

Abstract
In the late stage of human immunodeficiency virus-1 (HIV) infection, a subset of individuals develops HIV associated neurocognitive disorders (HAND), which in its severe form, is characterized by motor and cognitive dysfunction. Dendritic pruning, synaptic abnormalities and neuronal apoptosis are observed in these patients. There are numerous advances in our understanding of HIV interactions with cells of the central nervous system. However, the underlying causes of neurological symptoms and pathological alterations observed in HIV positive subjects are poorly understood. Moreover, little is still known about the molecular mechanisms by which HIV induces synaptic dysfunction and degeneration. HAND resembles other common neurological diseases such as Alzheimer’s and Huntington’s diseases. These neurodegenerative disorders are characterized by accumulation of toxic proteins such as tau and huntingtin, respectively, which promote axonal degeneration by impairing axonal transport. Axonal degeneration precedes neuronal death. Therefore, a better understanding of the mechanisms whereby HIV triggers axonal degeneration has potential implications for developing therapeutic compounds to prevent synaptic failure in HAND. This article highlights and reviews evidence showing that neuronal accumulation of viral proteins promotes axonal damage.
Keywords: Alzheimer’s disease, axonal degeneration, brain-derived neurotrophic factor, gp120, HAND, HIV, microtubules, neurocognitive impairments
INTRODUCTION
The human immunodeficiency virus type 1 (HIV) promotes a progressive depletion of T cells, causing acquired immune deficiency syndrome (AIDS). In addition, during the first few weeks after initial systemic infection, HIV invades the central nervous system (CNS) through migrating myeloid and lymphoid cells [1–4], and establishes a persistent infection in perivascular macrophages and microglia [5]. Infection, which is often associated with glial and endothelial activation and is referred to as neuroAIDS, promotes inflammation and degenerative insults that lead to cognitive and motor impairments termed HIV-associated neurocognitive disorders (HAND). HAND includes three degrees of neurocognitive disorders, asymptomatic neurocognitive impairments, mild neurocognitive disorders and HIV-associated dementia (HAD) [6].
HIV infection, replication and AIDS are significantly reduced by the combined antiretroviral therapy (cART), which includes a cocktail of nucleotide reverse transcriptase inhibitors, non-nucleotide reverse transcriptase inhibitors, protease inhibitors, or integrase inhibitors. Before the introduction of the cART, a substantial proportion (20–50%) of HIV positive subjects exhibited HAD. This severe form of dementia was characterized as a progressive disabling disorder that manifested as loss of attention and concentration, notable motor slowing, and severe behavioral impairments [7]. cART has shown positive outcome in reversing and preventing HAD. In fact, as treatment was initiated, patients showed reliable improvement on timed motor tasks and obvious clinical neurological progress, although a substantial proportion of them exhibited mild (33%) or moderate (12%) neurocognitive impairments [8, 9]. One of the reasons why neurocognitive impairments persist is attributed to the fact that these patients have detectable levels of HIV RNA in their cerebrospinal fluid even when it is undetectable in blood [10]. Thus, it is plausible to suggest that the brain concentration of drugs in cART is not sufficient to remove HIV replication in the brain. There is also evidence that the CNS of some individuals acts as an HIV sanctuary site unless a highly CNS-penetrating antiretroviral regimen is initiated [11]. Unfortunately the chemical characteristics of most antiretroviral drugs prevent them from crossing the blood brain barrier. On the other hand, there are other potential pathogenic mechanisms that may explain the neurologic symptoms of HAND including the neurotoxicity of antiretroviral drugs and the persistent activation of microglia cells [12]. Indirect effects from comorbid conditions such as cardiovascular and cerebrovascular diseases [13] as well as hepatitis C co-infection, which alone affect cognitive function [14] could also play a role in predisposing HIV subjects to cognitive alterations.
In this article we provide experimental data on how HIV may cause neuronal degeneration. In particular, we present and discuss new evidence suggesting that HIV infection of the brain promotes aggregation of viral proteins, which similar to the inclusion of proteins in other chronic neurodegenerative diseases, causes neural injury.
HIV accelerates the aging of the brain
cART has increased the survival of AIDS patients [15]. A comparative analysis assessing HIV-positive and -negative patients from the pre-cART and post-cART eras has shown that in the HIV-positive cohort impairments in motor skills, cognitive speed, and verbal fluency predominated in the pre-cART era, whereas impairments in memory and learning, and executive function predominated in the post-cART era [16]. Subtle cognitive changes in HIV patients in the post-cART era are also seen in prospective memory, or the ability to ‘remember to remember’, which could affect function in the workplace and impair adherence to medication. Arterial spin labeling, which measures cerebral blood flow, has shown that soon after seroconversion cerebral blood flow in HIV+ individuals is equivalent to HIV-seronegative people who are 15–20 years older [17]. Thus, it appears that HIV accelerates the neurodegenerative process to a degree similar to what has been observed in aging.
CNS volumetric analysis of HAND patients has shown that HIV infection is associated with a significant reduction in volume in selected subcortical regions including the amygdala, the striatum and the corpus callosum [18, 19]. Basal ganglia are particularly vulnerable in severely immunosuppressed subjects [20]. The pathology of the basal ganglia, as assessed by a global atrophy of the caudate nucleus and putamen, persists even in the era of cART [18] and correlates with cognitive impairment [21]. Moreover, cortical areas such as the frontal and superior temporal gyri as well as the cingulate areas also exhibit reduction of volume [22]. Neuronal atrophy and hypoactivity in the frontal cortex is also seen in other chronic degenerative diseases such as Alzheimer’s disease (AD) [23]. The earliest clinical signs of AD include memory impairment as well as deficit in language and executive functioning. A similar pattern of cognitive decline is now reported in HAND subjects although they are younger than HIV negative patients with AD. Clinical preliminary data support a link between HAND and AD. In fact, a sixty-three year-old HAND patient receiving cART has lower glucose accumulation, a marker of hypometabolism, measured by positron emission tomography (PET) using the glucose analogue [18F]-fluorodeoxyglucose, in several cortical areas, including parietal, frontal and temporal lobes. Voxel-based statistical analysis comparing this subject with a normal population (Figure 1) has revealed pathological features consistent with loss of function and cortical gray matter typical of AD. Additional PET scan with biomarkers of AD will further support a diagnosis of AD. The same patient exhibited a hypermetabolism in the caudate/putamen (Figure 1), consistent with HAND. It is possible, but not proven, that the patient may have both AD and HAND diagnoses for his mild clinical dementia. Moreover, functional magnetic resonance imaging has shown that HIV promotes a pattern of degradation in brain function similar to aging [24]. These data are consistent with the notion that HIV positive individuals demonstrate an age-decrease decline in brain activation and that aging has a deleterious effect in HIV positive subjects [25], possibly placing older patients who are HIV positive to an increased risk for developing cognitive impairment.
