

Abstract
Although the intracellular Cl−/H+ exchanger Clc-5 is expressed in apical intestinal endocytic compartments, its pathophysiological role in the gastrointestinal tract is unknown. In light of recent findings that CLC-5 is downregulated in active ulcerative colitis (UC), we tested the hypothesis that loss of CLC-5 modulates the immune response, thereby inducing susceptibility to UC. Acute dextran sulfate sodium (DSS) colitis was induced in Clcn5 knockout (KO) and wild-type (WT) mice. Colitis, monitored by disease activity index, histological activity index, and myeloperoxidase activity were significantly elevated in DSS-induced Clcn5 KO mice compared with those in WT mice. Comprehensive serum multiplex cytokine profiling demonstrated a heightened Th1–Th17 profile (increased TNF-α, IL-6, and IL-17) in DSS-induced Clcn5 KO mice compared with that in WT DSS colitis mice. Interestingly, Clcn5 KO mice maintained on a high vitamin D diet attenuated DSS-induced colitis. Immunofluorescence and Western blot analyses of colonic mucosa validated the systemic cytokine patterns and further revealed enhanced activation of the NF-κB pathway in DSS-induced Clcn5 KO mice compared with those in WT mice. Intriguingly, high baseline levels of IL-6 and phospho-IκB were observed in Clcn5 KO mice, suggesting a novel immunopathogenic role for the functional defects that result from the loss of Clc-5. Our studies demonstrate that the loss of Clc-5 1) exhibits IL-6–mediated immunopathogenesis, 2) significantly exacerbated DSS-induced colitis, which is influenced by dietary factors, including vitamin D, and 3) portrays distinct NF-κB–modulated Th1–Th17 immune dysregulation, implying a role for CLC-5 in the immunopathogenesis of UC.
Although characterized by distinct clinical and histopathological features, the etiology and pathogenesis of Crohn’s disease (CD) and ulcerative colitis (UC), the two major forms of inflammatory bowel disease (IBD), have not yet been fully defined (1, 2). The mucosal immune system is the central effector of intestinal inflammation, with inflammatory mediators, primarily cytokines, playing a central role in modulating innate and adaptive immune responses in IBD (3, 4). We and others have shown from studies of human patients and animal IBD models that both CD and UC have specific mucosal-damage pathways, characterized by the dysregulation of distinct Th1 and Th2 cytokine profiles at different stages of the disease process (4–6). These studies have demonstrated UC to be a prototypic Th2-type disorder (mediated by IL-4, IL-5, and IL-10) and CD to be primarily associated with Th1–Th17-type responses mediated by TNF-α, IL-12, IFN-γ, and IL-17 (4, 6, 7).
Genes associated with IBD are generally categorized into those affecting immune response and microbial recognition and those affecting ion and water transport (8, 9). Diarrhea (altered fluid transport) is one of the most prevalent symptoms in patients with IBD (10). Impaired colonic salt and water transport in IBD occur as a result of decreased Na+ absorption and increased Cl− secretion and have been described to be major pathogenic factors in IBD-associated diarrhea (11, 12). The dysregulation of several membrane transporters in different models have been linked to IBD-associated diarrhea, including Na+/K+ ATPase (13–15), the epithelial Na+ channel (16), Na+/H+ exchangers 1 and 3 (NHE1,3; in cell models only) (17, 18), and Na+/K+/2Cl− (13, 16). Recently, we demonstrated the coordinated downregulation of several Na+ transporters in sigmoid mucosal biopsies of patients with active IBD and mice with experimental colitis, including that of the chloride channel CLC-5, NHE1,3 (but not NHE2), epithelial Na+ channel, Na+/K+ ATPase, and Na+/H+ exchanger regulatory factor 1 (19).
The chloride channel CLC gene family encodes nine known isoforms in mammals, the mutations and/or disruptions of some of which have been shown to underlie human diseases and pathology, including Bartter syndrome (with or without deafness), Dent disease, lysosomal storage diseases, myotonia, blindness, male infertility, defective endocytosis, osteopetrosis, leukodystrophy, and neurodegeneration (20–25). One of these isoforms, a 746-aa protein CLC-5, encoded by the CLCN5 gene is a voltage-dependent Cl−/H+ exchanger (20, 26). Mutations in the CLCN5 gene are associated with X-linked renal tubulopathy of Dent disease, with functional defects in both patients and mouse that are characterized by low-m.w. proteinuria, aminoaciduria, glycosuria, phosphaturia, hypercalciuria, nephrolithiasis, and progressive renal failure (27–35). In general, CLC channels have been demonstrated to contribute to a host of biological and cellular processes, including cell migration, proliferation, and apoptosis (36). Only CLC-3 has been shown to play a critical role in TGF-β–induced apoptosis of human airway epithelial cells and has recently been shown to be involved in the recruitment and activation of immune cells in the respiratory tract (36, 37). However, the specific role of CLC-5 and/or cellular mediators that modulate important immune functions to trigger downstream signaling pathways has not yet been defined.
Identifying changes of CLC-5 and associated modulators in IBD may lead to a better understanding of the molecular causes for IBD-associated diarrhea. Because IFN-γ inhibits intestinal transport by downregulating Na+/K+ ATPase and Na+/K+/2Cl−, we and others previously suggested that IBD-associated inflammatory cytokines may play a role (6, 13). In experimental colitis models, we recently demonstrated that diarrhea was associated with significant elevation of various cytokines in colonic mucosa (6). In the acute dextran sulfate sodium (DSS) colitis model, Th1–Th2 cytokines (IL-6, IFN-γ, and IL-17) were increased, whereas in chronic colitis models IL-6 and IFN-γ (but not IL-12 p40/70 and IL-17) were elevated. Our studies also suggested that, although there are clear differences in the production of specific cytokines between DSS and trinitrobenzene sulfuric acid models of experimental colitis, these cytokine differences result in a similar clinical consequence: diarrhea (6). The cytokine network in IBD is a complex and dynamic system in which cellular and humoral cytokines, chemokines, and growth factors regulate the initiation and perpetuation of inflammation (4, 6). Given the significance of CLC-5 and that CLC-5 is downregulated in sigmoid mucosal biopsies of most patients with active UC (19), we tested the hypothesis that the loss of CLC-5 modulates the immune response, thereby inducing susceptibility to UC. Herein, we demonstrate that the loss of Clc-5 1) significantly exacerbates DSS-induced colitis, 2) is associated with elevated baseline levels of IL-6 and phospho-IκB, and 3) is influenced by dietary factors, such as vitamin D and NF-κB–mediated distinct Th1–Th17 immune dysregulation, implying the role of downregulated CLC-5 in the immunopathogenesis of UC.
Materials and Methods
Animals and diet
C57BL/6 Clcn5 knockout (KO) mice, created by deletion of exon VI of Clcn5 (35), and wild-type (WT) C57BL/6 age-matched adult male mice (6–8 wk old) were group-housed at Johns Hopkins Animal Facility under controlled temperature (25°C) and photoperiods (12:12 h light–dark cycle). Care and experimentation of mice were performed in accordance with institutional guidelines under protocols approved by the Institutional Animal Care and Use Committee. Mice were fed on Harlan Teklad Diet (H-diet; Harlan Laboratories, Madison, WI). In studies of dietary effects on DSS-induced colitis, subgroups of mice were also fed with NIH-31 modified open formula diet (Z-diet; Zeigler Brothers, Gardners, PA) or a vitamin D (4.18 IU/g)-supplemented H-diet (Vit D-enriched H-diet).
