

Abstract
Background
Malignant melanoma has an increasing incidence rate and the metastatic disease is notoriously resistant to standard chemotherapy. Loss of cell cycle checkpoints is frequently found in many cancer types and makes the cells reliant on compensatory mechanisms to control progression. This feature may be exploited in therapy, and kinases involved in checkpoint regulation, such as Wee1 and Chk1/2, have thus become attractive therapeutic targets.
Methods
In the present study we combined a Wee1 inhibitor (MK1775) with Chk1/2 inhibitor (AZD7762) in malignant melanoma cell lines grown in vitro (2D and 3D cultures) and in xenografts models.
Results
Our in vitro studies showed that combined inhibition of Wee1 and Chk1/2 synergistically decreased viability and increased apoptosis (cleavage of caspase 3 and PARP), which may be explained by accumulation of DNA-damage (increased expression of γ-H2A.X) - and premature mitosis of S-phase cells. Compared to either inhibitor used as single agents, combined treatment reduced spheroid growth and led to greater tumour growth inhibition in melanoma xenografts.
Conclusions
These data provide a rationale for further evaluation of the combination of Wee1 and Chk1/2 inhibitors in malignant melanoma.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-015-1474-8) contains supplementary material, which is available to authorized users.
Keywords: Malignant melanoma, Wee1, MK1775, Chk1/2, AZD7762, Cancer therapy, Cell cycle inhibitors
Background
Malignant melanoma is the deadliest form of skin cancer, in addition to having one of the most increasing incidence rates of all cancer forms [1]. Although curable by surgical excision at an early stage, patients diagnosed with metastatic melanoma (stage IV) have had a median survival of 6–10 months [2]. Despite recent year’s advances leading to new treatment options such as Ipilimumab, Vemurafenib, Trametinib and Dabrafenib, there are still no curative treatment alternatives for the majority of the patients with advanced disease [3, 4].
In response to DNA-damage, the dividing cell is arrested through activation of checkpoint mechanisms in order to allow time for DNA-repair to be completed. If the damage is too severe, apoptosis or senescence is induced to ensure that unrepaired DNA-damage is not passed on to future generations of cells. Loss of checkpoint mechanisms is frequently found in cancer, a trait which can be exploited in cancer therapy. Wee1 is a kinase involved in checkpoint regulation that in response to DNA-damage or replication stress can halt the cell cycle progression in S- and G2 phases by adding inhibitory phosphorylations (Tyr15) on cyclin-dependent kinases CDK2 and CDK1, respectively [5]. In our previous study we found that Wee1 was up-regulated in human melanomas as compared to benign nevi, and that high expression of Wee1 was associated with poor disease-free survival [6]. Likewise, over-expression of Wee1 (protein and/or mRNA) has been reported in osteosarcoma, glioblastoma and ovarian- and vulvar squamous cell carcinomas, thus emphasizing its potential as a therapeutic target in cancer [6–10]. Furthermore, targeting Wee1, either by siRNA mediated silencing or inhibitors, has in several studies, including ours, been shown to lead to increased DNA-damage and apoptosis [6, 11, 12].
Although mono-targeting of Wee1 has shown anti-tumour effect in some cancer cell lines, a stronger effect has been observed when combining Wee1 inhibitors with for instance DNA-damaging agents, Heath Shock Protein 90 inhibitors and more recently inhibitors of other cell cycle regulatory proteins such as Chk1/2 [13–16]. The Chk1/2 kinases are key regulators of DNA-damage surveillance pathways and DNA repair. Chk2 is a protein that is stably expressed throughout the cell cycle and is activated in response to DNA-damage; in particular DNA double-strand breaks through the ATM-Chk2-p53-p21 pathway. The expression of Chk1, on the other hand, is primarily found in S- and G2 phases, where it is active even in the unperturbed cell cycle. During normal cell cycle progression, Chk1 has been shown to regulate replication forks during DNA replication and entry into mitosis. Chk1 activation in response to DNA-damage is preferentially triggered through the ATR-Chk1-CDC25 pathway, although some cross-talk between Chk1 and ATM has been reported (reviewed in [17]. Chk1 has been shown to activate Wee1 in Xenopus extracts and yeast; however such a relationship has not been recapitulated in higher eukaryotes [18, 19]. Previous studies have shown an advantageous effect of combining Wee1 and Chk1/2 inhibitors as compared to mono-targeting in a variety of cancer cell lines, and points to complementary functions of the kinases [13, 20, 21].
In the present study we investigated the combined use of the Wee1 inhibitor MK1775 with a Chk1/2 inhibitor (AZD7762) in a panel of metastatic melanoma cell lines. While both inhibitors had an effect as mono-agents, combined administration gave a stronger anti-tumour effect both in vitro and in xenografts models. Co-treatment led to increased dephoshorylation of CDK1, DNA-damage, premature mitosis and apoptosis. In summary, our results warrant further evaluation of combined use of Wee1 and Chk1/2 inhibition in malignant melanoma.
Methods
Cell lines and growth conditions
The human metastatic melanoma cell lines WM239, WM45.1, WM983B and WM1366 were kindly provided by Prof. Meenhard Herlyn (the Wistar institute, Philadelphia, USA) [22, 23]. The FEMX-1 cell line was established at the Radium hospital [24]. The ‘Patient 3’ cell line was a kind gift from Prof. Peter Hersey (Royal North Shore Hospital, Sydney, Australia) [25]. All cell lines were maintained in RPMI-1640 medium (LONZA, Verviers, Belgium) supplemented with 5 % Fetal Calf Serum (Biochrom, KG, Berlin, Germany) and 2 mM L-glutamine (LONZA, Verviers, Belgium). The cells were grown in culture at 37 °C in humidified conditions containing 5 % CO2, either as monolayer cultures in 75 cm2 bottles or in 96 flat-bottom well plates. Normal human melanocytes (FOMA4) and fibroblasts (FF144sc) were isolated from human foreskin and cultured in 254CF (Invitrogen corporation, CA, USA) and DMEM 10 % FBS medium, respectively, as previously described [6].
