

Microtubules have been studied extensively since the buffer conditions for in vitro polymerization were first described. As a result, the biochemistry and biophysics of microtubule dynamics is relatively well understood in the test tube. However, our understanding of microtubules in situ is limited at best. In cells, microtubules exhibit considerable diversity at the molecular level, including tubulin isotypes, posttranslational modifications, and associated proteins. Microtubules in different cell types or even in different subcellular compartments may exhibit strikingly different properties with regard to dynamics, composition and function. This heterogeneity is particularly striking in neurons, where the bulk of the microtubules are not associated with the microtubule-organizing center, yet may exhibit exceptional stability. The answers to questions about the functional diversity of neuronal microtubules may be critical for understanding many aspects of neuronal development, function and pathology.
One obstacle to characterizing specific populations of neuronal microtubules is the complexity of nervous tissue. Separating neuronal microtubules from glial microtubules, dendritic microtubules from axonal or cell body microtubules is effectively impossible when using brain tissue as a source, so any studies on the biochemistry and biophysics of neuronal microtubules from brain reflect the properties of a mixed pool. The problem is compounded by the fact that a large fraction of neuronal tubulin is lost during standard preparations of brain tubulin and this population of stable microtubules has received little attention, despite representing more than 50% of axonal tubulin in mature neurons.
Isolated axoplasm from the squid giant axon provides a unique model system for studying exclusively axonal microtubules both in situ and in vitro. Although isolated axoplasm has not been widely used, experiments using this model have provided novel insights into the axonal cytoskeleton and studies on axoplasm have the potential to produce additional insights. Here, we describe the preparation of isolated axoplasms, the use of physiological buffers that more accurately reflect intracellular environments and examples of experiments that can only be done in this model system (Figure 1).
Figure 1.

Flow chart for preparation of axoplasms.
Preparation of Axoplasm
The procedures described here are based on our continuing studies using axons from the Atlantic Longfin Squid, Loligo pealeii and update previous descriptions (Brady, Lasek, & Allen, 1985; Brady, Richards, & Leopold, 1993; Leopold, Lin, Sugimori, Llinas, & Brady, 1994). Large to medium squid (0.3–0.5 meters in length) are preferred as they have axons 300µm~500µm in diameter. Suitable squid are seasonally available (April-October typically) at the Marine Biological Laboratory in Woods Hole, MA, but protocols should be readily adapted for other species of squid with suitable axons (>300µm in diameter). Smaller squid and smaller axons are not suitable as the viscoelastic properties of the axoplasm will lead to disruption of the axoplasm from smaller axons (<150µm) (unpublished data) (Brady et al., 1993).
A healthy squid with a translucent body is chosen and decapitated to begin the dissection. The head and tentacles are discarded. The mantle is cut along the dorsal midline to a sheet of muscle that is placed skin side down on the dissecting light table with running seawater. The viscera are removed and the clear pen is carefully pulled away from the mantle, taking care to avoid tearing the nerve fibers that are along each side of the pen. The mantle is turned over and the skin is peeled away to improve visualization of the nerves, then the mantle is pinned to the table with the nerves on the upper surface.
Dissect the nerve bundle containing the largest giant axon (paralleling the pen on both sides) free from the mantle using fine dissection scissors (i.e. 4.5 inch with 10 mm cutting edge, spring action Castroviejo curved scissors, available George Tiemann or Fine Science Tools) and Dumont 5–45 forceps. Start adjacent to the stellate ganglion and proceed distally to the point where the nerve moves deeper into the muscle, approximately 3 cm (Figure 2). This is rough dissection, taking care to leave connective tissue and smaller axons surrounding the giant axon. These will be removed at the fine dissection step. Leaving them at this stage reduces the chance of damaging the giant axon. When the nerve fibers are freed from the mantle, tie off the two ends with cotton thread to facilitate handling. To ensure that orientation is maintained, proximal and distal ends are ligated with cotton thread (black for distal and white for proximal ends). The ganglion is freed from the mantle and the distal end of the nerve beyond the ligation. The nerve should be handled by the threads to avoid damage. Axons from Loligo with mantles >0.3 m typically run 400–500µm in diameter and will produce ~2.5µl per centimeter of giant axon.
Figure 2.