Abs
Abs used include: IL-6 mAb (Transduction Laboratories, Lexington, KY), IL-17 and NF-κB polyclonal Abs (pAbs) (Santa Cruz Biotechnology, Santa Cruz, CA), IL-12 p40/70 pAb (BioSource International, Camarillo, CA), phospho-IκB pAb (Abcam, Cambridge, MA), and actin pAb (Sigma-Aldrich, St. Louis, MO). Clc-5 pAb was obtained as previously described (35).
Induction of colitis
Acute colitis of C57BL/6 mice was induced by feeding mice (n ≥ 7 mice in each group) with 2.5% (w/v) DSS (molecular mass 40 kDa; ICN Biochemicals, Aurora, OH) as described previously (6).
Evaluation of colitis
Animals were observed twice daily for weight, water/food consumption, morbidity, stool consistency, piloerection, and the presence of gross blood in feces and at the anus. Disease activity index (DAI) was calculated as described previously (6). At day 7 following induction with DSS, animals were sacrificed by CO2 inhalation, rapidly dissected, and the entire colon was quickly excised, photographed, and gently cleared of feces with 4°C saline. Small segments of the colon taken for histopathology and immunohistochemistry were fixed in 10% normal buffered formalin as described previously (6). Sections (4 μm) were stained with H&E (Richard Allen Scientific, Kalamazoo, MI), histological scores were blindly determined with minor modifications from Obermeier et al. (38), and histological activity index (HAI) was calculated as described previously (6). The activity of the enzyme myeloperoxidase (MPO), a marker of polymorphonuclear neutrophil primary granules, was also determined in colonic mucosa as described previously (6, 39)
Isolation of colonic mucosa and extraction of proteins for SDS-PAGE and Western blot analysis
At 4°C, the mucosa was scraped from the muscle layer of the colon, and samples were snap frozen and stored at −80°C for the remaining experiments. Frozen tissue samples were homogenized in homogenization buffer (50 mM Tris-HCl [pH 7.2]) containing Na3VO4 and a protease-inhibitor mixture (Sigma-Aldrich) using an Omni TH homogenizer (Omni International, Marietta, GA). Following sonication, the homogenate was centrifuged at 2000 × g for 10 min. Supernatants were collected as total mucosal proteins, and protein concentrations were measured using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). Protein extraction, SDS-PAGE, and Western blots were performed as described previously (6, 19, 40).
Serum collection and biometric multiplex cytokine profiling
Blood was collected by cardiac puncture in endotoxin-free, silicone-coated tubes without additive. Blood samples were allowed to clot at room temperature for 30 min before centrifugation (2200 × g, 4°C, 10 min), and the serum was collected and stored at −80°C until analyzed. A multiplex sandwich immunoassay from the Bio-Plex Protein Array System (Bio-Rad), which contains fluorescently labeled microspheres conjugated with mAbs specific for 16 target cytokines, was used as described previously (6, 41). Analytes measured include IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, IL-17, IFN-γ, TNF-α, GM-CSF, interferon-inducible protein 10, keratinocyte-derived chemokine (KC), MCP-1, monokine-induced by IFN-γ, and MIP-1α (source of Abs: Invitrogen, Carlsbad, CA).
Isolation of peritoneal macrophages and PBMCs
Peritoneal macrophages were prepared as previously described (42). Briefly, 2 ml thioglycollate broth was injected into the peritoneal cavities of mice to elicit macrophages. After 3 d, peritoneal cells were recovered by washing the peritoneal cavity with 10 ml DMEM. Peritoneal macrophages were then purified by adhesion to tissue culture plastic (4 d after initial culture). Mouse PBMCs were isolated from whole blood by Ficoll gradient. Briefly, whole blood from three mice (~2–3 ml) was mixed with an equal volume of HBSS and then overlaid onto 9 ml Ficoll. After centrifugation (with brake off) at 2500 rpm for 30 min, the PBMC fraction (buffy coat layer) was collected and diluted two times with HBSS. PBMCs were recovered by centrifugation at 1500 rpm for 10 min.
Immunocytochemistry
Immunocytochemical detection was determined in 4-μm paraffin-embedded colonic sections as described previously (6) using Alexa Fluor 488 (Invitrogen) goat anti-mouse IgG (MCP-1 and IL-17) or Alexa Fluor 568 (Invitrogen) goat anti-mouse IgG (IL-6).
Statistical analysis
Statistical analysis (for all of the data with the exception of cytokine experiments) within and between each groups was done using a Student t test, and descriptive results are presented as mean ± SD. p values <0.05 were considered statistically significant.
Statistical analysis of cytokine profiles
Analyte concentrations were quantified by fitting using a calibration or standard curve. A five-parameter logistic regression analysis was performed to derive an equation that allowed concentrations of unknown samples to be predicted. Statistical differences in measured values were assessed by a Mann-Whitney U test. Data are presented with mean, median, upper, and lower quartile values. p values <0.05 were considered statistically significant.
Multidimensional scaling
Multidimensional scaling (MDS) is an iterative process to detect meaningful underlying dimensions to explain observed similarities or dissimilarities between the groups studied (43). This analysis uses correlational matrices to construct configurations of the data in a lower-dimensional matrix, such that the relative distances between the groups are similar to those in the higher-dimensional matrix. The degree of correspondence between the distances and the matrix input by the user is measured (inversely) by a stress function defined by Φ = Σ[dij − f(δij)]2, where dij is the Euclidean distance and δij is the observed distance. The proximities and distances are then represented on a two-dimensional Shepard diagram scatter plot, which facilitates visualization and the interpretation of patterns. All of the statistical analyses for MDS were performed with R software, version 2.8 (44).
Results
Clcn5 KO mice exhibit increased susceptibility to DSS-induced colitis
We recently demonstrated the coordinated downregulation of several transporter proteins, including CLC-5, in the sigmoid colon of patients with active IBD, suggesting its potential contribution to IBD-associated diarrhea (19). To investigate whether the loss of Clc-5 would induce susceptibility to colitis, we subjected Clcn5 KO mice to the well-established DSS chemical model of mucosal inflammation. Oral DSS administration for 7 d induced more significant acute colitis in Clcn5 KO mice when compared with that in WT mice (n = 7 mice per group) (Fig. 1). This was characterized by a significant increase in DAI scores (p = 0.034), as demonstrated by greater weight loss, earlier appearance of diarrhea/loose feces, and fecal blood in DSS-induced Clcn5 KO mice when compared with those in DSS-induced WT mice, as early as 5 d following induction with DSS (Fig. 1A). Morphological examination at day 5 and day 7 of induction revealed significant reduction in colon length and increased loose bloody stools in DSS-induced Clcn5 KO mice when compared with those in DSS-induced WT mice (Fig. 1B). Histological examination of acute DSS colitis in Clcn5 KO mice was characterized by greater loss of architecture, fewer goblet cells, and increased inflammatory cell infiltration at the areas of lesions when compared with those in DSS-induced WT mice (Fig. 1C). This was also demonstrated by the significant increase in HAI colitis scores for DSS-induced Clcn5 KO mice relative to those for DSS-induced WT mice (p = 0.039) (Fig. 1D). Neutrophil infiltration, as demonstrated by increased MPO activity, was also significantly elevated in Clcn5 KO mice induced with DSS colitis when compared with that in DSS-induced WT mice, indicative of an increased susceptibility to DSS-induced colitis (p = 0.028). The colons of Clcn5 KO mice uninduced with DSS colitis appeared normal and not different from those of control WT mice (Fig. 1). Our data therefore demonstrate that the loss of Clc-5 significantly exacerbates DSS-induced colitis.
FIGURE 1.