Figure 1.

Statistical analysis of a HAND patient FDG PET/CT compared to a reference normal population. A. Tridimensional maps of Voxel-Based analysis. The voxels that show significant changes in hypometabolism in the HAND subject compared to normal are shown in blue, those showing significant hypermetabolism are shown in red. B. Table of the regional analysis showing the corresponding standard deviations from the normal population for all the regions showing statistically significant changes in the 3D map. BG = Basal ganglia; FL = Frontal lobe; PC = Posterior cingulate; PL = Parietal lobe, TL = Temporal lobe.
Although there is no evidence linking AD to HIV infection, there are clinical and pathological similarities between HAND and AD as well as in the mechanisms shared by these diseases that can potentially explain the selective loss of neurons. One similarity concerns the apoprotein E ε4 allele (APOEε4), which encodes for a protein important for shuttling fatty substances throughout the body and regulates cholesterol metabolism. This gene is a major risk factor for impaired neurocognitive function in AD and it has been used as a major genetic indicator of AD [26]. Younger HIV positive APOEε4 carriers exhibit loss of putamen and cerebral white matter volume to a degree similar to older HIV positive non-APOEε4 carriers [27]. In addition more disruption of the frontal and motor connections is seen in older HIV APOEε4 carriers than non-carriers [28]. Other studies have indicated that HIV positive patients exhibit a higher deposition of β-amyloid protein than HIV negative patients [29]. This protein is an abundant component of amyloid plaques that are found throughout the AD brain and used to diagnose pathological AD. Although the correlation between the presence of β-amyloid and APOEε4 and the cause of HAND and AD are still under investigation, the similarities between HAND and AD could create challenges in the differentiation of these diseases in elderly patients with HIV. Overall, it appears that HIV accelerates brain atrophy and alteration of synaptodendritic network typically seen during the ageing process even during cART. These clinical data form a rationale of why we need to know more about the mechanisms whereby HIV infection leads to cognitive dysfunction in order to develop new treatments.
HIV infection and chemokine receptors
HIV belongs to a family of primate lentiviruses (a subgroup of retroviruses). Like other retroviruses, HIV contains a viral capsid, the diploid single-stranded RNA genome, and the three viral enzymes, protease, reverse transcriptase, and integrase. Reverse transcriptase is the hallmark of a retrovirus and this enzyme is capable of transcribing its genomic RNA into double-stranded DNA. The virion envelope consists of a lipid bilayer membrane, derived from the host cell, and a virally encoded tetrameric envelope protein complex, of which each subunit consists of two non-covalently-linked membrane proteins, gp120 and gp41. The envelope protein complex facilitates viral entry by binding to the receptor CD4, the main cellular receptor for all primate lentiviruses, via the outer envelope protein gp120 [30]. The transmembrane protein gp41 is involved in the fusion of viral envelope with cellular membrane. In addition to these structural proteins found in all retroviruses, HIV contains a series of accessory proteins such as Tat and Nef. The function of the Tat protein is to up-regulate transcription from the viral promoter whereas Nef is required for efficient replication in vivo.
Initial viral binding to cell membrane and subsequent entry into host cells are catalyzed by a high affinity interaction between gp120 and the host cellular surface antigen CD4. CD4 is found on the surface of immune cells such as T helper, monocytes, macrophages, and dendritic cells. The first evidence that implicated CD4 as the primary HIV receptor was the ability of anti-CD4 monoclonal antibodies to inhibit virus infection [31]. However, CD4 is necessary but not sufficient to support virus entry. Indeed, the interaction between gp120 and CD4 causes gp120 to undergo a conformational change, exposing the co-receptor binding site [32]. The chemokine receptors CCR5 and CXCR4 are the most utilized co-receptors [33–36]. Different strains of HIV can use either CCR5 or CXCR4 or both to enter cells [37, 38]. R5 viruses use CD4 and CCR5 to infect and replicate efficiently in macrophages and microglia, whereas X4 viruses use CD4 and CXCR4 to enter and replicate in T lymphocytes. X4 virus can also infect astrocytes in a CD4-independent manner [39]. Dual tropism viruses that use both co-receptors are designated R5X4. The presence of R5 or X4 strains varies during the course of the infection. R5 tropic viruses predominate during the initial phase of infection and are transmitted with greater efficiency, whereas the X4 viruses emerge later during the disease and are associated with rapid progression to AIDS [40].
HIV tropism in the brain
Brain-derived HIV isolates differ from those typically found in systemic circulation [41, 42]. Moreover, comparisons between peripheral blood-derived viruses and those derived from multiple brain compartments in the same patient have suggested segregated evolution of viral strains present within differing brain regions [43]. Comparative molecular and biological analyses of HIV isolates derived from both HAD and non-HAD patients have suggested that differences in viral tropism can discriminate between patients with and without dementia, indicating that neurotropic HIV variants evolve independently within the brain and contribute to neuropathogenesis [44]. In addition, brain-derived HIV sequences from HAD patients differ from those isolated from non-HAD patients, primarily within the V1 and V3 region of gp120. The V3 loop is considered the major viral determinant for co-receptor usage [45, 46], whereas the V1 loop is crucial for evading antibody-mediated neutralization [31], suggesting that specific HIV envelope sequences may be associated with clinical onset of dementia [47, 48]. One specific envelop variant N283, which is present in the CD4-binding site of gp120, enhances the affinity of gp120 for CD4 by decreasing the dissociation rate between these two molecules [49], thus enabling HIV to utilize lower levels of CD4 for binding and entry and subsequently enhancing viral replication within macrophages and microglia. This variant has been identified at high frequency in brain from HAD patients (41%) compared with non-HAD patients (8%), [49] suggesting that R5 HIV may participate in causing the neurologic impairment. Nevertheless, data from HIV RNA concentration in the cerebral spinal fluid (to measure HIV infection of the CNS) suggest that while HIV infection of the CNS indicates neuronal damage, HIV RNA concentration does not correlate with impaired cognitive function [50].
Mechanisms of HIV-mediated neuronal injury
Productive HIV infection of the CNS is limited to specific cell types such as perivascular macrophages and microglia cells [51]. Very few astrocytes are infected and most of these astrocytes constitute the end feet of the blood brain barrier [52]. Thus, the vulnerability of synapses in HAND does not result from direct intrinsic viral infection of neurons. These data have prompted studies to characterize mechanisms of HIV neurotoxicity that are independent from direct viral infection and have led to the discovery of host factors, cytotoxins, and viral proteins as major causes of neuronal injury [53]. HIV produces nine proteins that play a role in the viral life cycle. At least six of these proteins can damage neurons; these include gp120, gp41, Tat, Nef, Vpr, and Rev. The proteins may evoke neuronal injury by promoting the release of pro-inflammatory cytokines and chemokines from microglia and astrocytes [54–58], thus supporting the notion that HIV neurotoxicity is caused by cytotoxins released from non-neuronal cells. In addition, Tat and gp120 promote the activation of neurotoxic mechanisms involved in glutamate-mediated excitotoxicity, and oxidative stress [59]. Such mechanisms are also been suggested to cause other neurodegenerative diseases but so far therapies to block excitotoxicity have failed to produce a positive clinical outcome. Therefore, excitotoxicity may not explain all pathological features observed in HAND.