Dissection of giant axons. A diagram illustrating the position of the squid giant axons after removal of the internal organs and pen. The squid mantle is roughly conical after the head and tentacles are removed. At that point, the mantle is cut along the dorsal midline to create flat sheet of muscle. The viscera can then be removed along with clear siff pen located in the center of the ventral side of the mantle, between the two largest giant axons. All axons exist in pairs on the right and left side of the midline. The largest diameter axons extend the paralleling the midline and are the longest axons. The other smaller axons are not used because they are not typically large enough to extrude. The two largest giant axons are dissected free from the surrounding muscle and threads are tied at the distal (green) and proximal ends (red, near the cell bodies in the stellate ganglion). These
The dissected nerve is transferred to a 100mm Petri dish containing cold filtered seawater (use 0.45µm pore filters and maintain at 0–4°C) for fine dissection. We prefer natural seawater to reveal axon damage that may affect transport, but some use Ca2+-free artificial seawater for the fine dissection. The nerve bundle is fixed place by wrapping the thread around 18g hypodermic needles inserted into a small amount of dental wax (Surgident Periphery Wax) placed at opposite edges of the Petri dish.
Using dark-field illumination and a stereo dissection microscope, small axons and connective tissue are gently teased away from the giant axon with Vannas-style iris scissors and No. 5 Dumont forceps, taking care to avoid damaging the membrane of the giant axon. We recommend starting proximal to the stellate ganglion and proceeding distally. Particular caution is needed to avoid cutting small collateral branches that extend from the giant axon occasionally. They may be detectable as slight protrusions on the axon surface and the giant axon may be slight reduced distal to the branch. These collateral branches need to be cut at least 0.1 mm away from the giant axon. After removal of extraneous tissues, the axon is inspected for the presence of small holes, which can be identified as white patches due to the influx of Ca2+ from the seawater. Axons with significant white patches should be discarded as the Ca2+ activates proteases and may disrupt axoplasmic organization.
Axons to be extruded are removed from the seawater and rinsed briefly in a suitable intracellular buffer (Buffer X, see below). Holding the axon by the distal thread, the axon is placed briefly on filter paper to remove excess fluid and cut adjacent to the proximal thread. The axon is placed on a glass slide or 0 thickness 24×60mm coverslip to extrude for assay of vesicle motility or many biochemical and radiolabeling studies (Brady et al., 1985; Brady et al., 1993; Leopold et al., 1994). To extrude, a section of PE190 tubing is used to compress the distal gently but firmly and the axon is pulled using the distal thread. This leaves a cylinder of axoplasm behind on the coverslip (Figure 3). Typically, a cylinder of 2–2.5cm in length and roughly 0.4mm in diameter is used for microscopy, but longer extrusions may be useful for biochemical studies. For imaging, spacers are made by cutting 22×22 mm 0 thickness coverslips into 5×22 mm sections with a diamond tipped pen and positioning them on either side of the axoplasm secured by a thin coating of Compound 111 silicon grease (Dow Corning). Compound 111 is stable in the seawater and non-toxic. Do not use high vacuum silicon grease, which is not designed to seal against aqueous solutions and will be extracted. A top 22×22 0 thickness coverslip is placed to create a sealed chamber. The top coverslip may be secured with 1:1:1 mix of Vaseline:lanolin:paraffin (VALAP) kept fluid at 50–60°C, and this creates a micro-incubation chamber that will maintain the axoplasm for hours. The ends of the chamber are normally kept open to allow perfusion. Chambers prepared in this way have a volume of approximately 25µl, permitting perfusion of axoplasm with buffers a defined ratio of 1:1 to 1:5, minimizing dilution of axoplasm and allowing introduction of reagents at defined concentrations.
Figure 3.
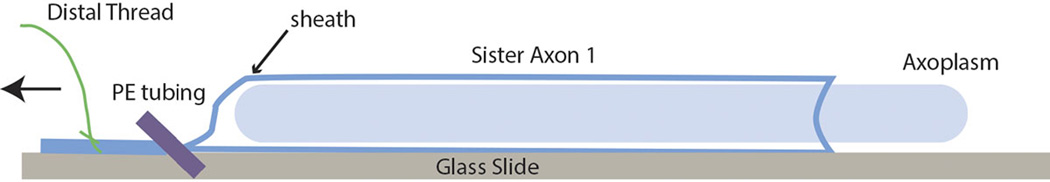
Extrusion of axoplasm. Once the axon is dissected and cleaned of surrounding small axons/connective tissue, the axoplasm must be extruded. Briefly, the axon is handled by the threads and rinsed in intracellular buffer (buffer X) to remove Ca2+ containing sea water. The axon is then blotted on filter paper and cut adjacent to the proximal thread. Using the distal thread, the axon is placed on the coverslip or glass slide. If the goal is to extrude an axoplasm on the surface, one places pressure on the axon close to the thread with a short piece of PE190 tubing and pulls the axon with the thread holding the tubing in place. To extrude into a buffer chamber, the axon is held stationary and the tubing is moved toward the proximal end.