Administration of DSS demonstrated increased susceptibility to experimental colitis in Clcn5 KO mice. Results presented are representative of seven mice from each group. A, DAI was scored from WT and Clcn5 KO mice for weight loss, stool consistency, and bleeding. B, Clinical assessment of acute DSS colitis in WT and Clcn5 KO mice. C, Histological analysis of acute DSS colitis in WT and Clcn5 KO mice by H&E-stained colonic sections (original magnification ×20). D, For detailed histological analysis, colonic sections of WT and Clcn5 KO mice were scored in a blinded fashion as described in Materials and Methods. E, MPO activities in colon from WT and Clcn5 KO mice were determined as described in Materials and Methods. Clcn5 KO mice induced with colitis demonstrated statistically significant elevations in DAI, HAI, and MPO relative to those of WT mice, demonstrating increased susceptibility to acute experimental colitis. *p < 0.05. e, epithelial disruption; i, inflammatory infiltrate; m, lamina muscularis mucosae; s, submucosal edema.
Unique systemic immune profiles in Clcn5 KO mice at both basal conditions and when induced with DSS colitis
It is not known whether Clc-5 modulates the immune response to trigger distinct signaling inflammatory pathways. To analyze the influence of the cytokine patterns in the increased susceptibility of Clcn5 KO mice to DSS, we performed systemic multiplex serum cytokine profiling in DSS-induced Clcn5 KO and WT mice. The levels of 16 systemic cytokines covering a broad spectrum of immune and inflammatory mechanisms were measured in parallel following induction of colitis. Acute DSS-induced Clcn5 KO mice demonstrated a cellular, cytotoxic, and chemotactic profile with significant elevated levels of IL-12 and IL-17 (p < 0.05), as well as IFN-γ, KC, MIP-1α, and MCP-1 (p < 0.001), when compared with those in DSS-induced WT mice (Fig. 2A, 2B). This predominantly chemotactic cellular immune profile is distinct and is unlike any of our prior observations from several autoimmune conditions (6, 41, 45–47), suggesting the unique immunomodulatory role of Clc-5 in DSS colitis. Interestingly, the baseline levels of IL-6 were significantly elevated in Clcn5 KO mice when compared with those in WT mice (Fig. 2C), suggesting the likelihood that IL-6 may function as a proinflammatory mediator in Clcn5 KO mice.
FIGURE 2.
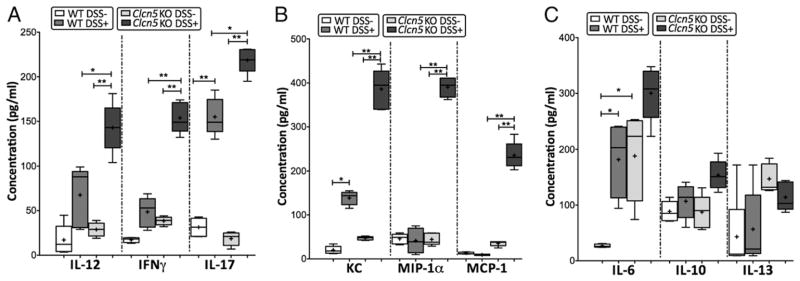
Distinct cellular cytotoxic and chemotactic patterns identified in Clcn5 KO mice induced with acute experimental colitis. Levels of 16 cytokines were measured simultaneously using a biometric multiplex assay from serum of WT and Clcn5 KO mice with and without acute DSS colitis. Data is presented with mean (+), median (−), and upper and lower quartile values. *p < 0.05; **p < 0.001. A and C, Cytokine pattern in acute colitis of Clcn5 KO mice is represented by a Th1–Th17-polarized (IL-17, IL-12, and IFN-γ) and a strong chemotactic pattern (MIP-1α, KC, and MCP-1) when compared with that of acute colitis in WT mice. B, No significant differences were observed in levels of humoral cytokines and other cellular cytokines and chemokines between WT and Clcn5 KO mice. Of particular significance are the statistically increased levels of IL-6 in untreated Clcn5 KO mice. At least seven mice were used in each group.
MDS analyses reveal unique categorical cytokine networks in Clcn5 KO mice
Complementary multivariate analytical methods provide a vivid picture of the biological significance of the immune profile network. MDS provides a means of identifying correlational configurations of statistically significant cytokines and allows for a visual representation of the pattern of proximities within the groups studied (43). As depicted in Fig. 3, MDS analysis of the cytokine patterns in DSS-induced Clcn5 KO mice identified a strong positive cluster between IL-12 and IL-17 (r = 0.742, p = 0.041), whereas those clusters were weaker in DSS-induced WT mice (r = 0.558, p = 0.057) (Fig. 3A). DSS-induced Clcn5 KO mice also showed a significantly tight positive cluster among KC, MIP-1α, and MCP-1 (r = 0.713, p = 0.027), whereas those clusters were absent in DSS-induced WT mice (Fig. 3A). However, DSS-induced WT mice showed a strong positive correlation between IL-12 and IFN-γ (r = 0.758, p = 0.03), one that was not present in DSS-induced Clcn5 KO mice (Fig. 3B). Interestingly, even at basal conditions (uninduced) Clcn5 KO mice showed a strong positive correlation between IL-6 and IL-10 (r = 0.722, p = 0.039), which was absent among all other groups studied (Fig. 3C). These unique representations provide a visual inspection of similarities and differences between cytokine changes among the group, indicating the intricate but distinct immune network associated with Clc-5 expression.
FIGURE 3.

Unique categorical cytokine networks identified in Clcn5 KO mice induced with acute experimental DSS colitis. MDS analysis was used to generate dimensions that can interpret statistically significant differences between cytokine networks in DSS-induced WT and Clcn5 KO mice. A, Two strong positive clusters, 1) between IL-12 and IL-17 and 2) among MPO, KC, MIP-1β, and MCP-1, were identified in DSS-induced Clcn5 KO mice, when compared with those in DSS-induced WT mice (marked by circles). B, DSS-induced WT mice showed a strong positive correlation between IL-12 and IFN-γ when compared with those in DSS-induced Clcn5 KO mice. C, Interestingly, uninduced Clcn5 KO mice showed a strong positive correlation between IL-6 and IL-10 (r = 0.722, p = 0.039), which was absent among all of the other groups studied. These unique representations provide a visual inspection of similarities and differences between cytokine changes among the group, indicating the intricate but distinct immune network in Clcn5 KO mice.
Alteration of cytokine profiles in colon validate and correlate with that of systemic cytokines
To determine whether the observed systemic cytokine profiles in acute experimental colitis of Clcn5 KO mice correlated with that of local levels seen within the colonic mucosa, immunoblots and immunofluorescence analysis of colons from DSS mice were performed. Proteins were extracted from mucosa scraped from freshly excised colons, and samples were analyzed by SDS-PAGE Western blots using primary Abs for IL-6, IL-12 p40/70, and IL-17. As shown in Fig. 4A, acute DSS-induced Clcn5 KO colitis had significantly higher IL-6, IL-12 p40/70, and IL-17 protein expression in the colon than that in WT DSS-induced colitis, suggesting similar patterns of changes to those observed in the systemic levels, thereby validating the correlation of the systemic immune response to that observed in local tissue.
FIGURE 4.