Other experimental data have favored the hypothesis of HIV neurotoxicity that occurs by a direct interaction of viral proteins with neuronal pathways/stimuli that promote cell death. This notion is mostly inferred from animal studies and should be viewed with caution because, as mentioned before, hepatitis C co-infection, antiretroviral drug toxicity and cerebrovascular disease may play a role in CNS damage in HAND. Nevertheless, even in the absence of hepatitis or other co-morbidities, viral proteins are neurotoxic. Some HIV proteins, especially Tat [60] and gp120 [61] can be released from infected cells and accumulate inside rodent neurons in vitro and in vivo [61, 62]. Such accumulation occurs without a productive infection (Figure 2) and appears to activate intracellular mechanisms that are neurotoxic because neurons that are gp120-positive exhibit signs of synaptic atrophy. In addition, Tat promotes mitochondrial dysfunction, dendritic loss and cell death [63] at concentrations lower than those needed to support viral replication. Remarkably, both HIV and gp120 are toxic to rodent neurons, similarly to human CNS cultures [64–66]. Moreover, gp120 alone is sufficient to cause neurite pruning, synaptic simplification, and apoptosis in primary neuronal cultures, similar to HIV [67]. In vivo models corroborated these findings. In fact, gp120 transgenic mice exhibit synaptodendritic injury and loss of pyramidal neurons, in several brain areas [68]. In addition, several subtypes of neurons are sensitive to the neurotoxic action of gp120. These include hippocampal [69], cortical [70] and striatal neurons [71]. Loss of neurons in the striatum is followed by a selective degeneration of dopaminergic synapses and neurons in the substantia nigra (SN) [72]. Neurodegeneration can occur in the presence [73, 74] or absence [75] of inflammatory responses, including activated microglia. The ability of gp120 to promote neuronal loss in the hippocampus and the basal ganglia may explain why some HAND subjects experience memory deficits while others develop parkinsonian symptoms.
Figure 2. HIV sheds gp120.
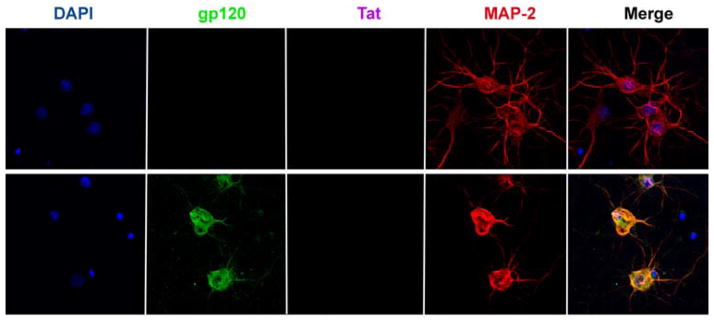
Rat cortical neurons were exposed to (upper panel) heat inactivated HIV or (lower panel) T-lymphotropic HIV (AIDS Research and Reference Reagent program, NIAID, NIH, Bethesda, MD) for 24 h. Neurons were then fixed and stained for gp120, Tat and MAP-2. Please note that Tat was undetectable and that neurons exposed to gp120 exhibit shorter processes.
Neurotrophic factors
There are other mechanisms that could explain the profound loss of synaptic connection in HAD as these subjects exhibit a decrease in neurotrophic factors, which follow or parallel cellular loss and atrophy in brain [76]. Adult neurons rely on the availability of neurotrophic factors for their maintenance and support. Neurotrophic factors are also crucial for proper development of synapses and to promote survival. Lack of trophic support often leads to axonal and dendritic degeneration [77]. One of the most abundant and potent trophic factors in the adult CNS is brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family of growth factor, which includes nerve growth factor, neurotrophin-3, and neurotrophin 4/5.
BDNF possesses a variety of properties that are crucial for cognitive [77], motor [78] and endocrine functions [79]. It is important to note that BDNF modulates synaptic activity involved in learning and memory [80]. Likewise, the lack of BDNF appears to be a common pathological feature in other chronic neurodegenerative diseases. For instance, a deficiency in BDNF synthesis has been described in postmortem brains [81] and cerebrospinal fluid [82] of patients with AD. BDNF expression is also decreased in nigrostriatal dopamine neurons in PD [83, 84], and cortical neurons in schizophrenia [85]. A decrease in BDNF gene expression has been linked to the etiology of Huntington’s disease (HD) [86]. A similar reduction in BDNF levels has been observed in HIV infected T-lymphocytes [87]. Thus, HIV may produce synaptic-dendritic degeneration by altering the endogenous mechanisms of neuronal protection. On the other hand, BDNF has been shown to down-regulate the expression and function of the co-receptor CXCR4, suggesting that BDNF may also reduce the intrinsic neurotoxic mechanisms of gp120 that depend on the activation of this receptor. In fact, experimental data have shown that gp120 is more toxic in BDNF heterozygous (+/−) mice, which express 50% less BDNF than wild type [72].
BDNF binds to two distinct classes of receptors, TrkB, which contains a tyrosine kinase motif and mediates the neurotrophic activity of BDNF, and p75NTR, a member of the tumor necrosis factor family of receptors [88]. The p75NTR contains a death motif [89] and promotes cell death when TrkB is not activated. Examples of neurotoxic action of p75NTR include the death of oligodendrocytes [90], the axonal degeneration in the peripheral [91] and CNS [92], and the mediation of beta-amyloid induced cell death [93]. Cell death induced by p75NTR appears to occur through c-Jun N-terminal kinase [94, 95], a pathway that is also activated by HIV [96]. BDNF binds to p75NTR with low affinity; however, the high molecular weight precursor protein of BDNF (proBDNF) has high affinity for p75NTR and when it activates p75NTR causes cell death [97, 98]. Thus, proBDNF is considered a negative modulator of synaptic plasticity. This issue is important in HAND because gp120 inhibits the processing of proBDNF [76], which in turn induces neurotoxicity [76, 99]. These experimental data suggest that unless HIV seropositive subjects receive antagonists of the p75NTR, their neurons are at high risk to die due to activation of p75NTR by gp120.
