009-9082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison PF, Rosenberg Z, Bowcut J. Topical microbicides for disease prevention: Status and challenges. Clin Infect Dis. 2003;36:1290–1294. doi: 10.1086/374834. [DOI] [PubMed] [Google Scholar]
- 12.Isaacs CE. The antimicrobial function of milk lipids. Adv Nutr Res. 2001;10:271–285. doi: 10.1007/978-1-4615-0661-4_13. [DOI] [PubMed] [Google Scholar]
- 13.Isaacs CE, Thormar H. The role of milk-derived antimicrobial lipids as antiviral and antibacterial agents. Adv Exp Med Biol. 1991;310:159–165. doi: 10.1007/978-1-4615-3838-7_19. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Estes JD, Schlievert PM, Duan L, Brosnahan AJ, Southern PJ, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458:1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strandberg KL, Peterson ML, Lin YC, Pack MC, Chase DJ, Schlievert PM. Glycerol monolaurate inhibits Candida and Gardnerella vaginalis in vitro and in vivo but not Lactobacillus. Antimicrob Agents Chemother. 2010;54:597–601. doi: 10.1128/AAC.01151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skinner MC, Kiselev AO, Isaacs CE, Mietzner TA, Montelaro RC, Lampe MF. Evaluation of WLBU2 peptide and 3-O-octyl-sn-glycerol lipid as active ingredients for a topical microbicide formulation targeting Chlamydia trachomatis. Antimicrob Agents Chemother. 2010;54:627–636. doi: 10.1128/AAC.00635-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moncla BJ, Pryke K, Isaacs CE. Killing of Neisseria gonorrhoeae,Streptococcus agalactiae (group B streptococcus), Haemophilus ducreyi, and vaginal Lactobacillus by 3-O-octyl-sn-glycerol. Antimicrob Agents Chemother. 2008;52:1577–1579. doi: 10.1128/AAC.01023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chamilos G, Luna M, Lewis RE, Chemaly R, Raad II, Kontoyiannis DP. Effects of liposomal amphotericin B versus an amphotericin B lipid complex on liver histopathology in patients with hematologic malignancies and invasive fungal infections: A retrospective, nonrandomized autopsy study. Clin Ther. 2007;29:1980–1986. doi: 10.1016/j.clinthera.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 19.Vyas SP, Quraishi S, Gupta S, Jaganathan KS. Aerosolized liposome-based delivery of amphotericin B to alveolar macrophages. Int J Pharm. 2005;296:12–25. doi: 10.1016/j.ijpharm.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Patel NA, Patel NJ, Patel RP. Design and evaluation of transdermal drug delivery system for curcumin as an anti-inflammatory drug. Drug Dev Ind Pharm. 2009;35:234–242. doi: 10.1080/03639040802266782. [DOI] [PubMed] [Google Scholar]
- 21.Hong RL, Tseng YL. Phase I and pharmacokinetic study of a stable, polyethylene-glycolated liposomal doxorubicin in patients with solid tumors: The relation between pharmacokinetic property and toxicity. Cancer. 2001;91:1826–1833. [PubMed] [Google Scholar]
- 22.Panwar P, Pandey B, Lakhera PC, Singh KP. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int J Nanomedicine. 2010;5:101–108. doi: 10.2147/ijn.s8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han HD, Lee A, Hwang T, Song CK, Seong H, Hyun J, et al. Enhanced circulation time and antitumor activity of doxorubicin by comblike polymer-incorporated liposomes. J Control Release. 2007;120:161–168. doi: 10.1016/j.jconrel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 24.Pollock S, Nichita NB, Böhmer A, Radulescu C, Dwek RA, Zitzmann N. Polyunsaturated liposomes are antiviral against hepatitis B and C viruses and HIV by decreasing cholesterol levels in infected cells. Proc Natl Acad Sci USA. 2010;107:17176–17181. doi: 10.1073/pnas.1009445107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malavia NK, Zurakowski D, Schroeder A, Princiotto AM, Laury AR, Barash HE, et al. Liposomes for HIV prophylaxis. Biomaterials. 2011;32:8663–8668. doi: 10.1016/j.biomaterials.2011.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zidan AS, Spinks C, Fortunak J, Habib M, Khan MA. Near-infrared investigations of novel anti-HIV tenofovir liposomes. AAPS J. 2010;12:202–214. doi: 10.1208/s12248-010-9177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker MT, Heriot W. Method and Apparatus for Liposome Production. USA patent US99/26738. 1999 [Google Scholar]
- 28.Patton DL, Cosgrove-Sweeney YT, Rabe LK, Hillier SL. The pig-tailed macaque rectal model: Microflora and chlamydial infection. Sex Transm Dis. 2001;28:363–366. doi: 10.1097/00007435-200107000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Patton DL, Sweeney YT, Paul KJ. A summary of preclinical topical microbicide rectal safety and efficacy evaluations in a pigtailed macaque model. Sex Transm Dis. 2009;36:350–356. doi: 10.1097/OLQ.0b013e318195c31a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isaacs CE, Thormar H, Pessolano T. Membrane-disruptive effect of human milk: Inactivation of enveloped viruses. J Infect Dis. 1986;154:966–971. doi: 10.1093/infdis/154.6.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reed LJ, Muench M. A simple method of estimating 50 percent end points. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 32.Rohan LC, Ratner D, McCullough K, Hiller SL, Gupta P. Measurement of anti-HIV activity of marketed vaginal products and excipients using a PBMC-based in vitro assay. Sex Transm Dis. 2004;31:143–148. doi: 10.1097/01.olq.0000114655.79109.ed. [DOI] [PubMed] [Google Scholar]
- 33.Moncla BJ, Hillier SL. Why nonoxynol-9 may have failed to prevent acquisition of Neisseria gonorrhoeae in clinical trials. Sex Transm Dis. 2005;32:491–494. doi: 10.1097/01.olq.0000170444.13666.e9. [DOI] [PubMed] [Google Scholar]
- 34.Food and Drug Administration (FDA) US Department of Health and Human Services Guidance for industry:Nonsterile semisolid dosage forms. Scale-Up and postapproval changes: Chemistry manufacturing, and controls; In vitro release testing and in vivo bioequivalence documentation. 1997 Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070930.pdf.Accessedon21January2011.