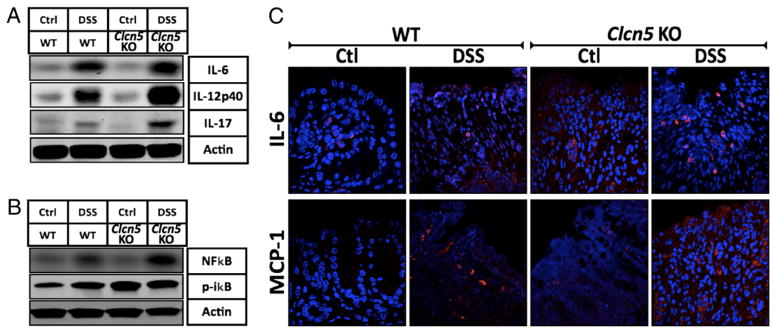
Correlation of cytokine profiles in colon with that of systemic levels in Clcn5 KO mice. Western blot and immunofluorescence analyses of cytokines from mucosal scrapings and tissue of colon in DSS-induced and uninduced WT and Clcn5 KO mice. A, WB show significantly higher IL-6, IL-12 p40/70, and IL-17 protein expression in acute DSS-induced Clcn5 KO colon when compared with that in WT DSS-induced colon. B, WB show significantly higher levels of colonic NF-κB and phospho-IκB protein expression in acute DSS-induced Clcn5 KO colitis when compared with those in WT DSS-induced colitis. C, IF shows higher IL-6 and MCP-1 protein in lamina propria of acute DSS-induced Clcn5 KO colitis when compared with those in WT DSS-induced colitis. These data validate the observed systemic cytokine profiles with those of local levels seen within tissue. Representatives of at least three independent experiments are shown in A and B.
Acute experimental colitis of Clcn5 KO mice is characterized by an NF-κB–mediated immunomodulatory profile. DSS-induced Clcn5 KO colitis had significantly higher NF-κB and phospho-IκB protein expression in the colon than that in WT DSS-induced colitis, suggesting the role of the NF-κB proinflammatory pathway in the distinct immune regulation associated with Clc-5 expression (Fig. 4B). Intriguingly, the baseline levels of phospho-IκB (no DSS treatment) were also significantly elevated in Clcn5 KO mice compared with those in WT mice, suggesting the likelihood that phospho-IκB, along with IL-6, may function as a proinflammatory mediator in Clcn5 KO mice.
Changes in mucosal cytokine levels are consistent with systemic levels as observed by immunofluorescence. As shown in Fig. 4C, IL-6 and MCP-1 were elevated in Clcn5 KO mice with DSS-induced colitis, predominantly associated with lamina propria infiltrating mononuclear cells, demonstrating the immune modulatory potential of inflammatory infiltrates and further validating the observed systemic cytokine profiles with that of local levels seen within tissue.
Elevated levels of IL-6 protein expression identified in the kidney, but not the colon, of Clcn5 KO mice
Although Clcn5 KO mice had significantly high systemic IL-6 levels relative to those of WT controls, the colonic mucosa from Clcn5 KO mice did not exhibit elevated levels of IL-6 protein expression in Western blots compared with those of WT controls (Supplemental Fig. 1A, 1B). Because mutations in the CLCN5 gene are associated with functional defects characterized by nephrolithiasis and progressive renal failure (27, 29, 31–33, 48–50), we investigated the protein expression of IL-6 in the kidney cortex of Clcn5 KO mice and WT mice. SDS-PAGE and Western blot studies demonstrated a significant increase in IL-6 protein expression in the kidney cortex of Clcn5 KO mice relative to that in WT mice, suggesting the novel finding that IL-6 may contribute to an immunopathogenic role (Supplemental Fig. 1A). To further define the expression and localization of renal IL-6 expression in Clcn5 KO mice, we performed immunofluorescence analyses on frozen sections of kidney cortex of WT and Clcn5 KO mice. Immunofluorescence analyses showed that IL-6 was significantly elevated in the kidney cortex of Clcn5 KO mice compared with that in WT mice (Supplemental Fig. 1B). In WT kidney, a low level of IL-6 expression was localized to the tubular basement membrane. However, in the Clcn5 KO kidney, there was increased expression of IL-6 in the basement membrane and upregulation in the interstitium, surrounding renal tubules, as well as epithelial cells of the proximal tubules, which was not observed in WT kidney sections. Our data demonstrate, for the first time, the involvement of novel immunomodulatory effects in a mouse model of Dent disease.
Clc-5 is highly expressed in the macrophages and weakly expressed in the colonic mucosa of WT mice but not in PBMCs
Because the immune profiles of Clcn5 KO mice are distinctly different from those of WT mice both under the basal condition (baseline level) and in DSS-induced colitis, we speculate that Clc-5 may be expressed in the immune cells, where it could directly affect the function of these cells. Peritoneal macrophages and PBMCs, including lymphocytes and monocytes, were isolated from both WT and Clcn5 KO mice. Surprisingly, Clc-5 was highly expressed in the macrophages (comparable to that in kidney), but not in PBMCs (Supplemental Fig. 2). Low levels of Clc-5 expression were also observed in the colonic mucosa of WT mice. These data provide the first evidence that Clc-5 is expressed in immune cells.
Protective effect of diet and vitamin D on Clcn5 KO DSS-induced colitis
A case-control study to evaluate the etiological role of dietary factors in UC identified dietary patterns that were associated with an increased risk to develop UC (51). To evaluate the influence of diet on the development of experiment colitis in Clcn5 KO mice, we subjected a group of mice (n = 5 per group) to a separate diet: Z-diet (Supplemental Fig. 3). The diet was initiated from birth (in both mother and F1 generation used in these experiments) and contains greater amounts of vitamin D, vitamin B12, selenium, and choline (all >1.5-fold) and lower amounts of iodine, vitamin K3, inositol, pantothenic acid, vitamin B2, vitamin B6, vitamin B7, and vitamin E (all <1.5-fold) when compared with the H-diet, which is the regular diet used at our mouse facility (Supplemental Fig. 3). Interestingly, a significant increase in colitis, as demonstrated by DAI scores, was observed in DSS-induced Clcn5 KO mice maintained on H-diet, when compared with that of Clcn5 KO DSS-induced mice on the Z-diet (p = 0.047) (Fig. 5A). Among the differences in dietary components between H-diet and Z-diet, vitamin D has been reported to play a role in preventing or ameliorating the IBD in the colitis model of IL-10 KO mice (52). Because Z-diet contains 2-fold higher level of vitamin D than that in the H-diet (Supplemental Fig. 1), we hypothesized that the high level of vitamin D in the Z-diet might be an important reason why Z-diet exhibited protective effect on colitis of Clcn5 KO mice. Because we did not observe the beneficial effect of Z-diet on DSS colitis of WT mice, we decided to test the effect of vitamin D supplement on DSS colitis in Clcn5 KO mice by giving the mice Vit D-enriched H-diet, which was specially formulated H-diet containing a higher level of vitamin D (4.18 IU/g), which is the same as that found in the Z-diet. As shown in Fig. 5B and 5C, DSS-treated Clcn5 KO mice on Vit D-enriched H-diet for 4 wk exhibited a more significant reduction of disease activity than those on regular H-diet. Interestingly, this effect depends on the duration of the diet change: Clcn5 KO mice on Vit D-enriched H-diet for 1 wk exhibited only marginal improvement of DSS colitis compared with that of the mice on the same diet for 4 wk (Fig. 5B). There was no significant difference in colitis DAI (and MPO scores, data not shown) of DSS-induced WT mice between the mice fed with the H-diet and the mice fed the Z-diet (Fig. 5A). This suggests that the loss of Clc-5 significantly exacerbates DSS-induced colitis and is influenced by the dietary factors, such as vitamin D. Further quantitative studies are warranted to more fully investigate whether the loss of Clc-5 is a true risk factor in the development of UC.
FIGURE 5.