There are other growth factors that exhibit neuroprotective properties and may help reduce the neurotoxic activity of HIV; these include glial-derived neurotrophic factor (GDNF), platelet-derived growth factor [100] and fibroblast growth factors [101]. Each growth factor acts on different neuronal populations. However, GDNF appears to have a unique profile of activity. GDNF belongs to a family of trophic factors consisting of four members: GDNF, Neurturin, Artemin, and Persephin. The most important neurotrophic property of this family of growth factor that have a therapeutic importance is the neuroprotection of dopaminergic neurons of the nigrostriatal system [102, 103]. This pathway degenerates in PD. The trophic property of GDNF could also be of a particular importance for HAND, especially at a stage in which HIV seropositive subjects exhibit clinical features that resemble those found in PD, such as the postural instability, involuntary movements, bradykinesia, and impairment in fine motor skills [20]. In addition, the SN of HAD subjects exhibit lower expression of GDNF than HIV subjects without dementia [104]. A similar scenario has been observed in experimental animals injected with gp120 in the striatum [105]. These results, while they suggest that neurotrophic factors can be a useful therapy for HAND, also introduce a new view of the cause of HAND based on the notion that the lack or loss of trophic support could lead to neuronal degeneration in these subjects. Thus, in addition to cART, future clinical trials should contain a therapy based also on neurotrophic factors. Nevertheless, a neurotrophic factor-based therapy should consider that the efficiency of neurotrophic factors to cross the blood brain barrier is less than optimal and that the pharmaceutical composition of BDNF cannot be contaminated by proBDNF.
Intracellular accumulation of gp120
Loss of selective neuronal population is a common feature of many neurodegenerative disorders. However, prominent axonal pathology often precedes neuronal loss in the form of ‘dying back’, in which axons from the synaptic regions gradually degenerate toward the cell body. These phenomena have been observed in HIV patients [106] as well as in gp120-treated animals [68, 105]. Thus, mechanisms leading to the pathology of these diseases could help in discovering and characterizing the cause of HAND.
AD [107], PD [108] and HD [109] are associated with cytoplasmic inclusion of aggregated proteins such as β-amyloid, α-synuclein, and huntingtin, respectively. Misfolded proteins can generate from genetic mutations, inappropriate protein assembly, aberrant post-translational modification, as well as environmental stress that affect their biogenesis. How these inclusions lead to neurodegeneration is beyond the scope of this article. More than simply being nonfunctional, misfolded proteins are prone to forming aggregates that can interfere with normal cellular function and physiological homeostasis [110] by forming insoluble plaques that can lead to a progressive synaptic pruning and neuritic injury. Thus, misfolded proteins are closely monitored, processed, and degraded to prevent their accumulation in cells because aggregated proteins are toxic. The exact mechanisms underlying the etiology about how these proteins create the diseases are unclear. However, drugs that inhibit aggregation and accumulation of misfolded proteins show promising therapeutic effects in animal models of PD, AD [111] and HD [112]. These drugs work mostly on the endogenous mechanism of protein clearance. Under non-injury conditions, neurons maintain a balance between synthesis, transport and degradation of cellular components through the process of autophagy, a regulatory catabolic mechanism that degrades proteins and other components in lysosomes. The degradation of proteins is recognized and often polyubiquitinated by a complex network of proteins. However, once aggregated, misfolded proteins are not polyubiquitinated efficiently and therefore are not degraded by proteasome machinery. Thus, when autophagy is impaired, proteins aggregate and accumulate in the cytoplasm, leading to cytotoxicity [113].
Aggregation of viral proteins is observed in HIV positive subjects as a result of virus replication. However, it is important to distinguish which cells accumulate these viral proteins in order to link accumulation of viral proteins to toxic events that may explain HAND. In human brain, gp120 is seen in multinucleated giant cells and microglia [114]. This is expected because these cells express both CD4 and the co-receptors and are the primary cell types in the brain that are infected with HIV and harbor viral particles intracellularly, reflecting their potential as a reservoir. Astrocytes can also be infected, although their infection occurs primarily by a CD4-independent endocytosis [39, 115]. In astrocytes the endosomal system degrades HIV [116]; thus, little accumulation of viral proteins occurs in these cells. Neurons are not infected; nevertheless, both gp120 and Tat, according to animal models, accumulate inside neurons [61, 62, 117]. Moreover, few and isolated gp120 positive neurons can be detected in postmortem brain of HAD subjects (Figure 3), suggesting that neurons cannot dispose gp120 or are highly inefficient in degrading the viral protein. It is unclear whether neuronal gp120 is misfolded or forms insoluble aggregates. However, this aggregation could be toxic. An important question is why neurons accumulate gp120 in a manner that could be due to an impairment of autophagy. Experimental data have shown that although gp120 activates autophagy [118] this activation last only for a few hours [119], suggesting that autophagy remains inefficient in gp120-treated neurons. There is no easy answer to the question of why autophagy is impaired by gp120, though, it is important to note the ubiquitin proteasome system, which is required to initiate autophagy, is altered in HAD subjects [120]. These considerations would be in line with the notion that chronic neurodegenerative diseases may start when autophagy becomes impaired. If so, a therapy for HAND should include a way to dispose aggregated gp120. However, more experimental data should be obtained to determine the role gp120 accumulation plays in neuronal degeneration.
Figure 3. Gp120 immunoreactivity in human brains.

Paraffin-embedded sections from the midbrain of HIV positive (no HAD) and HAD subjects (National NeuroAIDS Tissue Consortium) were immunostained with an antibody against gp120 (AIDS Research and Reference Reagent program, NIAID, NIH, Bethesda, MD). The figure is an example (n = 3) of a neuron positive for gp120 immunoreactivity (arrow) in HAD. No immunoreactivity was detected in HIV subjects.
Gp120 and microtubules
The accumulation of gp120 may initiate mechanisms of axonal degeneration similar to those described for β-amyloid, α-synuclein or other toxic proteins. How aggregates lead to neuronal loss is reviewed elsewhere [107]; however we will present examples to illustrate the toxic effect of aggregated proteins. For instance, β-amyloid accumulation induces hyperphosphorylation of tau [121], which promotes neurotoxicity by depletion of mitochondria in synapses. Aggregated mutant huntingtin evokes neurodegeneration by inhibiting axonal transport of BDNF as well as increasing mitochondria fragmentation [122]. Additional information about mechanisms whereby aggregated proteins induce neurodegeneration can be found elsewhere [123, 124]. A major difference between gp120 and β-amyloid or other proteins is that gp120 is not produced endogenously by neurons but HIV carries it into the brain. HIV or infected cells then shed gp120, which in turn, is endocytosed in astrocytes and neurons most likely by binding to mannose lectin [125]. Nevertheless, the toxic properties of gp120 are only seen in neurons, suggesting that accumulation of gp120 in cells per se may not be sufficient to cause neural impairment; rather, gp120 must alter intracellular organelles. For instance, aggresome formation and pathological inclusions containing misfolded proteins can lead to disruption of the normal architecture of the Golgi apparatus. Fragmentation of the Golgi apparatus is observed in neurons from AD brains as well as from other neurodegenerative diseases [126]. On the other hand, the Golgi apparatus is a dynamic organelle whose organization depends on continuous microtubule-based vesicular traffic. One critical experimental result has demonstrated that gp120 travels along microtubules (MTs) in neurons [61]. MTs are found in both axons and dendrites where they play a role in neurite elongation, intracellular transport of mitochondria and vesicles. Thus, binding of gp120 to MTs can have profound significance for HIV-mediated neuronal degeneration. However, little is known about how gp120 interacts with MTs.