Spheroids were generated by plating suspended cells (500–4000 cells/well, dependent on the cell line) in Corning® 96 Well Clear Round Bottom Ultra Low Attachment Microplates (Corning, MA, USA). Spheroid formation was allowed for 3 days prior to treatment. Images of spheroids were obtained using phase-contrast on an Olympus IX81 microscope with a 4× objective. Spheroid volume was calculated using Olympus Soft Imaging Solution Gm6H software. A minimum of two independent biological experiments were conducted, where each experiment contained at least four parallels of the individual treatment options.
Chemical inhibitors
Wee1 inhibitor MK1775 and Chk1/2 inhibitor AZD7762 were purchased from Selleck Chemicals (TX, USA) and used for time intervals and concentration indicated in the text.
Small interfering RNA (siRNA) transfection
All cell lines were plated in either 6-well plates (1.5 × 105 cells/well) or in 96-well plates (5 × 103 cells/well) 24 h in advance of the transfection. The cells were transfected with 10nM siRNA targeting Wee1 (OligioID; ‘VHS50841’), Chk1 (OligioID: ‘VHS40226’) or RNAi negative control duplexes (Negative Control LOW GC, 12935–200) using Lipofectamine™ RNAiMAX transfection reagents (all reagents from Invitrogen corporation, CA, USA). Transfection was allowed for 5 h before the medium was replaced with RPMI w/5 % FCS and 2 mM L-glutamine. Transfected cells were harvested after 48 h for further analysis.
Viability assays
Four thousand cells per well were seeded in 96-well plates and left to attach overnight, before treatment with MK1775 and/or AZD7762 for 48 h. The growth inhibitory effects of mono- and combined treatments were measured using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay (Promega, WI, USA). Absorbance was measured at 490 nm using ASYS UVM340 96-well plate reader. Alternatively, viability was assessed using the CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega) following the manufacturer's protocol. Luminescence was measured using GloMax® Luminometer (Promega). Viability of treated cells was normalized to the untreated control cells. Each experiment was performed with three parallel observations and repeated at least three times.
Calcusyn analysis
Synergy was determined by the Chou and Talalay Combination Index (C.I.) [26] for non-exclusive treatments (treatments affecting different targets or sites of the same target), and calculated by Calcusyn software (BioSoft, Feruson, MO, USA). Of note, this method requires that a dose effect curve for each drug is made, in which the data-points give a good r-value (>0.90 for cell systems) [27]. Given the variation in dose effect of the drugs in the different cell lines, the concentrations of the inhibitors were adjusted for the individual cell lines (Additional file 1: Figure S1) in order to abide to the requirements of the method.
Western blot analysis
Cells were harvested and western blot conducted as previously described [6]. Caspase 3 (#9662/#9664 (even mix)), Caspase 8 (#9746), Caspase 9 (#9502), PARP (#9532) and Wee1 (#4936S) primary antibodies were purchased from Cell Signaling (Beverly, MA). α-tubulin (DMIB) was acquired from Calbiochem (Nottingham, UK), whereas Cyclin A (sc-751) and p53 (sc-126) antibodies were obtained from Santa Cruz (Santa Cruz, CA). γ-H2AX (#05-636) and pCDK1Tyr15 (ab47594) antibodies were acquired from Millipore and Abcam (Cambridge, England), respectively. Following primary hybridization, membranes were washed 3 × 10 min in TBST and hybridized with an appropriate secondary antibody (HPR-conjugated anti-rabbit or anti-mouse IgG antibodies (Promega)) for 1 h at room temperature, with gentle agitation. Membranes were then washed in TBST for 3 × 10 min before being incubated in ECL-plus (GE Healthcare, Chalfont St Gils, UK) for 5 min. Protein bands were visualized by exposing the membranes to X-ray films.
Flow cytometry and barcoding
Cells were harvested by trypzination and fixated in 70 % ice-cold ethanol at −20 °C for a minimum of 24 h. In order to eliminate variation in antibody staining, control as well as treated cells were labelled with different concentrations of Pacific Blue (0.125, 0.031, 0.0062 and 0.00078 ng/μL, respectively) for 30 min at RT in the dark, in accordance with the fluorescent cell barcoding (FCB) technique [28]. All samples were subsequently mixed in one tube and incubated with primary antibodies (mouse anti-γH2AX (1:500, 05–636, Millipore) and rabbit anti-phospho Histone H3 (ser10) (1:500, CS#9701, Cell Signaling)) diluted in detergent buffer (0.1 % Igepal CA-630, 6.5 mM Na2HPO4, 1.5 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, 0.5 mM EDTA [pH 7.5]) containing 4 % nonfat milk for 1 h at RT, and afterwards with secondary antibodies; anti-rabbit Alexa488 and anti-mouse Dyelight549 (1:1000, Invitrogen) for 30 min at RT in the dark. Cells were finally incubated with PBS containing Cell Cycle 633/Fx cycle™ far red stain (1 μL/mL,) and PureLink RNAse A (5 μL/mL) (Invitrogen) for 30 min at 4 °C in the dark. Flow cytometric analysis was performed using a LSRII flow cytometer (BD Biosciences) with Diva software, and the four samples were gated based on the Pacific Blue signal before analyzes of cell cycle distribution, γH2AX and phospho Histone H3 (ser10) expression.
In vivo studies
‘Patient 3’ cells (2 × 106) were subcutaneously injected on each side of the dorsa of nude female mice (athymic nude foxn1 nu) and tumour bearing mice were subsequently divided into groups of 7 mice each. MK1775 and AZD7762 were dissolved and administered as recommended by the manufacturer (Selleck Chemicals). Briefly, 30 mg/kg MK1775 were given orally whereas 25 mg/kg AZD7762 were administered intravenously both as single agents and in combination. The treatments were given 3 times a week for 2 weeks. Control mice received no treatment as we previously have observed no effect of the vehicles alone. Tumour sizes were measured regularly using a calliper, and the volume V was calculated as follows: V = W2 × L × 0.5 (where W and L are tumour width and length, respectively). The experimental protocol was evaluated and approved by the National Animal Research Authority and conducted in accordance with regulations of the European Laboratory Animals Science Association.
Statistical analysis
All statistical analyzes were conducted using SPPS PASWStatistics version 18. Comparison of tumour volume at day 48 was performed with one-way between-groups ANOVA. Post-hoc comparisons were performed using the Tukey HSD tests.