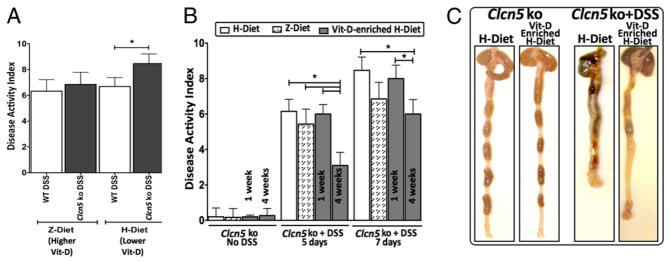
Protective effect of vitamin D supplement on DSS-induced colitis in Clcn5 KO mice. A, Clcn5 KO mice maintained on the H-diet exhibited a significant increase in DSS-induced colitis, as demonstrated by DAI scores, when compared with Clcn5 KO DSS-induced mice on the Z-diet (p = 0.047). There is no significant difference in colitis DAI and MPO scores of DSS-induced WT mice between the mice fed with the H-diet and the mice fed with the Z-diet. Clcn5 KO mice (6–7 wk old) were fed either continuously on regular H-diet or on Vit D (4.18 IU/g)-enriched H-diet for 1 or 4 wk. A total of 2.5% DSS was given in drinking water for 7 d to induce colitis. Disease activity was monitored daily by DAI as described in Fig. 1. B, DAI score 5 and 7 d after DSS nduction: Mice fed on Vit D-enriched H-diet exhibited significantly less disease activity than those fed on regular H-diet, whereas 1 wk Vit D-enriched H-diet improved DSS colitis only marginally. C, Morphology of the colons in Clcn5 KO mice treated with DSS for 7 d: mice on regular H-diet show severe colitis characterized by shortened colon and dark-colored blood in the stool throughout the colon and cecum when compared with that of control (no DSS). Furthermore, mice on Vit D-enriched H-diet for 4 wk exhibited marked improvement of colitis when compared with those on regular H-diet alone, including normal appearance of stool pellets in the colon (although less stool pellets in general), much less blood in the colon, and less colon shortening. *p < 0.05.
Discussion
Diarrhea continues to be a frequent symptom in UC patients, the underlying mechanisms of which depend on various factors, including, but not limited to, the location, extent, and severity of the inflammation, altered motility, associated infections, and iatrogenic factors (10, 53). Furthermore, diarrhea in UC is associated with fluid and electrolyte imbalance, indicative of impaired gastrointestinal epithelial ion transport (54). Recently, we demonstrated the coordinated downregulation of CLC-5 among several Na+ transporters in sigmoid mucosal biopsies of patients with active IBD and mice with experimental colitis (19). However, the specific role of CLC-5 in UC has not yet been defined.
Previous studies have demonstrated that the Guggino Clcn5 KO mice develop classical signs of Dent disease with fluid and electrolyte imbalance manifested by polyuria, low-m.w. proteinuria, aminoaciduria, glycosuria, and hypercalciuria (28–30, 35). Although fluid, sodium, and chloride measurements have not yet been performed on the stool from these mice, Clcn5 KO mice afford an ideal model to investigate whether the loss of CLC-5 would induce susceptibility to UC. In so doing, we present here the first evidence that the loss of Clc-5 significantly exacerbates both clinical and histopathological signs of DSS-induced UC-like colitis. It is important to infer the mechanisms of the phenotypic abnormalities in intestinal mucosa as a result of the lack of Clc-5 function. Clc-5 is an intracellularly localized Cl−/H+ exchanger that has been suggested to play an important role in regulation of protein trafficking in epithelial cells (54). Our recent studies have shown that, in mouse mucosa, both Nhe3 and Clc-5 comigrated in the same endosomal pool isolated by OptiPrep gradient fractionation (data not shown). Because trafficking of Nhe3 between plasma membrane and endosomal compartments is a major regulatory mechanism of Nhe3 (55), our data suggest that Clc-5 may be involved in Nhe3 trafficking. Lack of Clc-5 causes alkalinization of the early and perhaps recycling endosmes (56) but not late endosomes. Because Clc-5 acts to provide the neutralizing negative charge for the proton moved by the H-ATPase that acidifies this compartment, the lack of Clc-5 increases the pH in the endosmal compartment. The lack of Clc-5, and consequent alkaline endosomal pH, causes trafficking defects in the apical membrane proteins Na/phosphate cotransporter 2a (Npt2a), Nhe3, and megalin (55). In proximal tubules devoid of Clc-5, a significantly reduced apical membrane expression of megalin (57) results in low-m.w. proteinuria. Furthermore, both Npt2a and Nhe3 are mislocalized to a subapical compartment in the proximal tubule of Clcn5 KO mice. The reduced apical membrane localization of Npt2a and Nhe3 decreases transport activity and thus results in hyperphosphaturia and sodium loss, respectively. By analogy, we expect that the same physiological outcome would be present in the intestine where Nhe3 is well known to traffic on and off of the plasma membrane (58). The idea of less surface Nhe3 caused by defective Nhe3 trafficking in the Clcn5 KO mice would be compatible with other mouse KO studies that have shown the lack of colonic Nhe3 or its regulatory factor Na+/H+ exchanger regulatory factor 1 (data not shown) causes diarrhea and susceptibility to IBD (59). Given these considerations and the studies reported herein, our data further substantiate the hypothesis that IBD-associated diarrhea manifests as a result of the coordinated downregulation of multiple Na+ transporters and related regulatory proteins, including NHE3 and CLC-5.
Our studies herein also ascertained that the exacerbated colitis from the loss of Clc-5 was influenced by specific immune-mediated dysregulation, implying the immunopathogenic role of downregulated CLC-5 in UC. The activation of the innate immune system has been shown to provide the source of cytokines, which includes IL-12, that then trigger the adaptive CD4+ T cellular and humoral immune response in IBD (2). Costimulated and activated T cells then secrete a distinct set of cytokines that perpetuate the disease process. In UC, there are specific mucosal-damage pathways characterized by dysregulated cytokine profiles at different stages of the disease process (2). We have previously shown that acute experimental DSS-induced colitis was represented by a distinct set of Th1–Th17 cytokine profiles that involved altered expression of TNF-α, IL-6, IL-17, and KC (6). Our current studies of experimental colitis using Clcn5 KO mice demonstrated a further exacerbation of the Th1–Th17 immune response, characterized by elevated levels of TNF-α, IL-6, and IL-17, suggesting the immune-mediated pathophysiological role of downregulated CLC-5 in UC.
The inflammatory response to an antigenic stimulus in UC is primarily manifested by the recruitment of distinct chemokines characteristic for the activation that function as critical players in the regulation of the immune response (6, 60). Our profiles identified a significant positive cluster among KC, MIP-1β, and MCP-1 as active chemokines in the signaling network of the acute experimental colitis in Clcn5 KO mice. It is likely that these distinct chemokines drive the initial acute innate immune response, which is consistent with the finding that MPO levels, a marker of innate neutrophil activity, were significantly elevated in the colonic tissues of acute experimental colitis in Clcn5 KO mice. Chemokines involved in the recruitment of CD4+ T cells are also expressed in colonic epithelial cells, and it has been shown that in patients with UC colonic enterocytes are the major source for neutrophil-directing chemokines, such as MCP-1 (61, 62), which is in agreement with our observations.
The mechanisms of the innate–adaptive interface in IBD have been well-demonstrated to primarily involve the TLR pathway, which generates inflammatory factors modulated by the NF-κB pathway (63). This is initiated by the stimulation of intermediate kinases, leading to phosphorylation of the inhibitor of B kinase and subsequent release of NF-κB, which then translocates to the nucleus, where it activates transcription of proinflammatory genes (63). Our studies in experimental colitis using Clcn5 KO mice also show increased activation of phospho-IκB and increased dissociation of NF-κB, suggesting the role of the TLR–NF-κB pathway mediated inflammatory mechanisms in the acute experimental colitis of Clcn5 KO mice.