MTs are copolymers assembled from tubulin heterodimers comprised of repeats of α- and β-tubulin, which are conserved among all eukaryotic species [127]. Both α- and β-tubulin consist of numerous isotypes, which differ in amino acid sequence and cellular and tissue distribution. The secondary structure of gp120 contains three α-helix motifs that can determine associations between different proteins [128]. Neuronal specific tubulin-β3 (TUBB3) also contains multiple α helix motifs. Analysis of sequence of these α-helices by Wenxiang diagram (Figure 4) revealed that the α-helix of gp120 near the V3 loop (Figure 5) could form a dimer with the α helix of TUBB3 located in its carboxyl terminal tail (CTT). Such binding may alter the stability and function of neuronal MTs. Local changes in the integrity and dynamics of MTs are sufficient to alter axon and dendrite specification and their normal function, especially the rate of transport. When the normal functions of axons and dendrites are compromised, a distinct neurodegeneration arises. Indeed, impaired MTs is a signature for several neurological disorders. As discussed above, in AD, hyperphosphorylation of tau, one of the complement of MT-associated protein, disrupts the normal function of axons resulting in an inhibition of mitochondrial function and activation of inflammatory responses [129]. In HD, mutant huntingtin binds to mitochondrial fission GTPase and causes mitochondrial fragmentation [130]. Thus, gp120 by binding to neuronal specific tubulin may impair the function of MTs and, consequently, inhibit their ability to act as transporters of cargoes.
Figure 4. Amino acid sequences of gp120 α-helix and the carboxy terminal tail of TUBB3 plotted on a helical wheel.
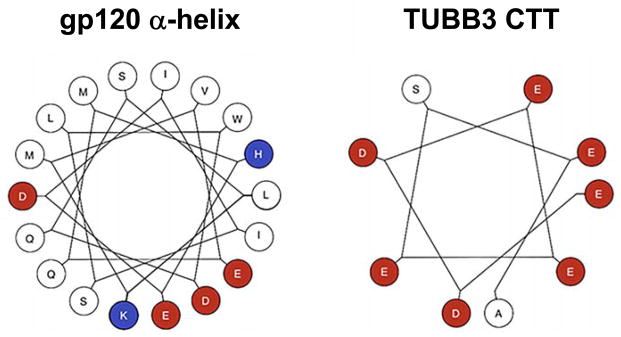
A Wenxiang diagram with the axis direction set from the amino-terminal to the carboxy-terminal was used to show the properties of gp120 α helix and the carboxy terminal tail (CTT) of TUBB3. The hydrophobic sides are in red and hydrophilic sides in blue. The α helix motif of gp120 has only two hydrophilic amino acids at approximately 120 degrees from each other.
Figure 5. Synopsis of the proposed mechanisms of HIV-mediated neuronal injury.

HIV sheds gp120, which is endocytosed and accumulates inside neurons. The α helix of gp120 binds to the CTT of TUBB3 (enlargement) leading to destabilization of MTs, which, in turn, become impaired in their ability to transport, both retrogradely and anterogradely, mitochondria and other organelles.
CONCLUSIONS
Neurological alterations in HIV positive subjects remain a current problem that is not solved by current antiretroviral therapy. In addition, some antiretroviral medications also cause CNS and peripheral nerve adverse effects, and induce the immune restoration inflammatory syndrome [131]. The incomplete understanding of the pathogenic mechanisms of HIV neurotoxicity has constituted a major roadblock to the development of new therapies. HAND shares some similarities with other chronic neurodegenerative disorders. Among others, neurons of HIV-infected patients exhibit inclusion of gp120. This viral protein is endocytosed and accumulates inside neurons by inhibiting autophagy. Most importantly, gp120 may bind to TUBB3 and may alter the function of MTs as axonal transporters of organelles and vesicles, including neurotrophic factors (Figure 5). Thus, accumulated gp120 could decrease axonal transport similar to other toxic proteins such as α-synuclein, mutated huntingtin or β-amyloid, which cause the breakdown of axonal cytoskeleton and the progression of neuronal loss seen in PD, HD and AD, respectively. Without intervention, gp120 continues to accumulate and predisposes neurons to inflammatory responses, which amplify the progression of the clinical pathology.
Acknowledgments
This work was supported by HHS grant NS079172-01. We thank the National NeuroAIDS Tissue Consortium for human brain sections and the AIDS Research and Reference Reagent program for reagents.