Results
Synergistic effect of combined treatment with Wee1 inhibitor MK1775 and Chk1/2 inhibitor AZD7762 in melanomas
In order to study the effects of targeting Wee1 and Chk1/2, a panel of six metastatic melanoma cell lines were treated with increasing concentrations (0,063 μM to 1 μM) of the commercially available inhibitors MK1775 (Wee1) and AZD7762 (Chk1/2) for 48 h (Fig. 1a and b). Mono-treatment with either inhibitor reduced the viability in all tested cell lines, though the effect differed in a dose- and cell line dependent manner. The most pronounced effect was observed in WM45.1 (IC50: 0.4 μM (MK1775), 0.15 μM (AZD7762)) and WM983B (IC50: 0.35 μM (MK1775), 0.04 μM (AZD7762)) cells. In the other cell lines neither MK1775 nor AZD7762 were able to reduce the viability by 50 % following treatment with concentrations up to 1 μM of either drug (Fig. 1a and b).
Fig. 1.
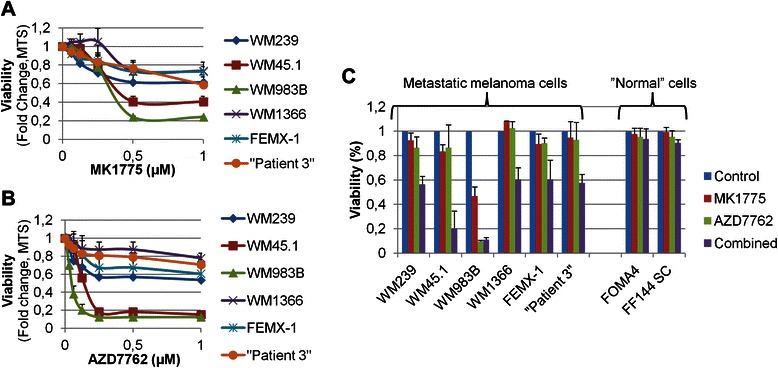
Increased anti-tumour effect of combined inhibition of Wee1 and Chk1/2. a and b, Melanoma cell lines grown as monolayer cultures were treated with increasing concentrations (0.063–1 μM) of MK1775 (a) or AZD7762 (b) for 48 h, and viability was measured by MTS analysis. c. Metastatic melanoma cell lines and primary cultures of human melanocytes (FOMA4) and fibroblasts (FF144 SC) were exposed to 200nM MK1775 and/or 100nM AZD7762 for 48 h. Cell viability was determined by MTS analysis. Data presented are the mean of at least three independent biological experiments and presented with positive standard deviation
We next investigated if combining the drugs could further decrease the cell viability as compared to single-agent treatments. The concentrations of the inhibitors used for combinational studies were chosen to be within the target-specific range of the compounds [29, 30]. While exposure to either 200nM MK1775 or 100nM AZD7762 for 48 h only led to a modest reduction, co-treatment resulted in a marked reduction in cell viability (Fig. 1c). For WM983B cells, however, 100nM AZD7762 alone decreased the viability by 90 % and co-treatment with MK775 did not enhance the effect. When reducing the AZD7762 concentration, combined treatment with MK1775 was superior to single-agent treatment also in this cell line (Additional file 1: Figure S1). Of note, primary cultures of melanocytes (FOMA4) and fibroblasts (FF144SC) were only modestly affected when exposed to either mono- or combinational treatment (Fig. 1c). Finally, in order to verify that the effect of the inhibitors could be assigned to targeting Wee1 or Chk1 we performed siRNA mediated knockdown of the proteins in three of the cell lines (WM239, WM45.1, WM983B) (Additional file 2: Figure S2A and 2B).
To further address the anti-tumour effect of combining the two inhibitors, drug interaction was determined using the Chou-Talalay method for quantifying synergy or antagonism [26]. Co-treatment with different concentrations of MK1775 and AZD7762 resulted in Combination Index (C.I.) values of less than 1 in the majority of cases, indicating a synergistic interaction of the drugs (Table 1 and Additional file 1: Figure S1).
Table 1.
The Combination Index (C.I.) values after combined treatment with MK1775 and AZD7762
| Cell line | Average C.I. value* | Standard deviation |
|---|---|---|
| WM239 | 0.3 | ±0.1 |
| WM45.1 | 0.9 | ±0.3 |
| WM983B | 0.7 | ±0.4 |
| WM1366 | 0.6 | ±0.4 |
| FEMX-1 | 0.4 | ±0.1 |
| “Patient 3” | 1.0 | ±0.2 |
*C.I. values are estimated from the dose response effects presented in Additional file 1: Figure S1A and 1B
Together, our results demonstrate an increased anti-tumour effect of co-targeting Wee1 and Chk1/2 in melanoma cell lines.
Combined inhibition of Wee1 and Chk1/2 leads to reduced viability and irreversibly inhibits growth of melanoma 3D-cultures
Three-dimensional tumour cell cultures have in several studies been reported as superior to monolayer cultures in terms of reflecting in vivo conditions for testing drug delivery systems and efficacy [31–33]. Hence, our next aim was to study long-term effects of MK1775 and AZD7762 on cells grown as spheroids (3D-cultures). Cells cultured as spheroids for 3 days were exposed to 200nM MK1775 and/or 100nM AZD7762 for a total of 3 days, and followed for an additional 8 days (Fig. 2a). During the latter period, 50 % of the medium was replaced twice with drug-free medium. Whereas spheroids given mono-treatments appeared diminished on day 14, combinational inhibition led to a marked decrease in spheroid size (Fig. 2b and d). Moreover, daily monitoring of growth revealed that WM45.1 and WM983B spheroids treated with the drug combination were unable to expand in size following drug-removal, thus indicating an irreversible effect within the time-frame of the experiment (Fig. 2d). The volume of the WM239 spheroids, however, continued to increase following treatment, but remained markedly reduced compared to the control. In consistence with this, cell viability estimated by CellTiterGlo assay on day 14 was greatly reduced in spheroids treated with the drug-combination in contrast to those in the control or mono-treatment groups (Fig. 2c). In accordance with our observation in 2D models, the decrease in viability following combinational treatment was more profound in WM45.1 and WM983B spheroids, as compared to WM239 cells. Together, our results indicate that combined use of MK1775 and AZD7762 has an increased anti-tumour effect compared to mono-treatment also in 3D cultures, and prevents spheroid growth even after drug-removal.