Alternatively the proximal end may be placed in a small reservoir to extrude into a buffer of choice (Morris & Lasek, 1982). If the extrusion is into a reservoir, the axon is held in place with the proximal end in the fluid and the rest of the axon on a clean glass slide. The tubing is slid from the distal end towards the proximal end. In this case, the axoplasm will extrude into the reservoir while maintaining the form of the axon(Morris & Lasek, 1982).
After extrusion, we routinely incubate the axoplasms in a humidified chamber at 4°C for 15–30 min, which allows the axoplasm to “rest” after extrusion. Empirically, this produces more reproducible measurements.
Choice of buffers is critical for study of in situ microtubule properties. The axoplasm is unique in that the small molecular weight components of the cytoplasm can be directly measured (table 1) (G. J. Deffner & R. E. Hafter, 1960; Morris & Lasek, 1982). Standard buffers used for study of microtubules in vitro such as BRB80 (80mM PIPES, 1mM MgCl2, 1mM EGTA, ph6.8) and others (Borisy, Marcum, Olmsted, Murphy, & Johnson, 1975; Olmsted & Borisy, 1975; Weisenberg, 1973) diverge significantly from in vivo conditions. Analysis of axoplasm shows that in vivo conditions have high levels of organic anions (such as amino acids) and K+ is the major cation (Brady, Lasek, & Allen, 1982; Brady et al., 1985; Morris & Lasek, 1982). Axoplasm is a highly reducing environment, consistent with observations in other cell types. Although total Ca2+ is 3.5mM, free Ca2+ levels are much lower, on the order of 100 nM. The axoplasmic cytoskeleton is sensitive to both ionic composition and ionic strength ((Brady et al., 1985; Brown & Lasek, 1990, 1993).
Table 1.
Nonprotein Composition of Axoplasm
| CLASS | COMPONENT | mM |
|---|---|---|
| Amino Acids | Nonpolar AA (alanine) | 16.14 |
| Polar AA (glycine) | 18.41 | |
| Acidic AA (aspartate) | 100.29 | |
| Basic AA (arginine) | 6.06 | |
| Betaine | 73.7 | |
| Taurine | 106.7 | |
| Homarine | 20.4 | |
| Cysteic Acid | 4.9 | |
| Organic Metabolites | Isethionic Acid | 164.6 |
| Glycerol | 4.35 | |
| Others | 11.02 | |
| Carbohydrates | Glucose | 0.24 |
| Mannose | 0.92 | |
| Fructose | 0.24 | |
| Sucrose | 0.24 | |
| Inorganic Ions | Potassium | 344 |
| Chloride | 151.2 | |
| Sodium | 35 | |
| Phosphate | 17.8 | |
| Magnesium | 10 | |
| Sulfate | 7.5 | |
| Calcium | 3.5 | |
| Nucleoside Triphosphates | ATP | 1 |
Adapted from (G. G. Deffner & R. E. Hafter, 1960; Morris & Lasek, 1982)
An alternative buffer, Buffer X (Table 2), was developed which retained the major biochemical features of the axoplasmic milieu (Brady et al., 1985). Potassium aspartate serves as the major organic anion with glycine representing the other amino acids. Taurine is the major reducing agent and betaine serves as an organic osmolyte. EGTA is added to buffer the free Ca2+ at 50–100nM. Halides (F, Cl, Br, and I) may be problematic in many studies as they can alter protein-protein interactions significantly (Collins, 2004, 2006; Hearn, Hodder, & Aguilar, 1988; Westh, Kato, Nishikawa, & Koga, 2006) and disrupt the axoplasmic cytoskeleton (Baumgold, Gallant, Terakawa, & Pant, 1981; Brown & Lasek, 1993), so they are minimized in Buffer X. Extruded axoplasm placed in buffer X will retain its overall shape and organization for >24h (Morris & Lasek, 1982), and will maintain fast axonal transport for >3–4 hours (Brady et al., 1985; Brady et al., 1993).