ABBREVIATIONS
- AD
Alzheimer’s disease
- AIDS
acquired immune deficiency syndrome
- APOEε4
apoprotein E ε4 allele
- BDNF
brain-derived neurotrophic factor
- cART
combined antiretroviral therapy
- CNS
central nervous system
- CTT
carboxyl terminal tail
- GDNF
glial-derived neurotrophic factor
- HAD
HIV-associated dementia
- HAND
HIV-associated neurocognitive disorders
- HIV
human immunodeficiency virus type 1
- HD
Huntington’s disease
- MTs
microtubules
- PD
Parkinson’s disease
- SN
substantia nigra
- TUBB3
tubulin-β3
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
References
- 1.Janssen RS, Cornblath DR, Epstein LG, McArthur J, Price RW. Neurology. 1989;39:119. doi: 10.1212/wnl.39.1.119. [DOI] [PubMed] [Google Scholar]
- 2.Levy JA, Shimabukuro J, Hollander H, Mills J, Kaminsky L. Lancet. 1985;2:586. [PubMed] [Google Scholar]
- 3.Gabuzda DH, Ho DD, de la Monte SM, Hirsch MS, Rota TR, Sobel RA. Ann Neurol. 1986;20:289. doi: 10.1002/ana.410200304. [DOI] [PubMed] [Google Scholar]
- 4.Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Neurology. 1988;38:9. doi: 10.1212/wnl.38.1.9. [DOI] [PubMed] [Google Scholar]
- 5.Spudich S, Gonzalez-Scarano F. Cold Spring Harb Perspect Med. 2012;2:7120. doi: 10.1101/cshperspect.a007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nightingale S, Winston A, Letendre S, Michael BD, McArthur JC, Khoo S, Solomon T. Lancet Neurol. 2014;13:1139. doi: 10.1016/S1474-4422(14)70137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC. Neurology. 1998;50:1814. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- 8.Simioni S, Cavassini M, Annoni JM, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave JP, Giacobini E, Hirschel B, Du Pasquier RA. AIDS. 2010;24:1243. doi: 10.1097/QAD.0b013e3283354a7b. [DOI] [PubMed] [Google Scholar]
- 9.Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, Group C, Group H. J Neurovirol. 2011;17:3. [Google Scholar]
- 10.Letendre SL, Lanier ER, McCutchan JA. J Infect Dis. 1999;180:310. doi: 10.1086/314866. [DOI] [PubMed] [Google Scholar]
- 11.Masliah E, DeTeresa RM, Mallory ME, Hansen LA. AIDS. 2000;14:69. doi: 10.1097/00002030-200001070-00008. [DOI] [PubMed] [Google Scholar]
- 12.Garden GA. Glia. 2002;40:240. doi: 10.1002/glia.10155. [DOI] [PubMed] [Google Scholar]
- 13.Wright EJ, Grund B, Robertson K, Brew BJ, Roediger M, Bain MP, Drummond F, Vjecha MJ, Hoy J, Miller C, Penalva de Oliveira AC, Pumpradit W, Shlay JC, El-Sadr W, Price RW, Group ISS. Neurology. 2010;75:864. doi: 10.1212/WNL.0b013e3181f11bd8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fletcher NF, McKeating JA. J Viral Hepat. 2012;19:301. doi: 10.1111/j.1365-2893.2012.01591.x. [DOI] [PubMed] [Google Scholar]
- 15.Bhaskaran K, Mussini C, Antinori A, Walker AS, Dorrucci M, Sabin C, Phillips A, Porter K, Collaboration C. Ann Neurol. 2008;63:213. doi: 10.1002/ana.21225. [DOI] [PubMed] [Google Scholar]
- 16.Clifford DB, Ances BM. Lancet Infect Dis. 2013;13:976. doi: 10.1016/S1473-3099(13)70269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ances BM, Vaida F, Yeh MJ, Liang CL, Buxton RB, Letendre S, McCutchan JA, Ellis RJ. J Infect Dis. 2010;201:336. doi: 10.1086/649899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker JT, Sanders J, Madsen SK, Ragin A, Kingsley L, Maruca V, Cohen B, Goodkin K, Martin E, Miller EN, Sacktor N, Alger JR, Barker PB, Saharan P, Carmichael OT, Thompson PM, Multicenter ACS. Brain Imaging Behav. 2011;5:77. [Google Scholar]
- 19.Ances BM, Ortega M, Vaida F, Heaps J, Paul R. J Acquir Immune Defic Syndr. 2012;59:469. doi: 10.1097/QAI.0b013e318249db17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger JR, Nath A. Intervirology. 1997;40:122. doi: 10.1159/000150539. [DOI] [PubMed] [Google Scholar]
- 21.Chiang MC, Dutton RA, Hayashi KM, Lopez OL, Aizenstein HJ, Toga AW, Becker JT, Thompson PM. Neuroimage. 2007;34:44. doi: 10.1016/j.neuroimage.2006.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Towgood KJ, Pitkanen M, Kulasegaram R, Fradera A, Kumar A, Soni S, Sibtain NA, Reed L, Bradbeer C, Barker GJ, Kopelman MD. Cortex. 2012;48:230. doi: 10.1016/j.cortex.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Tekin S, Mega MS, Masterman DM, Chow T, Garakian J, Vinters HV, Cummings JL. Ann Neurol. 2001;49:355. [PubMed] [Google Scholar]
- 24.Thomas JB, Brier MR, Snyder AZ, Vaida FF, Ances BM. Neurol. 2013;80:1186. doi: 10.1212/WNL.0b013e318288792b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang L, Holt JL, Yakupov R, Jiang CS, Ernst T. Neurobiol Aging. 2013;34:1240. doi: 10.1016/j.neurobiolaging.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michaelson DM. Alzheimers Dement. 2014;10:861. doi: 10.1016/j.jalz.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 27.Chang L, Andres M, Sadino J, Jiang CS, Nakama H, Miller E, Ernst T. Neuroimage. 2011;58:1017. doi: 10.1016/j.neuroimage.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jahanshad N, Valcour VG, Nir TM, Kohannim O, Busovaca E, Nicolas K, Thompson PM. Brain Connect. 2012;2:335. doi: 10.1089/brain.2012.0105-Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. AIDS. 2005;19:407. doi: 10.1097/01.aids.0000161770.06158.5c. [DOI] [PubMed] [Google Scholar]
- 30.Clapham PR, McKnight A. Br Med Bull. 2001;58:43. doi: 10.1093/bmb/58.1.43. [DOI] [PubMed] [Google Scholar]
- 31.Moore JP, McKeating JA, Weiss RA, Sattentau QJ. Science. 1990;250:1139. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- 32.Raja A, Venturi M, Kwong P, Sodroski J. J Virol. 2003;77:713. doi: 10.1128/JVI.77.1.713-718.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. Nature. 1996;381:667. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 34.Scarlatti G, Tresoldi E, Bjorndal A, Fredriksson R, Colognesi C, Deng HK, Malnati MS, Plebani A, Siccardi AG, Littman DR, Fenyo EM, Lusso P. Nat Med. 1997;3:1259. doi: 10.1038/nm1197-1259. [DOI] [PubMed] [Google Scholar]
- 35.He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D. Nature. 1997;385:645. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- 36.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Nature. 1996;381:661. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 37.Berson JF, Long D, Doranz BJ, Rucker J, Jirik FR, Doms RW. J Virol. 1996;70:6288. doi: 10.1128/jvi.70.9.6288-6295.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. Cell. 1996;85:1149. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, Blum J, He JJ. J Virol. 2004;78:4120. doi: 10.1128/JVI.78.8.4120-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P, Gabuzda D. Curr HIV Res. 2006;4:267. doi: 10.2174/157016206777709500. [DOI] [PubMed] [Google Scholar]