Fig. 2.
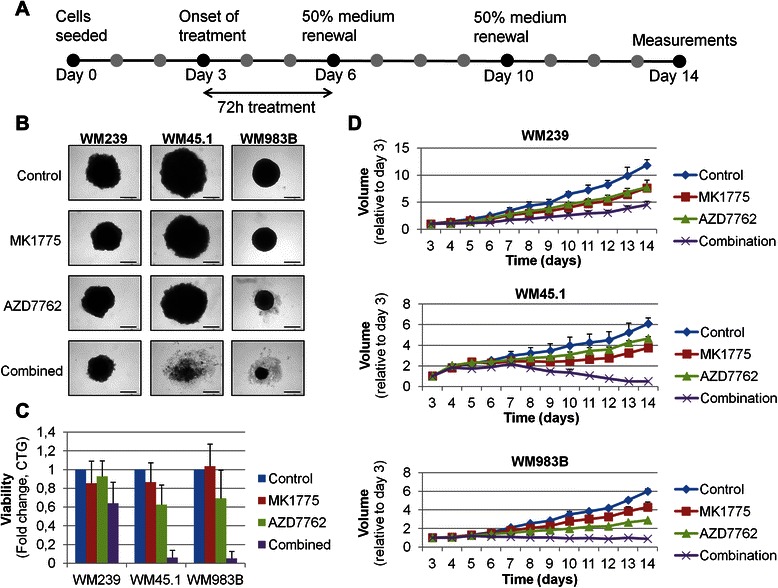
Cytotoxic effects of MK1775 and AZD7762 are not reversed by medium renewal in 3D cultures. a. Experiment outline; spheroid formation was allowed prior to treatment (day 0–3), inhibitors (MK1775: 200nM, AZD7762: 100nM) were added on day 3. On day 6 and 10 50 % of the medium was substituted by fresh medium containing no inhibitors. b. Spheroids were visually assessed on day 14 using light microscopy; scales in bottom right corners show 500 μm. c. Spheroid viability was estimated by CellTiterGlow analysis on day 14, and presented as the average of four individual experiments. d. Spheroid volume (Y-axis) was measured/estimated using Olympus Soft Imaging Solution Gm6H software. Changes in volume were estimated for each spheroid relative to its size on day 3. The graphs represent the average of two individual experiments presented with positive standard deviations
Combinatorial therapy reduces tumour growth in melanoma xenografts
Based on the promising in vitro results, we next investigated the in vivo efficacy of treatment with MK1775 and AZD7762 in melanoma xenografts. For this study we chose a cell line named ‘Patient 3’, that was recently established from a metastasis from a patient undergoing treatment with the selective BRAF inhibitor PLX4720, and previously DTIC [25]. Mono-treatment with either inhibitor resulted in reduction of tumour sizes as compared to the control group, and a tendency to a more prominent effect in the group given a combination of the two drugs was observed (Fig. 3). A statistically significant difference (p < 0.05) in tumour volume was found by ANOVA. Post-hoc comparisons using the Tukey HSD test indicated that the mean score of the control group (M = 14.47, SD = 2.87) was significantly different (p < 0.024) from the combination group (M = 6.47, SD = 3.88). No direct immediate toxic effects were observed during the experiment with the given dosages.
Fig. 3.
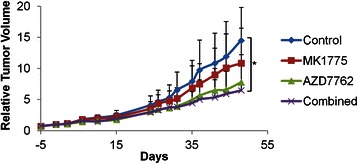
Combinatorial treatment with MK1775 and AZD7762 reduces tumour growth in melanoma xenografts. “Patient 3” xenografts were treated with MK1775 (30 mg/kg, three times per week, orally), AZD7762 (25 mg/kg, three times per week, i.v. injection) or a combination of theinhibitors, for two weeks (7 mice per group). The tumour volumes were measured twice a week using a caliper, and are presented as tumour volume relative to the volume of the tumour at the initiation of the treatment. Standard bars represent positive standard deviation. Comparison of tumour volume at day 48 was performed with one-way between groups ANOVA, and post-hoc comparisons with the Tukey HSD tests (*p < 0.024 untreated vs. combination group)
Inhibition of Wee1 and Chk1/2 lead to DNA-damage and apoptosis
To elucidate the mechanisms by which MK1775 and AZD7762 affect melanoma cell viability, we first examined the effect on Tyr15 phosphorylation of CDK1 in WM239, WM45.1 and WM983B cells after being exposed to 200nM MK1775 and/or 100nM AZD7762 for 48 h. Whereas Wee1 inhibition decreased pCDK1 expression in all cell lines, the combination of drugs resulted in an even stronger reduction, as assessed by western blot analysis (Fig. 4a). Down-regulation in pCDK1 expression following treatment was also observed after shorter exposure times (1, 3, 7 and 24 h, data not shown). Interestingly, treatment with the Chk1/2 inhibitor, alone or in combination with Wee1 inhibition, decreased the total level of CDK1 in WM983B cells. Similar patterns were observed following siRNA transfections targeting Wee1 and Chk1, although combinational treatments with siRNA had less effect than what was observed with the inhibitors (Additional file 2: Figure S2C).
Fig. 4.