IL-6 plays an important role in differentiation and growth of hematopoietic progenitor cells and lymphocytes and in the generation of the Th17 immune population (64). IL-6 contributes to increased T cell survival, which then accumulates in the lamina propria, leading to perpetuation of inflammation (65). Elevated proinflammatory levels of IL-6 have been identified in several chronic inflammatory conditions, including UC (1, 64), which is consistent with the increased systemic levels of IL-6 in acute experimental colitis of Clcn5 KO mice. Interestingly, we identified elevated baseline systemic levels of IL-6 in Clcn5 KO mice when compared with those in WT mice. We also demonstrated that Clcn5 KO mice exhibit elevated basal activation of IκB in colon, indicating the possibility that increased IκB and IL-6 activation at basal conditions may sensitize the Clcn5 KO mice to DSS-induced colitis. Further examination also identified elevated levels of IL-6 in proximal tubules, suggesting the likelihood that IL-6 may function as a proinflammatory mediator in Clcn5 KO mice. IL-6 has been previously demonstrated to enhance osteoclastogenesis by the induction of the NF-κB ligand in osteoblastic cells (66) and has also shown to be the central pathogenic player in arthritis. Given the multifacted role of IL-6, our findings therefore suggest that the increased production of IL-6 under basal conditions in the kidney may explain the development of both hypercalciuria and nephrolithiasis in Clcn5 KO mice, although a direct correlation has not been established (67, 68). Moreover, elevated IL-6 is involved in defective bone mineralization, which may also explain the sporadic presentation of rickets/osteomalacia in patients with Dent disease (27, 51, 69–71).
Why does the lack of Clc-5 expression lead to alteration of both basal and DSS-induced immunological responses? Our preliminary study demonstrates that Clc-5, a Cl−/H+ exchanger previously known to be expressed only in renal and intestinal epithelial cells (20, 28–30), is highly expressed in macrophages. This finding implicates an interesting possibility that Clc-5 may play an important and previously unrecognized role in regulating innate immunity. It is therefore necessary to further study this potentially novel function of Clc-5 in macrophages.
Diet is also suspected to influence the severity of IBD symptoms by influencing the microbial flora and directly modulating the mucosal immune response of the host(72,73). We observed attenuation of DSS colitis in Clcn5 KO mice maintained on a separate diet (Z-diet) containing high vitamin D, vitamin B12, and selenium (and low iodine, vitamin K3, vitamin B2, vitamin B6, and vitamin B7, among others), when compared with those maintained on a regular diet (H-diet). Because this was specifically not observed in the WT mice fed with the Z-diet, our data suggest that genetic predisposition appears to provide the substrate for such effects, implying the potential of a double-hit mechanism in the immunopathogenesis of UC, a disease contributed by both genetic and environmental factors. This therefore implies that diet can impact the severity of UC symptoms, particularly in patients genetically predisposed to either mutations or downregulation of CLC5. Among the dietary factors that may be involved in modulation of DSS colitis, we identified vitamin D as a key factor contributing to the protective effect of DSS-induced colitis on Clcn5 KO mice. vitamin D, recognized for a long time to play a significant immunomodulatory role, exerts a marked inhibitory effect on both adaptive and innate immune systems (74). Vitamin D has been reported to play a role in preventing or ameliorating IBD in the colitis model of IL-10 KO mice (53, 75). Furthermore, it was demonstrated that vitamin D receptor-deficient mice were more sensitive to DSS-induced colitis, implicating a beneficial effect of dietary vitamin D in the therapy of colitis (76). Our present data provide additional strong evidence that supports a protective role of vitamin D in IBD, particularly in genetically or immunologically compromised subjects.
Because the recurrent manifestation of diarrhea can have a significant impact on fluid and electrolyte status and given the varied pathophysiologic mechanisms of diarrhea in IBD and the heterogeneous nature of the disease, individually tailored treatments are essential for effective management (10, 53). Our studies demonstrate that the loss of Clc-5 both 1) exhibits IL-6–mediated immunopathogenesis and 2) significantly exacerbates DSS-induced colitis mediated by NF-κB-modulated Th1–Th17 immune dysregulation, which is influenced by dietary factors, particularly dietary vitamin D. These studies imply the role of downregulated CLC-5 in the immunopathogenesis of UC with a better understanding of the molecular causes for UC-associated diarrhea and provide rational strategies to pursue new therapeutic targets for the management of diarrhea in UC patients.
Supplementary Material
Acknowledgments
This work was supported by the Broad Medical Research Program (IBD-0119R), National Institute of Diabetes and Digestive and Kidney Diseases (R21 DK077064), National Institutes of Health Ruth L. Kirschstein National Research Service Award, National Institutes of Health (5U19AI062629, P20RR15577, and P01-DK-072084), and in part by Oklahoma Center for the Advancement of Science and Technology Oklahoma Applied Research Support (AR061-015 and AR081-006).
Abbreviations used in this paper
- CD
Crohn’s disease
- DAI
disease activity index
- DSS
dextran sulfate sodium
- e
epithelial disruption
- HAI
histological activity index
- H-diet
Harlan Teklad Diet
- i
inflammatory infiltrate
- IBD
inflammatory bowel disease
- KC
keratinocyte-derived chemokine
- KO
knockout
- m
lamina muscularis mucosae
- MDS
multidimensional scaling
- MPO
myeloperoxidase
- NHE
Na+/H+ exchanger
- Npt2a
Na/phosphate cotransporter 2a
- pAb
polyclonal Ab
- s
submucosal edema
- UC
ulcerative colitis
- Vit D-enriched H-diet
vitamin D-supplemented H-diet
- WT
wild-type
- Z-diet
NIH-31 modified open formula diet
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.O’Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pizarro TT, Cominelli F. Cytokine therapy for Crohn’s disease: advances in translational research. Annu Rev Med. 2007;58:433–444. doi: 10.1146/annurev.med.58.121205.100607. [DOI] [PubMed] [Google Scholar]
- 5.Elson CO, Cong Y, Lorenz R, Weaver CT. New developments in experimental models of inflammatory bowel disease. Curr Opin Gastroenterol. 2004;20:360–367. doi: 10.1097/00001574-200407000-00010. [DOI] [PubMed] [Google Scholar]
- 6.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 8.Russell RK, Nimmo ER, Satsangi J. Molecular genetics of Crohn’s disease. Curr Opin Genet Dev. 2004;14:264–270. doi: 10.1016/j.gde.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet. 2005;6:376–388. doi: 10.1038/nrg1607. [DOI] [PubMed] [Google Scholar]
- 10.Shah SB, Hanauer SB. Treatment of diarrhea in patients with inflammatory bowel disease: concepts and cautions. Rev Gastroenterol Disord. 2007;7(Suppl 3):S3–S10. [PubMed] [Google Scholar]
- 11.Binder HJ, Sandle GI, Rajendran VM. Colonic fluid and electrolyte transport in health and disease. In: Philips SF, Pemberton JH, Shorter RD, editors. The Large Intestine: Physiology, Pathophysiology, and Disease. Raven Press; New York: 1991. pp. 141–168. [Google Scholar]