Table 2.
Buffer X for squid axoplasm experiments (Composition and Instructions)
| Chemical | MW |
M (in stock) |
grams per 25 ml stock |
mM in X |
ml stock in 25 ml Buffer X |
|---|---|---|---|---|---|
| K-aspartate | 171.2 | 1.0 | 4.28 | 350 | 8.75 |
| Taurine | 125.1 | add by wt. | add by wt. | 130 | 0.407 g |
| Betaine | 135.2 | 1.0 | 3.38 | 70 | 1.75 |
| Glycine | 75.07 | 1.0 | 1.88 | 50 | 1.25 |
| MgCl2·6H2O | 203.31 | 1.0 | 5.08 | 12.9 | 0.323 |
| K2·EGTA (adjust stock to pH 7.2 with KOH before bringing to final volume) |
380.4 | 0.1 | 0.9510 | 10 | 2.5 |
| HEPES (adjust stock to pH 7.2 with KOH before bringing to final volume) |
238.3 | 1.0 | 5.96 | 20 | 0.5 |
| CaCl2·2H2O | 147.02 | 1.0 | 3.68 | 3 | 0.075 |
| Glucose | 180.2 | 0.1 | 0.4505 | 1 | 0.25 |
| K2·ATP (adjust stock to pH 7.2 with KOH before bringing to final volume) |
583.4 | 0.2 | 2.92 (0.1167 g/ml) |
1.0 | 0.25 |
Combine aliquots of stock reagents as indicated in the table and check pH. Adjust pH to 7.2 with KOH if needed. Bring to final volume of 25 ml with 20 mM HEPES pH 7.2 and filter on 45µm Millipore filter or equivalent. Store in suitable aliquots at –20 °C until ready for use. We routinely prepare aliquots without ATP, bringing the stock to 20 ml rather than the 25 ml final volume and storing as 0.4 ml aliquots. Immediately prior to use, an aliquot is thawed and ATP added from a concentrate stock. Experimental agents can be added at this point and the aliquot brought to a final volume of 0.5 ml.
Analysis of Axoplasmic Microtubule Dynamics
The stability of axoplasmic organization in buffer X provides a unique model study of microtubule and actin dynamics in situ (Morris & Lasek, 1984). Tubulin dimers represents 22% of total axoplasmic protein, with a concentration of approximately 25µM (Morris & Lasek, 1984). Squid microtubules resemble mammalian microtubules in many ways with regard to its polymerization (although at optimal temperature as 25°C) and depolymerization (by Cold, Ca2+ ions, colchicine, etc) (Sakai & Matsumoto, 1978) Dilution of extruded axoplasm by 1000 fold in buffer X allows determination of stable polymer, soluble polymer and free tubulin dimer in axoplasm from a single axon based on the kinetics of extraction for tubulin into the media (Morris & Lasek, 1984). This approach defines a Kinetic Equilibration Paradigm (KEP), which provides a unique perspective on microtubule dynamics in situ.
The KEP method allows one to define discrete pools The amount of free dimer in this assay was significantly less than levels reported from in vitro assays of microtubule polymerization and the level of polymer remaining after 24h extraction was 15% of the total tubulin (Morris & Lasek, 1984). This is consistent with the observed stability of microtubules in isolated axoplasm with minimal dilution (Weiss, Langford, Seitz-Tutter, & Keller, 1988).
For analysis of depolymerization kinetics, axoplasm should be extruded into a reservoir as described above. The axoplasm will retain its form suspended in the buffer. At this point the axoplasm can be picked up with Dumont forceps, taking care to avoid shearing. Elution of tubulin from the axoplasm can be monitored either by taking aliquots of the media at suitable intervals or by physically transferring the axoplasm into a fresh reservoir of buffer (Figure 4, adapted from (Song, 2010)).
Figure 4.

Kinetic Equilibration Paradigm (KEP). This method was developed originally by Morris and Lasek (Morris & Lasek, 1982, 1984) for analysis of monomer-polymer equilibrium of cytoskeletal proteins in the axoplasm. In this diagram, green dots represent modified tubulin with increased stability and white dots represent unmodified labile tubulins (Song et al., 2013). After placing the axoplasms in buffer X, unassembled tubulin dimers would be extracted into the buffer by 5 minutes. Labile microtubules begin to depolymerize by this time and by 45 minutes, the tubulin from labile microtubules (soluble polymer) has been extracted. By 120 minutes only stable microtubules remain as an axoplasmic “ghost” and does not change over periods as long as 24 hours. These ghosts are are enriched in modified tubulins.