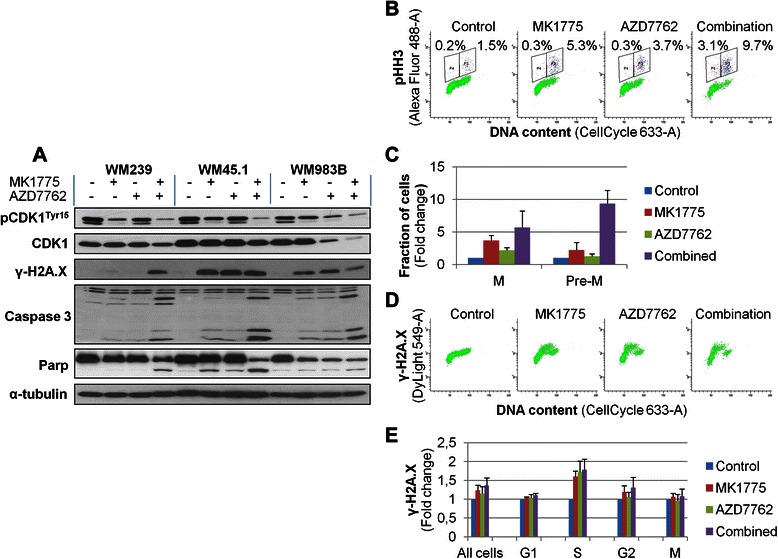
Combined inhibition of Wee1 and Chk1/2 leads to increased mitotic entry and DNA-damage in S-phase cells. a. Cells were treated with MK1775 (200nM) and/or AZD7762 (100nM) for 48 h. Expression of proteins involved in DNA-damage response and apoptosis was determined by western blot analysis, and α-tubulin was used as loading control. b, c, d and e. WM983B cells were treated with either DMSO (control) or high concentrations of MK1775 (2 μM) and/or AZD7762 (1 μM) for 3 h, and analyzed by Flow Cytometry B. Subpopulation gating of mitotic (blue) and pre-mitotic (purple) cells were based on their DNA content and expression of pHH3. The percentages of cells are indicated above the gated populations. c. The percentage of cells in mitosis (M) and pre-mitosis (Pre-M) are shown as fold change from the control cells. d. Expression of γ-H2A.X, indicative of DNA-damage, was assessed following treatment with inhibitors. e. The median expression of γ-H2A.X was determined for cells in the indicated phases of the cell cycle (for gating see Additional file 3: Figure S3), and shown as fold change from the control cells. Data are representative or average of four independent experiments
Furthermore, as seen in all three cell lines, both mono- and combinational treatment, as well as siRNA mediated knockdown, led to increased expression of γ-H2A.X, indicative of DNA double-strand breaks [34] (Fig. 4a and Additional file 3: Figure S3C). A further increase in γ-H2A.X was observed in WM239 cells following combined targeting of Wee1 and Chk1/2, as opposed to treatment with either inhibitor alone. Cleavages of Caspase 3 and PARP, associated with apoptosis, were observed in all three cell lines following treatment with the inhibitors. Combined inhibition led to a more pronounced cleavage product when compared to either single agent treatment, as assessed by immunoblotting (Fig. 4a). These findings indicate that simultaneous targeting of Wee1 and Chk1/2 leads to DNA-damage and apoptosis in melanoma cell lines.
Wee1 and Chk1/2 inhibitors induce premature mitosis and DNA damage in S-phase cells
To study how MK1775 and AZD7762 affect cells in different phases of the cell cycle, treatment with the respective drugs were allowed for three hours (Fig. 4b–e and Additional file 3: Figure S3). The short time frame serves as a compromise between allowing the drugs to work, while at the same time limiting cell cycle progression. Cells in different phases of the cell cycle were gated based on their relative amount of DNA and expression of the mitotic marker phospho-Histone H3 (pHH3), as analyzed by flow cytometry (Fig. 4b). Narrow gating for cells belonging to G1, S and G2 were used in order to avoid incorrect/uncertain assumptions of which cell cycle phase a cell belongs to (Additional file 3: Figure S3). Higher drug concentrations (2 μM MK1775/1 μM AZD7762) were used given the short exposure time, (Fig. 4b–e), however the same trends were also observed following treatment with lower doses of MK1775 (200nM) and AZD7762 (100nM) (Additional file 4: Figure S4). Whereas exposure to either inhibitor led to an increased amount of mitotic cells, this fraction was augmented by the drug combination. Moreover, combined treatment led to an increased fraction of S-phase cells co-expressing pHH3, indicative of cells having entered mitosis prematurely (Fig. 4b, c and Additional file 4: Figure S4).
To further study how the inhibitors affect cells in different phases of the cell cycle, we examined the median expression of γ-H2A.X in the gated sub-groups (Fig. 4c, (for gating of cells, see Additional file 3: Figure S3)) following short-time drug exposures. As seen in Fig. 4d and e, increased expression of γ-H2A.X was predominantly found in S-phase cells, indicating that cells undergoing DNA-replication are especially sensitive to DNA-damage. Our results therefore suggest that combined treatment with MK1775 and AZD7762 leads to premature mitosis and DNA-damage in S-phase cells.
Discussion
In our previous study we showed that high expression of Wee1 is associated with poor disease-free survival in patients with melanoma and that in vitro targeting of the kinase leads to decreased cell viability. To further evaluate Wee1 as a potential target in melanoma, we have in the present study combined a Wee1 inhibitor (MK1775) with a Chk1/2 inhibitor (AZD7762). Our hypothesis is that simultaneous inhibition of two cell cycle control proteins will introduce a high degree of DNA-damage incompatible with cell viability. The combined inhibition led to an increased anti-tumour effect both in vitro and in xenografts models. Furthermore, our in vitro studies showed that co-treatment synergistically decreased viability and increased apoptosis, which may be explained by DNA-damage- and premature mitosis of S-phase cells. Our results provide a rationale for further testing of the treatment regimen in melanoma models.
Mono-targeting of Wee1, either by siRNA mediated knockdown or inhibitors, has shown a potent anti-tumour effect in some cancer cell lines [6, 35] but only limited effect in other studies [7, 8]. In line with this, the efficacy of mono-targeting of Wee1 by MK1775 varied in the tested melanoma cell lines in the present study, where low concentrations (200nM) had little effect in some of the cell lines, such as WM1366. Interestingly, a stronger anti-tumour efficacy was observed in the same cell line following exposure to MK1775 in a recent study by Haarberg et al. [36], however the experimental conditions in our studies differ with regard to exposure time to the drug and assay for detecting viability.