- 12.Sandle GI. Salt and water absorption in the human colon: a modern appraisal. Gut. 1998;43:294–299. [Google Scholar]
- 13.Bertelsen LS, Eckmann L, Barrett KE. Prolonged interferon-gamma exposure decreases ion transport, NKCC1, and Na+-K+-ATPase expression in human intestinal xenografts in vivo. Am J Physiol Gastrointest Liver Physiol. 2004;286:G157–G165. doi: 10.1152/ajpgi.00227.2003. [DOI] [PubMed] [Google Scholar]
- 14.Musch MW, Clarke LL, Mamah D, Gawenis LR, Zhang Z, Ellsworth W, Shalowitz D, Mittal N, Efthimiou P, Alnadjim Z, et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J Clin Invest. 2002;110:1739–1747. doi: 10.1172/JCI15695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugi K, Musch MW, Field M, Chang EB. Inhibition of Na+,K+-ATPase by interferon gamma down-regulates intestinal epithelial transport and barrier function. Gastroenterology. 2001;120:1393–1403. doi: 10.1053/gast.2001.24045. [DOI] [PubMed] [Google Scholar]
- 16.Amasheh S, Barmeyer C, Koch CS, Tavalali S, Mankertz J, Epple HJ, Gehring MM, Florian P, Kroesen AJ, Zeitz M, et al. Cytokine-dependent transcriptional down-regulation of epithelial sodium channel in ulcerative colitis. Gastroenterology. 2004;126:1711–1720. doi: 10.1053/j.gastro.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 17.Amin MR, Malakooti J, Sandoval R, Dudeja PK, Ramaswamy K. IFN-gamma and TNF-alpha regulate human NHE3 gene expression by modulating the Sp family transcription factors in human intestinal epithelial cell line C2BBe1. Am J Physiol Cell Physiol. 2006;291:C887–C896. doi: 10.1152/ajpcell.00630.2005. [DOI] [PubMed] [Google Scholar]
- 18.Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. 2006;116:2682–2694. doi: 10.1172/JCI29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan S, Alex P, Dassopoulos T, Zachos NC, Iacobuzio-Donahue C, Donowitz M, Brant SR, Cuffari C, Harris ML, Datta LW, et al. Downregulation of sodium transporters and NHERF proteins in IBD patients and mouse colitis models: potential contributors to IBD-associated diarrhea. Inflamm Bowel Dis. 2009;15:261–274. doi: 10.1002/ibd.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devuyst O, Guggino WB. Chloride channels in the kidney: lessons learned from knockout animals. Am J Physiol Renal Physiol. 2002;283:F1176–F1191. doi: 10.1152/ajprenal.00184.2002. [DOI] [PubMed] [Google Scholar]
- 21.Jentsch TJ. Chloride channels: a molecular perspective. Curr Opin Neurobiol. 1996;6:303–310. doi: 10.1016/s0959-4388(96)80112-7. [DOI] [PubMed] [Google Scholar]
- 22.Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–215. doi: 10.1016/s0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 23.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 24.Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch TJ. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. 1991;354:304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- 25.Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bösl MR, Ruether K, Jahn H, Draguhn A, et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/s0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 26.Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 27.Dent CE, Friedman M. Hypercalcuric rickets associated with renal tubular damage. Arch Dis Child. 1964;39:240–249. doi: 10.1136/adc.39.205.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Souza-Menezes J, Morales MM, Tukaye DN, Guggino SE, Guggino WB. Absence of ClC5 in knockout mice leads to glycosuria, impaired renal glucose handling and low proximal tubule GLUT2 protein expression. Cell Physiol Biochem. 2007;20:455–464. doi: 10.1159/000107529. [DOI] [PubMed] [Google Scholar]
- 29.Guggino SE. Mechanisms of disease: what can mouse models tell us about the molecular processes underlying Dent disease? Nat Clin Pract Nephrol. 2007;3:449–455. doi: 10.1038/ncpneph0541. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Cai H, Cebotaru L, Hryciw DH, Weinman EJ, Donowitz M, Guggino SE, Guggino WB. ClC-5: role in endocytosis in the proximal tubule. Am J Physiol Renal Physiol. 2005;289:F850–F862. doi: 10.1152/ajprenal.00011.2005. [DOI] [PubMed] [Google Scholar]
- 31.Wrong OM, Norden AG, Feest TG. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87:473–493. [PubMed] [Google Scholar]
- 32.Fisher SE, Black GC, Lloyd SE, Hatchwell E, Wrong O, Thakker RV, Craig IW. Isolation and partial characterization of a chloride channel gene which is expressed in kidney and is a candidate for Dent’s disease (an X-linked hereditary nephrolithiasis) Hum Mol Genet. 1994;3:2053–2059. [PubMed] [Google Scholar]
- 33.Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, Harding B, Bolino A, Devoto M, Goodyer P, et al. A common molecular basis for three inherited kidney stone diseases. Nature. 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 34.Lloyd SE, Pearce SH, Günther W, Kawaguchi H, Igarashi T, Jentsch TJ, Thakker RV. Idiopathic low molecular weight proteinuria associated with hypercalciuric nephrocalcinosis in Japanese children is due to mutations of the renal chloride channel (CLCN5) J Clin Invest. 1997;99:967–974. doi: 10.1172/JCI119262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, Thakker RV, Guggino S, Guggino WB. Mice lacking renal chloride channel, CLC-5, are a model for Dent’s disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet. 2000;9:2937–2945. doi: 10.1093/hmg/9.20.2937. [DOI] [PubMed] [Google Scholar]
- 36.Cheng G, Ramanathan A, Shao Z, Agrawal DK. Chloride channel expression and functional diversity in the immune cells of allergic diseases. Curr Mol Med. 2008;8:401–407. doi: 10.2174/156652408785160934. [DOI] [PubMed] [Google Scholar]
- 37.Cheng G, Shao Z, Chaudhari B, Agrawal DK. Involvement of chloride channels in TGF-beta1-induced apoptosis of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1339–L1347. doi: 10.1152/ajplung.00121.2007. [DOI] [PubMed] [Google Scholar]
- 38.Obermeier F, Kojouharoff G, Hans W, Schölmerich J, Gross V, Falk W. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol. 1999;116:238–245. doi: 10.1046/j.1365-2249.1999.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin KW, Barrett KE. Mast cells are not essential to inflammation in murine model of colitis. Dig Dis Sci. 1994;39:513–525. doi: 10.1007/BF02088336. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Galli T, Leu S, Wade JB, Weinman EJ, Leung G, Cheong A, Louvard D, Donowitz M. Na+-H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: a role for rafts in trafficking and rapid stimulation of NHE3. J Physiol. 2001;537:537–552. doi: 10.1111/j.1469-7793.2001.00537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alex P, Szodoray P, Arthur E, Willis L, Hynd R, Flinn D, Centola M. Influence of intraarticular corticosteroid administration on serum cytokines in rheumatoid arthritis. Clin Rheumatol. 2007;26:845–848. doi: 10.1007/s10067-006-0419-7. [DOI] [PubMed] [Google Scholar]
- 42.Davies JQ, Gordon S. Isolation and culture of murine macrophages. Methods Mol Biol. 2005;290:91–103. doi: 10.1385/1-59259-838-2:091. [DOI] [PubMed] [Google Scholar]
- 43.Borg I, Groenen PJF. Modern Multidimensional Scaling Theory and Applications (Springer Series in Statistics) 2. Springer; New York: 2005. [Google Scholar]
- 44.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Statist. 1996;5:299–314. [Google Scholar]