- 41.Epstein LG, Kuiken C, Blumberg BM, Hartman S, Sharer LR, Clement M, Goudsmit J. Virol. 1991;180:583. doi: 10.1016/0042-6822(91)90072-j. [DOI] [PubMed] [Google Scholar]
- 42.Wong JK, Ignacio CC, Torriani F, Havlir D, Fitch NJ, Richman DD. J Virol. 1997;71:2059. doi: 10.1128/jvi.71.3.2059-2071.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang J, Jozwiak R, Wang B, Ng T, Ge YC, Bolton W, Dwyer DE, Randle C, Osborn R, Cunningham AL, Saksena NK. AIDS Res Hum Retrovir. 1998;14:25. doi: 10.1089/aid.1998.14.25. [DOI] [PubMed] [Google Scholar]
- 44.Smit TK, Wang B, Ng T, Osborne R, Brew B, Saksena NK. Virology. 2001;279:509. doi: 10.1006/viro.2000.0681. [DOI] [PubMed] [Google Scholar]
- 45.Xiang SH, Finzi A, Pacheco B, Alexander K, Yuan W, Rizzuto C, Huang CC, Kwong PD, Sodroski J. J Virol. 2010;84:3147. doi: 10.1128/JVI.02587-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Science. 2005;310:1025. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Power C, McArthur JC, Johnson RT, Griffin DE, Glass JD, Perryman S, Chesebro B. J Virol. 1994;68:4643. doi: 10.1128/jvi.68.7.4643-4649.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Marle G, Power C. J Neurovirol. 2005;11:107. doi: 10.1080/13550280590922838. [DOI] [PubMed] [Google Scholar]
- 49.Dunfee RL, Thomas ER, Gorry PR, Wang J, Taylor J, Kunstman K, Wolinsky SM, Gabuzda D. Proc Nat Acad Sci USA. 2006;103:15160. doi: 10.1073/pnas.0605513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brew BJ, Letendre SL. Int Rev Psychiatry. 2008;20:73. doi: 10.1080/09540260701878082. [DOI] [PubMed] [Google Scholar]
- 51.Gonzalez-Scarano F, Martin-Garcia J. Nat Rev Immunol. 2005;5:69. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 52.Eugenin EA, Clements JE, Zink MC, Berman JW. J Neurosci. 2011;31:9456. doi: 10.1523/JNEUROSCI.1460-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li W, Galey D, Mattson MP, Nath A. Neurotox Res. 2005;8:119. doi: 10.1007/BF03033824. [DOI] [PubMed] [Google Scholar]
- 54.Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Proc Natl Acad Sci USA. 1998;95:3117. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nath A. J Infect Dis. 2002;186:S193. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- 56.Kaul M, Garden GA, Lipton SA. Nature. 2001;410:988. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 57.van Marle G, Henry S, Todoruk T, Sullivan A, Silva C, Rourke SB, Holden J, McArthur JC, Gill MJ, Power C. Virology. 2004;329:302. doi: 10.1016/j.virol.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 58.Jones GJ, Barsby NL, Cohen EA, Holden J, Harris K, Dickie P, Jhamandas J, Power C. J Neurosci. 2007;27:3703. doi: 10.1523/JNEUROSCI.5522-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. J Neurochem. 2001;78:457. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- 60.Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A. J Neurosci. 2003;23:8417. doi: 10.1523/JNEUROSCI.23-23-08417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bachis A, Aden SA, Nosheny RL, Andrews PM, Mocchetti I. J Neurosci. 2006;26:6771. doi: 10.1523/JNEUROSCI.1054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. Nat Med. 2000;6:1380. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- 63.Mattson MP, Haughey NJ, Nath A. Cell Death Differ. 2005;12(Suppl 1):893. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- 64.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Proc Natl Acad Sci USA. 1998;95:14500. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kaul M, Lipton SA. Proc Natl Acad Sci USA. 1999;96:8212. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Y, Kulkosky J, Acheampong E, Nunnari G, Sullivan J, Pomerantz RJ. Proc Nat Acad Sci USA. 2004;101:7070. doi: 10.1073/pnas.0304859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avdoshina V, Bachis A, Mocchetti I. J Intern Med. 2013;273:454. doi: 10.1111/joim.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Nature. 1994;367:188. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 69.Bagetta G, Corasaniti MT, Aloe L, Berliocchi L, Costa N, Finazzi-Agro A, Nistico G. Proc Natl Acad Sci USA. 1996;93:928. doi: 10.1073/pnas.93.2.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maccarrone M, Bari M, Corasaniti MT, Nistico R, Bagetta G, Finazzi-Agro A. J Neurochem. 2000;75:196. doi: 10.1046/j.1471-4159.2000.0750196.x. [DOI] [PubMed] [Google Scholar]
- 71.Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. Brain Res. 2000;879:42. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- 72.Nosheny RL, Ahmed F, Yakovlev A, Meyer EM, Ren K, Tessarollo L, Mocchetti I. Eur J Neurosci. 2007;25:2275. doi: 10.1111/j.1460-9568.2007.05506.x. [DOI] [PubMed] [Google Scholar]
- 73.Corasaniti T, Maccarrone M, Nistico R, Malorni W, Rotiroti D, Bagetta G. J Neurochem. 2001;79:1. doi: 10.1046/j.1471-4159.2001.00537.x. [DOI] [PubMed] [Google Scholar]
- 74.Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. Nat Neurosci. 2001;4:702. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- 75.Ahmed F, MacArthur L, De Bernardi MA, Mocchetti I. Brain Behav Immun. 2009;23:355. doi: 10.1016/j.bbi.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bachis A, Avdoshina V, Zecca L, Parsadanian M, Mocchetti I. J Neurosci. 2012;32:9477. doi: 10.1523/JNEUROSCI.0865-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arancio O, Chao MV. Curr Opin Neurobiol. 2007;17:325. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 78.Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martin-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J. J Neurosci. 2004;24:7727. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marosi K, Mattson MP. Trends Endocrinol Metab. 2014;25:89. doi: 10.1016/j.tem.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. Cell. 2003;112:257. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 81.Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. Neuron. 1991;7:695. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 82.Laske C, Stransky E, Leyhe T, Eschweiler GW, Maetzler W, Wittorf A, Soekadar S, Richartz E, Koehler N, Bartels M, Buchkremer G, Schott K. J Psychiatr Res. 2007;41:387. doi: 10.1016/j.jpsychires.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 83.Nagatsu T, Mogi M, Ichinose H, Togari A. J Neural Transm Suppl. 2000;60:277. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 84.Howells DW, Porritt MJ, Wong JYF, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Exp Neurol. 2000;166:127. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 85.Durany N, Thome J. Eur Psychiatry. 2004;19:326. doi: 10.1016/j.eurpsy.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 86.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Science. 2001;293:493. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 87.Avdoshina V, Garzino-Demo A, Bachis A, Monaco MC, Maki PM, Tractenberg RE, Liu C, Young MA, Mocchetti I. AIDS. 2011;25:1126. doi: 10.1097/QAD.0b013e32834671b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chao MV, Hempstead BL. Trends Neurosci. 1995;18:321. [PubMed] [Google Scholar]