For the purpose of increasing the efficacy, inhibition of Wee1 (MK1775) has been combined with other therapeutic agents in clinical trials, such as gemcitabine and cisplatin [ClinicalTrials.gov]. Simultaneous inhibition of Wee1 and Chk1, on the other hand, has only been investigated in preclinical studies, but has been found as an eligible combination following siRNA high-throughput screens of acute myeloid leukemia, lung-, prostate- and ovarian cancer [13, 20, 21]. Combined inhibition of Wee1 and Chk1 using commercial inhibitors have been shown to synergistically enhance the therapeutic efficacy in several cancer cell lines, including the A2058 melanoma cell line [13, 16, 20, 37]. In agreement with these reports, combined targeting of Wee1 and Chk1/2 led to a synergistic reduction of viability in our cohort of melanoma cell lines. In addition, the same trends were observed for combined siRNA mediated knockdown of Wee1 and Chk1 compared to mono-targeting of either protein. However, siRNA mediated down-regulation and use of small molecular inhibitors are not always comparable [21]. The experiments using siRNA only targeted Chk1, whereas AZD7762 inhibits both Chk1 and Chk2. Still, a previous study by McNeeley et al. showed that the anti-tumour activity of AZD7762 following DNA damage is likely related to Chk1- rather than Chk2 inhibition [38].
Similar to what was observed in monolayer cultures, an increased effect of combined targeting was also found in multicellular spheroids. Likewise, a weak increase in efficacy was observed in melanoma xenografts following combined treatment versus either mono-treatment alone. A previous study showed that combinational inhibition of Wee1 (MK1775) and Chk1 (MK8776) led to increased in vivo efficacy in neuroblastoma xenografts [39], providing additional support for our results.
Studies have indicated that the response to Chk1 or Wee1 inhibitors is dependent on the p53 mutational status of the cells, in particular when combined with DNA-damaging agents, while others have reported no such effect [40, 41]. Partly supporting the first hypothesis, the anti-tumour effect was most pronounced in the p53 mutated WM983B and WM45.1 cells, and less in the p53WT cell line WM239. Although disabling point mutations in TP53 are only found in 10 % of melanomas, inactivation of the protein is found in approximately 90 % of the tumours (reviewed in [42]), suggesting that the effect of checkpoint inhibition in melanoma is not strictly dependent on the p53 mutation status. Furthermore, the level of genetic instability has been suggested as a predictor of the response to Wee1 and Chk1/2 inhibition [43, 44]. In this regard, melanomas have been shown to be one of the most genetically unstable tumour forms [45, 46]. Combined inhibition of Wee1 and Chk1/2 in primary cultures of melanocytes or fibroblasts had less effect on viability than observed in the melanomas. In accordance with these results, Carrassa et al. found no synergistic effects of combining Wee1 (MK1775) and Chk1 inhibitors PF-00477736 (or allegedly AZD7762) in the normal fetal human lung cell line MRC-5 [20]. While both Wee1 and Chk1 are known to have functions in the unperturbed cell cycle, normal cells are less proliferative and have a lower degree of genetic alterations- and instability than tumour cells, which may explain the difference in response to kinase inhibition. However, phase I clinical trials with AZD7762 have been terminated due to cardiac toxicity in patients [47, 48], and also clinical trials with other Chk1 and Chk1/2 inhibitors (reviewed in [49]) have been terminated. Concerns have been raised on whether targeting Chk1 may lead to toxicity in normal cells due the multiplicity of functions of the protein in the unperturbed cell cycle (Reviewed in [50]). Furthermore, embryonic lethality is associated with Chk1 depletion in mice [51]. On the other hand, the observed toxicities in patients following treatment with various Chk1 inhibitors have appeared as drug-specific rather than class-specific and may thus be a consequence of off-target effects (reviewed in [49]). Clinical studies with novel Chk1 (MK-8876/SCH 900776) and Chk1/2 (LY2606368) inhibitors with improved specificity are currently ongoing or recruiting (ClinicalTrials.gov).
When further investigating the effect of combined inhibition of Wee1 and Chk1/2, we found a stronger reduction in pCDK1 (tyr15) levels compared to treatment with either drug alone. In line with this, Wee1 has been shown to directly phosphorylate CDKs, and removal of Wee1 is thus expected to reduce such phosphorylations [52]. Chk1, on the other hand, phosphorylates the CDC25 phosphatases, leading to the latter proteins sequestration or degradation, thereby hindering activating dephosphorylation of pCDK1/2 (tyr15) [53]. CDK1/2 phosphorylation is thus indirectly maintained by Chk1, and removal of the latter may promote further reduction of pCDK1/2. Inhibition of Wee1 and Chk1 has previously been shown by Davis et al. to be accompanied by decreased expression of pCDK1 and a synergistic reduction in cell viability [13]. They further showed that the antiproliferative effect of the inhibitors was partially reversed after CDK inhibition (Roscovitine), suggesting that CDK hyperactivity may be a contributing factor, rather than the sole cause of the antiproliferative effect of Chk1 and Wee1 inhibition. Deregulated activity of CDKs, as a consequence of Wee1 depletion, has in a previous study been shown to induce replication stress and loss of genomic integrity through subsequent nucleotide-shortage and increased firing of replication origins [54]. In line with this, we observed that inhibition of Wee1 led to increased levels of DNA-damage and apoptosis, as assessed by expression of γ-H2A.X and cleavage of caspase 3 and PARP, respectively. The effect on apoptosis was amplified by combining the inhibitors, supporting a synergistic effect on viability. A further increase in DNA-damage following combined targeting of the kinases was seen in WM239 cells. Similarly, combined inhibition of Wee1 and Chk1 has in a previous study been demonstrated to induce a more intense and durable DNA-damage and anti-tumour effect than either drug alone [37]. Interestingly, treatment with the Chk1/2 inhibitor or Chk1 siRNA reduced the total protein level of CDK1 in the WM983B cell line. In agreement with this, transcriptional reduction of cell-cycle regulators, such as CDK1, has been observed after Chk1 depletion in somatic cells [55].