Alternatively, microtubule behavior can be monitored directly in chambers by imaging with video-enhanced contrast differential interference microscopy (Allen, Allen, & Travis, 1981; Weiss, Langford, Seitz Tutter, & Maile, 1991; Weiss et al., 1988). This is best done by use of lower ionic strength buffers (i.e. buffer X/2) that promote separation of cytoskeletal elements from the main axoplasm (Brown & Lasek, 1993; Weiss et al., 1988). Exogenous tubulin can be perfused into the chambers and changes in microtubule numbers and stability followed (Weiss et al., 1988). A variant on this approach would perfuse tubulin with a fluorescent tag either chemical (Texas red) or a protein (GFP, mCherry, etc.). Incorporation of fluorescent tubulin could be monitored and the effects of different experimental manipulations evaluated.
Biochemistry of Axoplasmic Microtubules
Since microtubules from extruded axoplasm are purely axonal and represent neuronal microtubule properties in this ex vivo setting, various biochemical experiments can be carried out to examine different tubulin isotype components, post-translational modifications of tubulins, distribution of microtubule associated proteins (MAPs), etc. In addition, experimental manipulation of microtubule modifications can be performed to understand how a particular modification may alter microtubule structure, its dynamics and stability and its affinity for motor proteins and MAPs(Song, 2010).
For biochemical experiments, two “sister” axoplasms of the same length from one squid are extruded on glass slides as described above, one serves as a control while the other is used for experimental manipulations (Figure 5). This ensures that the variability between individual squids is minimized. A circle is drawn around each of the two freshly extruded axoplasms using a Pad Pen (Zymed) or liquid blocker (Super Pap) to delineate the perfusion area before being placed in a humid chamber. Placing the slide on a piece of wet paper towel in the chamber may enhance humidity. After being incubated at 4°C for 10 minutes, a control axoplasm is perfused with 30µl of Buffer X/2+5mM ATP alone, while the experimental axoplasm is perfused with the appropriate effector mixed in the same buffer. Both axoplasms are incubated for sufficient amount of time at a temperature determined by specific experimental conditions. Axoplasms are transferred into microcentrifuge tubes where they are homogenized in 30µl of SDS (1%) or other proper buffers by trituration with a P200 pipette. The samples can be analyzed by SDS-PAGE, followed by immunoblot, autograph (if radio labeled) or direct gel documentation (if fluorescently labeled).
Figure 5.

Perfusion of axoplasms. Buffers of interest can be perfused into axoplasm for studies of microtubule biochemistry or imaging. Ideally, sister axoplasms (two axoplasms from a single squid) are used for comparison to reduce interanimal variability. For biochemistry, the axoplasms are perfused without a top coverslip while imaging studies require the top coverslip. In both cases, buffer volumes are kept small relative to the axoplasm volume to minimize dilution. Typical axoplasms are 5µl in volume, so buffer volumes are kept at 20–25µl. This is in contrast to conventional biochemical approaches where dilutions may be 103–105.
This procedure can also be combined with several other assays such as 1) the axoplasmic microtubule dynamics assay as described above to assess how specific modifications, pharmacological reagents, or MAPs alter microtubule dynamics. 2) Assays for microtubule pelleting, labeling tubulin, tubulin polymerization, etc (for detailed protocols on those, see http://mitchison.med.harvard.edu/protocols.html). 3) Immunoprecipitation assay to identify interacting proteins or 4) evaluation of pharmacological agents.
The pharmacology of axonal microtubules is of particular interest. For example, antimitotic drugs have been widely used in cancer therapy (Jordan & Wilson, 2004), many of which cause neurological side effects (Windebank & Grisold, 2008), but the underlying mechanism is not well understood. Since many of these drugs alter microtubule dynamics and stability (Jordan & Kamath, 2007), it is important to analyze microtubule properties in axons after the drug treatment. The ex vivo squid axoplasm system provides well-organized axonal microtubule structure that can be easily analyzed both morphologically (see below) and biochemically. Concurrent analysis of fast axonal transport in response to treatment with different drugs and biochemical reagents (Brady et al., 1993) provide an additional cell biological dimension to studies of microtubules in axoplasm.