- 35.Wang L, Schnaare RL, Dezzutti C, Anton PA, Rohan LC. Rectal microbicides: Clinically relevant approach to the design of rectal specific placebo formulations. AIDS Res Ther. 2011;8:12. doi: 10.1186/1742-6405-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halpern V, Ogunsola F, Obunge O, Wang CH, Onyejepu N, Oduyebo O, et al. Effectiveness of cellulose sulfate vaginal gel for the prevention of HIV infection: Results of a Phase III trial in Nigeria. PLoS ONE. 2008;3:e3784. doi: 10.1371/journal.pone.0003784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Damme L, Govinden R, Mirembe FM, Guédou F, Solomon S, Becker ML, et al. CS Study Group Lack of effectiveness of cellulose sulfate gel for the prevention of vaginal HIV transmission. N Engl J Med. 2008;359:463–472. doi: 10.1056/NEJMoa0707957. [DOI] [PubMed] [Google Scholar]
- 38.Abdool Karim SS, Richardson BA, Ramjee G, Hoffman IF, Chirenje ZM, Taha T, et al. HIV Prevention Trials Network (HPTN) 035 Study Team Safety and effectiveness of BufferGel and 0.5% PRO2000 gel for the prevention of HIV infection in women. AIDS. 2011;25:957–966. doi: 10.1097/QAD.0b013e32834541d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCormack S, Ramjee G, Kamali A, Rees H, Crook AM, Gafos M, et al. PRO2000 vaginal gel for prevention of HIV-1 infection (Microbicides Development Programme 301): A phase 3, randomised, double-blind, parallel-group trial. Lancet. 2010;376:1329–1337. doi: 10.1016/S0140-6736(10)61086-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Center for the AIDS Programme of research in South Africa (CAPRISA) Study of microbicide gel shows reduced risk of HIV & Herpes infection in women; XVIII International AIDS conference; Viennna, Austria. 2010. [Google Scholar]
- 41.Caron M, Besson G, Etenna SL, Mintsa-Ndong A, Mourtas S, Radaelli A, et al. Protective properties of non-nucleoside reverse transcriptase inhibitor (MC1220) incorporated into liposome against intravaginal challenge of Rhesus macaques with RT-SHIV. Virology. 2010;405:225–233. doi: 10.1016/j.virol.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Isaacs CE, Jia JH, Xu W. A lipid-peptide microbicide inactivates herpes simplex virus. Antimicrob Agents Chemother. 2004;48:3182–3184. doi: 10.1128/AAC.48.8.3182-3184.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavelic Z, Skalko-Basnet N, Jalsenjak I. Characterisation and in vitro evaluation of bioadhesive liposome gels for local therapy of vaginitis. Int J Pharm. 2005;301:140–148. doi: 10.1016/j.ijpharm.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 44.Karn PR, Vanic Z, Pepic I, Skalko-Basnet N. Mucoadhesive liposomal delivery systems: The choice of coating material. Drug Dev Ind Pharm. 2011;37:482–488. doi: 10.3109/03639045.2010.523425. [DOI] [PubMed] [Google Scholar]
- 45.Chen J, Ping QN, Guo JX, Chu XZ, Song MM. Effect of phospholipid composition on characterization of liposomes containing 9-nitrocamptothecin. Drug Dev Ind Pharm. 2006;32:719–726. doi: 10.1080/03639040500529077. [DOI] [PubMed] [Google Scholar]
- 46.Mehanna MM, Elmaradny HA, Samaha MW. Mucoadhesive liposomes as ocular delivery system: Physical, microbiological, and in vivo assessment. Drug Dev Ind Pharm. 2009;36:108–118. doi: 10.3109/03639040903099751. [DOI] [PubMed] [Google Scholar]
- 47.Qin J, Chen D, Lu W, Xu H, Yan C, Hu H, et al. Preparation, characterization, and evaluation of liposomal ferulic acid in vitro and in vivo. Drug Dev Ind Pharm. 2008;34:602–608. doi: 10.1080/03639040701833559. [DOI] [PubMed] [Google Scholar]
- 48.Garg T, Jain S, Singh HP, Sharma A, Tiwary AK. Elastic liposomal formulation for sustained delivery of antimigraine drug: In vitro characterization and biological evaluation. Drug Dev Ind Pharm. 2008;34:1100–1110. doi: 10.1080/03639040801965079. [DOI] [PubMed] [Google Scholar]
- 49.Lu XY, Hu S, Jin Y, Qiu LY. Application of liposome encapsulation technique to improve anti-carcinoma effect of resveratrol. Drug Dev Ind Pharm. 2011 doi: 10.3109/03639045.2011.602410. DOI: 10.3109/03639045.2011.602410. [DOI] [PubMed] [Google Scholar]
- 50.Ramana LN, Sethuraman S, Ranga U, Krishnan UM. Development of a liposomal nanodelivery system for nevirapine. J Biomed Sci. 2010;17:57. doi: 10.1186/1423-0127-17-57. [DOI] [PMC free article] [PubMed] [Google Scholar]