Similar to what has been reported by others [37, 56, 57], inhibition of Wee1 as well as Chk1 was shown to induce DNA-damage in the S phase of the cell cycle. These findings are in accordance with the proposed function of the kinases in restraining CDK activity during DNA replication (reviewed in [58]). Furthermore, it has been suggested that the cytotoxic effect of Chk1 inhibition in melanoma cells is most likely due to inhibition of Chk1 in S phase, which drives cells prematurely from late S phase into an aberrant mitosis [43]. In line with this, combinational treatment with Chk1/2 and Wee1 inhibitors increased the population of S-phase cells co-expressing the mitotic marker, pHH3. These are cells that have entered mitosis without having completed replication, most likely due to the compromised G2/M checkpoint in the absence of Wee1 and/or Chk1. Likewise, an increased proportion of mitotic cells was observed following mono-targeting, and to a further extent by the combination of the inhibitors. Most likely the G2 cells have abrogated the G2/M checkpoint and entered mitosis given the short timeframe of this experiment (3 h).
Conclusions
In summary, our results support an increased anti-tumour effect of combined inhibition of Wee1 and Chk1/2 and provide a rationale for further evaluation of the kinases as therapeutic targets in human melanomas.
Acknowledgements
This work was supported by grants from the Southern and Eastern Norway Regional Health Authorities (Helse Sør-Øst) and The Cancer Society (Kreftforreningen). We also wish to acknowledge the technical assistance of Petros Gebregziabher, and Prof. Peter Hersey for letting us use his cell line (“patient 3”).
Additional files
Synergistic effect of combining Wee1 and Chk1/2 inhibitors. A . Cells were treated with three different concentrations of MK1775 (200, 400 and 800nM) and/or AZD7762 (50, 100 and 200nM) as indicated. Cell viability was measured by MTS after 48 h. B. The combination Index (C.I.) values were calculated by CalcuSyn software based on the Chou Talalay method for measuring drug interaction, for each combination of inhibitors C and D. Drug concentrations were adjusted for WM983B (MK1775; 50, 100 and 200nM and/or AZD7762 (20, 40 and 80nM) and WM1366 (MK1775; 1, 2 and 4 μM and/or AZD7762 (0.25, 0.5 and 1 μM) cells in order to abide to Chou Talalay method requirements. Data are the mean of three independent experiments and presented with error bars showing positive standard deviation.
Increased anti-tumour effect of SiRNA mediated knock-down of Wee1 and Chk1. A and B. Cell viability was measured 48 h following SiRNA mediated knockdown by MTS (A) and CellTiterGlow (B) assays. C. Expression of proteins involved in DNA-damage response and apoptosis was determined by western blot analysis, and α-tubulin used as loading control. Data are the mean or representative of three independent experiments.
Example of sub-gating of cells in different phases of the cell cycle. Different samples (Control, MK1775, AZD7762, Combination) were separated by their relative expression of Pacific blue (See Methods for barcoding procedure). Cells of different phases of the cell cycle were sub-gated based on DNA content and expression of the mitotic marker pHH3 (dark green: G1, Blue: S, orange: G2 and purple: Mitotic cells). Median expressions of γ-H2A.X were determined by Diva software.
Combined inhibition of Wee1 and Chk1/2 leads to increased mitotic entry and DNA-damage in S-phase cells. Cells were treated with indicated concentrations of MK1775 (MK) and or AZD7762 (AZD) for 3 h, barcoded and analyzed by flowcytometry. A. Percentage of cells in mitosis (M) and pre-mitosis (Pre-M) were calculated relative to the control. B. Median expression of γ-H2A.X in cells of different cell-cycle phases, relative to the control. Data are the mean of four independent experiments.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GIM contributed to design, acquiring and interpretation of data as well as drafting of the manuscript. EE, and RF aquired the data. KGF performed the animal experiment, BE contributed to design and interpretation of animal experiments, VN-K contributed to conception, design and interpretation of flow cytometry analyses. AA and VAF contributed to design, interpretation of data and manuscript revision. All authors have read and approved the final manuscript.
Contributor Information
Gry Irene Magnussen, Email: Gry.Magnussen@rr-research.no.
Elisabeth Emilsen, Email: Elisabeth.Emilsen@rr-research.no.
Karianne Giller Fleten, Email: Karianne.Giller.Fleten@rr-research.no.
Birgit Engesæter, Email: Birgit.Engesater@rr-research.no.
Viola Nähse-Kumpf, Email: Viola.Naehse-Kumpf@curie.fr.
Roar Fjær, Email: Roar.Fjar@medisin.uio.no.
Ana Slipicevic, Email: Ana.Slipicevic@rr-research.no.
Vivi Ann Flørenes, Phone: +47 229 34 529, Email: vivi.ann.florenes@radiumhospitalet.no.
References
- 1.Rigel DS. Epidemiology of melanoma. Semin Cutan Med Surg. 2010;29:204–9. doi: 10.1016/j.sder.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Leung AM, Hari DM, Morton DL. Surgery for distant melanoma metastasis. Cancer J. 2012;18:176–84. doi: 10.1097/PPO.0b013e31824bc981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–91. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magnussen GI, Holm R, Emilsen E, Rosnes AK, Slipicevic A, Florenes VA. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: potential for targeted therapy. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnussen GI, Hellesylt E, Nesland JM, Trope CG, Florenes VA, Holm R. High expression of wee1 is associated with malignancy in vulvar squamous cell carcinoma patients. BMC Cancer. 2013;13:288. doi: 10.1186/1471-2407-13-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18:244–57. doi: 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.PosthumaDeBoer J, Wurdinger T, Graat HC, van Beusechem V, Helder MN, van Royen BJ, et al. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11:156. doi: 10.1186/1471-2407-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slipicevic A, Holth A, Hellesylt E, Trope CG, Davidson B, Florenes VA. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol Oncol. 2014;135:118–24. doi: 10.1016/j.ygyno.2014.07.102. [DOI] [PubMed] [Google Scholar]
- 11.Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122:347–57. doi: 10.1007/s10549-009-0571-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajeshkumar NV, De OE, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumour regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, et al. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther. 2011;12:788–96. doi: 10.4161/cbt.12.9.17673. [DOI] [PubMed] [Google Scholar]
- 14.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumour efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–22. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 15.Lokeshwar VB. Wee1-Hsp90 inhibitor combination treatment: molecular therapy with potentially broad applicability. Cell Cycle. 2012;11:3722–3. doi: 10.4161/cc.22119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-Phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012;2:524–539. [DOI] [PubMed]