Immunohistochemistry of Axoplasmic Microtubules
A unique feature of the extruded axoplasm is the well-preserved axonal microtubule structure that can be analyzed for morphological changes upon pharmacological treatment, the distribution of other proteins that may or may not interact with microtubules (such as motor proteins, MAPs and various kinases/phosphatases that modify microtubules and their associated proteins), and the incorporation of exogenous proteins perfused into the system.
Here we use one example to look at how different tubulin mutations may affect the incorporation of tubulins into microtubules. V5-tagged beta III tubulins harboring various mutations (Tischfield et al., 2010) can be synthesized in vitro using a rabbit reticulate system. “Sister” axoplasms are prepared as described in the biochemistry section, incubated with 30µl of Buffer X/2+5mM ATP alone as a control or mutant tubulin diluted in the same buffer for 50 minutes at room temperature. Buffer is removed at the end of incubation and extra solution around the axoplasm is carefully dried by placing a piece of Whatman paper close to the edge of the axoplasm. 50 µl of 4% PFA diluted in PBS is used to fix one axoplasm for 50 minutes at room temperature and then carefully removed as described above. Axoplasms together with the slide are carefully placed in a conical tube filled with 30ml PBS for 10 minutes each and repeated 3 times to wash away the fixative. 1% BSA and 0.1% Triton X100 in PBS is used to block for at least an hour at room temperature followed by incubation with primary antibodies (anti-V5 and DM1A) diluted in blocking buffer at 4 degrees Celsius for overnight. Same washing by passing through the conical tubes is repeated 3 times in PBS before incubation in fluorescent secondary antibodies (Alexa 488 anti-mouse and 594 anti-rabbit) diluted in PBS for an hour at room temperature. After the final washes to remove the secondary antibody, spacers made out of No.0 coverslips are coated on either side of the axoplasm on the glass with Compound 111 silicon grease and axoplasms are mounted with mounting medium (such as ProLong Gold Mounting medium) between the two spacers and covered with a No.1.5 coverslip. For long-term storage, nail polish can be applied to seal the edges. Images are taken and data analyzed by confocal microscopy. These approaches can also be modified to allow analysis of the axoplasmic microtubules by electron microscopy.
References
- Allen RD, Allen NS, Travis JL. Video-enhanced contrast, differential interference contrast (AVEC-DIC) microscopy: A new method capable of analyzing microtubule related movement in the reticulopodial netowork of Allogromia laticollaris. Cell Motil. 1981;1:291–302. doi: 10.1002/cm.970010303. [DOI] [PubMed] [Google Scholar]
- Baumgold J, Gallant P, Terakawa S, Pant H. Tetrodotoxin affects submembranous cytoskeletal proteins in perfused squid giant axons. Biochem Biophys Res Commun. 1981;103(2):653–658. doi: 10.1016/0006-291x(81)90500-3. [DOI] [PubMed] [Google Scholar]
- Borisy GG, Marcum JM, Olmsted JB, Murphy DB, Johnson KA. Purification of tubulin and associated high molecular weight proteins from porcine brain and characterization of microtubule assembly in vitro. Ann N.Y. Acad. Sci. 1975;253:107–132. doi: 10.1111/j.1749-6632.1975.tb19196.x. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD. Fast axonal transport in extruded axoplasm from squid giant axon. Science. 1982;218:1129–1131. doi: 10.1126/science.6183745. [DOI] [PubMed] [Google Scholar]
- Brady ST, Lasek RJ, Allen RD. Video microscopy of fast axonal transport in isolated axoplasm: A new model for study of molecular mechanisms. Cell Motil. 1985;5:81–101. doi: 10.1002/cm.970050203. [DOI] [PubMed] [Google Scholar]
- Brady ST, Richards BW, Leopold PL. Assay of vesicle motility in squid axoplasm. Meth. Cell Biol. 1993;39:191–202. doi: 10.1016/s0091-679x(08)60171-5. [DOI] [PubMed] [Google Scholar]
- Brown A, Lasek RJ. The cytoskeleton of the squid giant axon. In: Gilbert DL, Adelman JWJ, Arnold JM, editors. Squid as Experimental Animals. New York City: Plenum Publishing Corp.; 1990. pp. 235–302. [Google Scholar]
- Brown A, Lasek RJ. Neurofilaments move apart freely when released from the circumferential constraint of the axonal plasma membrane. Cell Motil. Cytoskel. 1993;26(4):313–324. doi: 10.1002/cm.970260406. [DOI] [PubMed] [Google Scholar]