- 45.Szodoray P, Alex P, Chappell-Woodward CM, Madland TM, Knowlton N, Dozmorov I, Zeher M, Jarvis JN, Nakken B, Brun JG, Centola M. Circulating cytokines in Norwegian patients with psoriatic arthritis determined by a multiplex cytokine array system. Rheumatology (Oxford) 2007;46:417–425. doi: 10.1093/rheumatology/kel306. [DOI] [PubMed] [Google Scholar]
- 46.Szodoray P, Alex P, Jonsson MV, Knowlton N, Dozmorov I, Nakken B, Delaleu N, Jonsson R, Centola M. Distinct profiles of Sjögren’s syndrome patients with ectopic salivary gland germinal centers revealed by serum cytokines and BAFF. Clin Immunol. 2005;117:168–176. doi: 10.1016/j.clim.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 47.Wright H, Alex P, Nguyen T, Bader T, Gurakar A, Sebastian A, Gonzales L, Wallis G, Naylor M, Dozmorov I, et al. Multiplex cytokine profiling of initial therapeutic response in patients with chronic hepatitis C virus infection. Dig Dis Sci. 2005;50:1793–1803. doi: 10.1007/s10620-005-2940-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cebotaru V, Kaul S, Devuyst O, Cai H, Racusen L, Guggino WB, Guggino SE. High citrate diet delays progression of renal insufficiency in the ClC-5 knockout mouse model of Dent’s disease. Kidney Int. 2005;68:642–652. doi: 10.1111/j.1523-1755.2005.00442.x. [DOI] [PubMed] [Google Scholar]
- 49.Guggino SE. Can we generate new hypotheses about Dent’s disease from gene analysis of a mouse model? Exp Physiol. 2009;94:191–196. doi: 10.1113/expphysiol.2008.044586. [DOI] [PubMed] [Google Scholar]
- 50.Silva IV, Cebotaru V, Wang H, Wang XT, Wang SS, Guo G, Devuyst O, Thakker RV, Guggino WB, Guggino SE. The ClC-5 knockout mouse model of Dent’s disease has renal hypercalciuria and increased bone turnover. J Bone Miner Res. 2003;18:615–623. doi: 10.1359/jbmr.2003.18.4.615. [DOI] [PubMed] [Google Scholar]
- 51.Fonseca JE, Santos MJ, Canhão H, Choy E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun Rev. 2009;8:538–542. doi: 10.1016/j.autrev.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 52.Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–2652. doi: 10.1093/jn/130.11.2648. [DOI] [PubMed] [Google Scholar]
- 53.Barrett KE, Dharmsathaphorn K. Pharmacological aspects of therapy in inflammatory bowel diseases: antidiarrheal agents. J Clin Gastroenterol. 1988;10:57–63. doi: 10.1097/00004836-198802000-00013. [DOI] [PubMed] [Google Scholar]
- 54.Beeken WL. Remediable defects in Crohn disease: a prospective study of 63 patients. Arch Intern Med. 1975;135:686–690. [PubMed] [Google Scholar]
- 55.Piwon N, Günther W, Schwake M, Bösl MR, Jentsch TJ. ClC-5 Cl−-channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408:369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- 56.Hara-Chikuma M, Wang Y, Guggino SE, Guggino WB, Verkman AS. Impaired acidification in early endosomes of ClC-5 deficient proximal tubule. Biochem Biophys Res Commun. 2005;329:941–946. doi: 10.1016/j.bbrc.2005.02.060. [DOI] [PubMed] [Google Scholar]
- 57.Christensen EI, Devuyst O, Dom G, Nielsen R, Van der Smissen P, Verroust P, Leruth M, Guggino WB, Courtoy PJ. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubilin in kidney proximal tubules. Proc Natl Acad Sci USA. 2003;100:8472–8477. doi: 10.1073/pnas.1432873100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donowitz M, Li X. Regulatory binding partners and complexes of NHE3. Physiol Rev. 2007;87:825–872. doi: 10.1152/physrev.00030.2006. [DOI] [PubMed] [Google Scholar]
- 59.Laubitz D, Larmonier CB, Bai A, Midura-Kiela MT, Lipko MA, Thurston RD, Kiela PR, Ghishan FK. Colonic gene expression profile in NHE3-deficient mice: evidence for spontaneous distal colitis. Am J Physiol Gastrointest Liver Physiol. 2008;295:G63–G77. doi: 10.1152/ajpgi.90207.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zimmerman NP, Vongsa RA, Wendt MK, Dwinell MB. Chemokines and chemokine receptors in mucosal homeostasis at the intestinal epithelial barrier in inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:1000–1011. doi: 10.1002/ibd.20480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keates S, Keates AC, Mizoguchi E, Bhan A, Kelly CP. Enterocytes are the primary source of the chemokine ENA-78 in normal colon and ulcerative colitis. Am J Physiol. 1997;273:G75–G82. doi: 10.1152/ajpgi.1997.273.1.G75. [DOI] [PubMed] [Google Scholar]
- 62.Blumberg R, Neurath MF, editors. Immune Mechanisms in Inflammatory Bowel Disease (Advances in Experimental Medicine and Biology) Vol. 579. Springer; New York: 2006. [Google Scholar]
- 63.Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut. 1998;42:477–484. doi: 10.1136/gut.42.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mudter J, Neurath MF. Il-6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflamm Bowel Dis. 2007;13:1016–1023. doi: 10.1002/ibd.20148. [DOI] [PubMed] [Google Scholar]
- 65.Neurath MF, Finotto S, Fuss I, Boirivant M, Galle PR, Strober W. Regulation of T-cell apoptosis in inflammatory bowel disease: to die or not to die, that is the mucosal question. Trends Immunol. 2001;22:21–26. doi: 10.1016/s1471-4906(00)01798-1. [DOI] [PubMed] [Google Scholar]
- 66.Moschen AR, Kaser A, Enrich B, Ludwiczek O, Gabriel M, Obrist P, Wolf AM, Tilg H. The RANKL/OPG system is activated in inflammatory bowel disease and relates to the state of bone loss. Gut. 2005;54:479–487. doi: 10.1136/gut.2004.044370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boonla C, Hunapathed C, Bovornpadungkitti S, Poonpirome K, Tungsanga K, Sampatanukul P, Tosukhowong P. Messenger RNA expression of monocyte chemoattractant protein-1 and interleukin-6 in stone-containing kidneys. BJU Int. 2008;101:1170–1177. doi: 10.1111/j.1464-410X.2008.07461.x. [DOI] [PubMed] [Google Scholar]
- 68.Ghazali A, Fuentés V, Desaint C, Bataille P, Westeel A, Brazier M, Prin L, Fournier A. Low bone mineral density and peripheral blood monocyte activation profile in calcium stone formers with idiopathic hypercalciuria. J Clin Endocrinol Metab. 1997;82:32–38. doi: 10.1210/jcem.82.1.3649. [DOI] [PubMed] [Google Scholar]
- 69.Dent CE. Rickets and osteomalacia of various origins. Birth Defects Orig Artic Ser. 1971;7:79–85. [PubMed] [Google Scholar]
- 70.Scheinman SJ. X-linked hypercalciuric nephrolithiasis: clinical syndromes and chloride channel mutations. Kidney Int. 1998;53:3–17. doi: 10.1046/j.1523-1755.1998.00718.x. [DOI] [PubMed] [Google Scholar]
- 71.Sylvester FA, Wyzga N, Hyams JS, Gronowicz GA. Effect of Crohn’s disease on bone metabolism in vitro: a role for interleukin-6. J Bone Miner Res. 2002;17:695–702. doi: 10.1359/jbmr.2002.17.4.695. [DOI] [PubMed] [Google Scholar]
- 72.Teahon K, Bjarnason I, Pearson M, Levi AJ. Ten years’ experience with an elemental diet in the management of Crohn’s disease. Gut. 1990;31:1133–1137. doi: 10.1136/gut.31.10.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winitz M, Adams RF, Seedman DA, Davis PN, Jayko LG, Hamilton JA. Studies in metabolic nutrition employing chemically defined diets. II. Effects on gut microflora populations. Am J Clin Nutr. 1970;23:546–559. doi: 10.1093/ajcn/23.5.546. [DOI] [PubMed] [Google Scholar]
- 74.Moro JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev Immunol. 2008;8:685–698. doi: 10.1038/nri2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Froicu M, Cantorna MT. Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol. 2007;8:5. doi: 10.1186/1471-2172-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, Bissonnette M, Li YC. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol. 2008;294:G208–G216. doi: 10.1152/ajpgi.00398.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.