- 17.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/S1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 18.Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell. 2001;12:551–63. doi: 10.1091/mbc.12.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–54. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrassa L, Chila R, Lupi M, Ricci F, Celenza C, Mazzoletti M, et al. Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumour growth inhibition in vivo. Cell Cycle. 2012;11:2507–17. doi: 10.4161/cc.20899. [DOI] [PubMed] [Google Scholar]
- 21.Chaudhuri L, Vincelette ND, Koh BD, Naylor RM, Flatten KS, Peterson KL, et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica. 2014;99:688–96. doi: 10.3324/haematol.2013.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Florenes VA, Lu C, Bhattacharya N, Rak J, Sheehan C, Slingerland JM, et al. Interleukin-6 dependent induction of the cyclin dependent kinase inhibitor p21WAF1/CIP1 is lost during progression of human malignant melanoma. Oncogene. 1999;18:1023–32. doi: 10.1038/sj.onc.1202382. [DOI] [PubMed] [Google Scholar]
- 23.Herlyn M, Balaban G, Bennicelli J, Guerry D, Halaban R, Herlyn D, et al. Primary melanoma cells of the vertical growth phase: similarities to metastatic cells. J Natl Cancer Inst. 1985;74:283–9. [PubMed] [Google Scholar]
- 24.Fodstad O, Kjonniksen I, Aamdal S, Nesland JM, Boyd MR, Pihl A. Extrapulmonary, tissue-specific metastasis formation in nude mice injected with FEMX-I human melanoma cells. Cancer Res. 1988;48:4382–8. [PubMed] [Google Scholar]
- 25.Lai F, Jiang CC, Farrelly ML, Zhang XD, Hersey P. Evidence for upregulation of Bim and the splicing factor SRp55 in melanoma cells from patients treated with selective BRAF inhibitors. Melanoma Res. 2012;22:244–51. doi: 10.1097/CMR.0b013e328353eff2. [DOI] [PubMed] [Google Scholar]
- 26.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 27.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–81. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 28.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Methods. 2006;3:361–8. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- 29.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumour cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 30.Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7:2955–66. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 31.Chitcholtan K, Sykes PH, Evans JJ. The resistance of intracellular mediators to doxorubicin and cisplatin are distinct in 3D and 2D endometrial cancer. J Transl Med. 2012;10:38. doi: 10.1186/1479-5876-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta G, Hsiao AY, Ingram M, Luker GD, Takayama S. Opportunities and challenges for use of tumour spheroids as models to test drug delivery and efficacy. J Control Release. 2012;164:192–204. doi: 10.1016/j.jconrel.2012.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinci M, Gowan S, Boxall F, Patterson L, Zimmermann M, Court W, et al. Advances in establishment and analysis of three-dimensional tumour spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012;10:29. doi: 10.1186/1741-7007-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–86. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 35.Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumour activity against sarcoma cells. Mol Cancer Ther. 2012;11:174–82. doi: 10.1158/1535-7163.MCT-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haarberg HE, Paraiso KH, Wood E, Rebecca VW, Sondak VK, Koomen JM, et al. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Mol Cancer Ther. 2013;12:901–12. doi: 10.1158/1535-7163.MCT-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guertin AD, Martin MM, Roberts B, Hurd M, Qu X, Miselis NR, et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumour activity upon pharmacologic inhibition. Cancer Cell Int. 2012;12:45. doi: 10.1186/1475-2867-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McNeely S, Conti C, Sheikh T, Patel H, Zabludoff S, Pommier Y, et al. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9:995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 39.Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, et al. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013;73:776–84. doi: 10.1158/0008-5472.CAN-12-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res. 2014;20:4274–88. doi: 10.1158/1078-0432.CCR-13-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29:6149–59. doi: 10.1038/onc.2010.343. [DOI] [PubMed] [Google Scholar]
- 42.Box NF, Vukmer TO, Terzian T. Targeting p53 in melanoma. Pigment Cell Melanoma Res. 2014;27:8–10. doi: 10.1111/pcmr.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brooks K, Oakes V, Edwards B, Ranall M, Leo P, Pavey S, et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 2013;32:788–96. doi: 10.1038/onc.2012.72. [DOI] [PubMed] [Google Scholar]
- 44.Vriend LE, De Witt Hamer PC, Van Noorden CJ, Wurdinger T. WEE1 inhibition and genomic instability in cancer. Biochim Biophys Acta. 1836;2013:227–35. doi: 10.1016/j.bbcan.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Bastian BC, Olshen AB, LeBoit PE, Pinkel D. Classifying melanocytic tumours based on DNA copy number changes. Am J Pathol. 2003;163:1765–70. doi: 10.1016/S0002-9440(10)63536-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sausville E, Lorusso P, Carducci M, Carter J, Quinn MF, Malburg L, et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumours. Cancer Chemother Pharmacol. 2014;73:539–49. doi: 10.1007/s00280-014-2380-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seto T, Esaki T, Hirai F, Arita S, Nosaki K, Makiyama A, et al. Phase I, dose-escalation study of AZD7762 alone and in combination with gemcitabine in Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol. 2013;72:619–27. doi: 10.1007/s00280-013-2234-6. [DOI] [PubMed] [Google Scholar]
- 49.Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today. 2012;17:194–202. doi: 10.1016/j.drudis.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 50.Goto H, Izawa I, Li P, Inagaki M. Novel regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good candidate for anti-cancer therapy? Cancer Sci. 2012;103:1195–200. doi: 10.1111/j.1349-7006.2012.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000;14:1439–47. [PMC free article] [PubMed] [Google Scholar]
- 52.Nakanishi M, Ando H, Watanabe N, Kitamura K, Ito K, Okayama H, et al. Identification and characterization of human Wee1B, a new member of the Wee1 family of Cdk-inhibitory kinases. Genes Cells. 2000;5:839–47. doi: 10.1046/j.1365-2443.2000.00367.x. [DOI] [PubMed] [Google Scholar]
- 53.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 54.Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, et al. Cyclin-Dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol. 2012;32:4226–36. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, et al. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell. 2008;132:221–32. doi: 10.1016/j.cell.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 56.Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188:629–38. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005;25:3553–62. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sorensen CS, Syljuasen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nuck Acids Res. 2011;40:477–86. [DOI] [PMC free article] [PubMed]