- Collins KD. Ions from the Hofmeister series and osmolytes: effects on proteins in solution and in the crystallization process. Methods. 2004;34(3):300–311. doi: 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Collins KD. Ion hydration: Implications for cellular function, polyelectrolytes, and protein crystallization. Biophys Chem. 2006;119(3):271–281. doi: 10.1016/j.bpc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Deffner GG, Hafter RE. Chemical investigations of the giant nerve fibers of the squid. III. Identification and quantitative estimation of free organic ninhydrin-negative constituents. Biochim Biophys Acta. 1960;42:189–199. doi: 10.1016/0006-3002(60)90780-0. [DOI] [PubMed] [Google Scholar]
- Deffner GJ, Hafter RE. Chemical investigations of the giant nerve fiber of the squid. Biochim. Biophys. Acta. 1960;42:200–205. doi: 10.1016/0006-3002(60)90781-2. [DOI] [PubMed] [Google Scholar]
- Hearn MT, Hodder AN, Aguilar MI. High-performance liquid chromatography of amino acids, peptides and proteins. LXXXVI. The influence of different displacer salts on the retention and bandwidth properties of proteins separated by isocratic anion-exchange chromatography. J Chromatogr. 1988;443:97–118. doi: 10.1016/s0021-9673(00)94786-1. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Kamath K. How do microtubule-targeted drugs work? An overview. Curr Cancer Drug Targets. 2007;7(8):730–742. doi: 10.2174/156800907783220417. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Leopold PL, Lin J-W, Sugimori M, Llinas R, Brady ST. The nervous system of Loligo pealei provides multiple models for analysis of organelle motility. In: Abbott NJ, Williamson R, Maddock L, editors. Cephalopod Neurobiology: Neuroscience studies in squid, octopus and cuttlefish. Oxford: Oxford Univ. Press; 1994. pp. 15–34. [Google Scholar]
- Morris J, Lasek RJ. Stable polymers of the axonal cytoskeleton: The axoplasmic ghost. J. Cell Biol. 1982;92:192–198. doi: 10.1083/jcb.92.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J, Lasek RJ. Monomer-polymer equilibria in the axon: Direct measurement of tubulin and actin as polymer and monomer in axoplasm. J. Cell Biol. 1984;98:2064–2076. doi: 10.1083/jcb.98.6.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted JB, Borisy GG. Ionic and nucleotide requirements for microtubule polymerization in vitro. Biochemistry. 1975;14(13):2996–3005. doi: 10.1021/bi00684a032. [DOI] [PubMed] [Google Scholar]
- Sakai H, Matsumoto G. Tubulin and other proteins from squid giant axon. [Comparative Study] J Biochem. 1978;83(5):1413–1422. doi: 10.1093/oxfordjournals.jbchem.a132051. [DOI] [PubMed] [Google Scholar]
- Song Y. Stabilization of Neuronal Microtubules by Polyamines and Transglutaminase: Its Roles in Brain Function. (Ph.D. ) Chicago, IL US: 2010. [Google Scholar]
- Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, Brady ST. Transglutaminase and Polyamination of Tubulin: Posttranslational Modification for Stabilizing Axonal Microtubules. Neuron. 2013 doi: 10.1016/j.neuron.2013.01.036. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, Engle EC. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberg RC. Microtubule formation in vitro from solutions containing low calcium concentrations. Science. 1973;177:1104–1105. doi: 10.1126/science.177.4054.1104. [DOI] [PubMed] [Google Scholar]
- Weiss DG, Langford GM, Seitz Tutter D, Maile W. Analysis of the gliding, fishtailing and circling motions of native microtubules. Acta Histochem.Suppl. 1991;41:81–105. [PubMed] [Google Scholar]
- Weiss DG, Langford GM, Seitz-Tutter D, Keller F. Dynamic instability and motile events of native microtubules from squid axoplasm. Cell Motil. Cytoskel. 1988;10:285–295. doi: 10.1002/cm.970100133. [DOI] [PubMed] [Google Scholar]
- Westh P, Kato H, Nishikawa K, Koga Y. Toward understanding the Hofmeister series. 3. Effects of sodium halides on the molecular organization of H2O as probed by 1-propanol. J Phys Chem A. 2006;110(5):2072–2078. doi: 10.1021/jp055036y. [DOI] [PubMed] [Google Scholar]
- Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13(1):27–46. doi: 10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]