- 89.Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E. Trends Biochem Sci. 1995;20:342. doi: 10.1016/s0968-0004(00)89070-2. [DOI] [PubMed] [Google Scholar]
- 90.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV. J Neurosci. 1999;19:3043. doi: 10.1523/JNEUROSCI.19-08-03043.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kenchappa RS, Zampieri N, Chao MV, Barker PA, Teng HK, Hempstead BL, Carter BD. Neuron. 2006;50:219. doi: 10.1016/j.neuron.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 92.Park KJ, Grosso CA, Aubert I, Kaplan DR, Miller FD. Nat Neurosci. 2010;13:559. doi: 10.1038/nn.2513. [DOI] [PubMed] [Google Scholar]
- 93.Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ. J Neurosci. 2008;28:3941. doi: 10.1523/JNEUROSCI.0350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salehi AH, Xanthoudakis S, Barker PA. J Biol Chem. 2002;277:48043. doi: 10.1074/jbc.M205324200. [DOI] [PubMed] [Google Scholar]
- 95.Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde YA. Science. 1996;272:542. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 96.Chen P, Flory E, Avots A, Jordan BW, Kirchhoff F, Ludwig S, Rapp UR. J Biol Chem. 2000;275:20382. doi: 10.1074/jbc.M001149200. [DOI] [PubMed] [Google Scholar]
- 97.Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. J Neurosci. 2005;25:5455. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jansen P, Giehl K, Nyengaard JR, Teng K, Lioubinski O, Sjoegaard SS, Breiderhoff T, Gotthardt M, Lin F, Eilers A, Petersen CM, Lewin GR, Hempstead BL, Willnow TE, Nykjaer A. Nat Neurosci. 2007;10:1449. doi: 10.1038/nn2000. [DOI] [PubMed] [Google Scholar]
- 99.Meeker RB, Poulton W, Feng WH, Hudson L, Longo FM. J Neuroimmune Pharmacol. 2012;7:388. doi: 10.1007/s11481-011-9325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yao H, Duan M, Yang L, Buch S. J Neurosci. 2012;32:9835. doi: 10.1523/JNEUROSCI.0638-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sanders VJ, Everall IP, Johnson RW, Masliah E. J Neurosci Res. 2000;59:671. doi: 10.1002/(SICI)1097-4547(20000301)59:5<671::AID-JNR10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 102.Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, Olson L. Nature. 1995;273:335. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- 103.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. Science. 1993;260:1130. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 104.Mocchetti I, Bachis A, Campbell LA, Avdoshina V. J Neuroimmune Pharmacol. 2014;9:80. doi: 10.1007/s11481-013-9488-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nosheny RL, Bachis A, Aden SA, De Bernardi MA, Mocchetti I. J Neurobiol. 2006;66:1311. doi: 10.1002/neu.20288. [DOI] [PubMed] [Google Scholar]
- 106.Pardo CA, McArthur JC, Griffin JW. J Peripher Nerv Syst. 2001;6:21. doi: 10.1046/j.1529-8027.2001.006001021.x. [DOI] [PubMed] [Google Scholar]
- 107.Frost B, Götz J, Feany MB. Trends Cell Biol. 2015;25:46. doi: 10.1016/j.tcb.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee HJ, Bae EJ, Lee SJ. Nat Rev Neurol. 2014;10:92. doi: 10.1038/nrneurol.2013.275. [DOI] [PubMed] [Google Scholar]
- 109.Martin DDO, Ladha S, Ehrnhoefer DE, Hayden MR. Trends Neurosci. 2015;38:26. doi: 10.1016/j.tins.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 110.Plemper RK, Wolf DH. Trends Biochem Sci. 1999;24:266. doi: 10.1016/s0968-0004(99)01420-6. [DOI] [PubMed] [Google Scholar]
- 111.Wenqiang C, Lonskaya I, Hebron ML, Ibrahim Z, Olszewski RT, Neale JH, Moussa CE-H. Human Mol Genet. 2014;23:4960. doi: 10.1093/hmg/ddu211. [DOI] [PubMed] [Google Scholar]
- 112.Yu S, Liang Y, Palacino J, Difiglia M, Lu B. Trends Pharmacol Sci. 2014;35:53. doi: 10.1016/j.tips.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 113.Nixon RA. Nat Med. 2013;19:983. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 114.Jones MV, Bell JE, Nath A. AIDS. 2000;14:2709. doi: 10.1097/00002030-200012010-00010. [DOI] [PubMed] [Google Scholar]
- 115.Chauhan A, Mehla R, Vijayakumar TS, Handy I. Virology. 2014;456–457:1. doi: 10.1016/j.virol.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vijaykumar TS, Nath A, Chauhan A. Virology. 2008;381:1. doi: 10.1016/j.virol.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Altmeyer R, Mordelet E, Girard M, Vidal C. Virology. 1999;259:314. doi: 10.1006/viro.1999.9780. [DOI] [PubMed] [Google Scholar]
- 118.Fields J, Dumaop W, Rockenstein E, Mante M, Spencer B, Grant I, Ellis R, Letendre S, Patrick C, Adame A, Masliah E. J Neurovirol. 2013;19:89. doi: 10.1007/s13365-012-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Passeri E, Mocchetti I, Moussa C. CNS Neurol Disord Drug Targets. 2014;13:1572. doi: 10.2174/1871527313666140806125841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kragh CL, Ubhi K, Wyss-Coray T, Masliah E. Brain Pathol. 2012;22:99. doi: 10.1111/j.1750-3639.2011.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.LaFerla FM. Biochem Soc Trans. 2010;38:993. doi: 10.1042/BST0380993. [DOI] [PubMed] [Google Scholar]
- 122.Pla P, Orvoen S, Saudou F, David DJ, Humbert S. Front Behav Neurosci. 2014;8:135. doi: 10.3389/fnbeh.2014.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Spillantini MG, Goedert M. Lancet Neurol. 2013;12:609. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- 124.Blokhuis A, Groen EN, Koppers M, van den Berg L, Pasterkamp RJ. Acta Neuropathol. 2013;125:777. doi: 10.1007/s00401-013-1125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Teodorof C, Divakar S, Soontornniyomkij B, Achim CL, Kaul M, Singh KK. Neurobiol Dis. 2014;69:54. doi: 10.1016/j.nbd.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gonatas NK, Stieber A, Gonatas JO. J Neurol Sci. 2006;246:21. doi: 10.1016/j.jns.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 127.Brouhard GJ, Rice LM. J Cell Biol. 2014;207:323. doi: 10.1083/jcb.201407095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bullock BN, Jochim AL, Arora PS. J Am Chem Soc. 2011;133:14220. doi: 10.1021/ja206074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Krstic D, Knuesel I. Nat Rev Neurol. 2013;9:25. doi: 10.1038/nrneurol.2012.236. [DOI] [PubMed] [Google Scholar]
- 130.Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E. Nat Med. 2011;17:377. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wilson EMP, Sereti I. Immunol Rev. 2013;254:343. doi: 10.1111/imr.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]