

Abstract
First established as a diagnosis by Nikolaus Friedreich in 1863, Friedreich’s ataxia (FA) is an autosomal recessive progressive neurodegenerative disorder cause by a trinucleotide repeat expansion. FA begins with the functional absence of the FXN gene product frataxin, a protein whose exact function still remains unknown. This absence results in impaired intracellular antioxidant defenses, dysregulation of iron-sulfur cluster proteins, depression of aerobic electron transport chain respiration, massive mitochondrial dysfunction, and ultimately cell death in the brain, spinal cord and heart. Herein, we review the molecular and cellular pathogenesis leading to widespread organ system dysfunction, as well as current therapeutic research aimed at preventing the debilitating effects of frataxin loss and preventing the signs and symptoms associated of FA. We also discuss the ongoing treatment strategies employed by our laboratory to prevent mitochondrial damage using synergistic effects of 17β-estradiol and methylene blue, previously shown by our group and others to have protective effects in human FA fibroblasts.
This article is part of a Special Issue entitled Hormone Therapy.
Keywords: Friedreich’s Ataxia, Therapeutic Strategies, Frataxin, 17β-estradiol, Methylene blue
1. Introduction, Symptoms and disease progression
First diagnosed by Nikolaus Friedreich in 1863 (Friedreich, 1863a, 1863b, 1863c, 1876, 1877), Friedreich’s ataxia (FA) affects 1 in 50,000 people worldwide with a carrier rate of 1 in 120 making it the most common type of inherited ataxia worldwide (Bradley et al., 2000; Campuzano et al., 1996; Harding, 1983; Leone et al., 1990; Pandolfo, 1998; Schulz et al., 2009). This disorder is autosomal recessive and found mainly in descendents of Mediterranean cultures in Europe, the Middle Eastern and North Africa (Labuda et al., 2000).
Although FA can be diagnosed with genetic tests before birth (Monros et al., 1995; Pandolfo and Montermini, 1998; Wallis et al., 1989), FA is usually diagnosed when the patient becomes symptomatic. Symptoms classically begin around puberty, although the onset is variable and can be from infancy to 25 years old (Dürr et al., 1996; Lodi et al., 2006). The presenting symptom is most often the ataxia and general gait instability from which the condition derives its name, with progressively worsening ataxia (Harding, 1981, 1983). The signs, symptoms and their severity are variable. Along with the worsening ataxia, other neurological symptoms predominate in FA, including positional and vibration loss, sensory neuropathy, motor weakness, loss of deep tendon reflexes, lower extremity spasticity, Babinski’s sign, sensorineural deafness, optic nerve degeneration, nystagmus, dysarthria and dysmetria (Harding, 1981; Pandolfo, 2009; Santos et al., 2010). Patients also typically require a wheel chair to ambulate within a decade after clinical diagnosis (Dürr et al., 1996; Harding, 1981, 1983; Montermini et al., 1997). Characteristic symptoms that are required for official diagnosis of FA are: an autosomal recessive inheritance pattern, onset of symptoms before 25 years old, gait ataxia, Babinski’s sign, loss of tendon reflexes and sensory neuropathy (Harding, 1981; Santos et al., 2010).
There are also a number of later-presenting non-neurological symptoms that are typically coexistent in FA. These include pes cavitus, lateral and kyphoscoliosis, a 10–20% incidence of type 1 diabetes, and a 20–40% incidence of insulin resistance and glucose intolerance (Al-Mahdawi et al., 2006; Dürr et al., 1996; Geoffroy et al., 1976; Harding, 1981; Harding, 1983). Cardiac symptoms occur in 66–91% of patients, including atrial fibrillation, left ventricular hypertrophy and hypertrophic cardiomyopathy with interstitial fibrosis, which is the leading cause of premature death among the FA population (Bradley et al., 2000; Harding, 1981, 1983; Leone et al., 1990; Santos et al., 2010).
2. Disease mechanism and molecular pathogenesis
The genetic basis of FA is a trinucleotide GAA repeat expansion in the first intron of the FXN gene on chromosome 9q13–21, which normally produces frataxin protein (Fujita et al., 1989; Hanauer et al., 1990). When this trinucleotide sequence grows beyond 100–200 repeats, a self-associating complex of triple helical DNA forms forcing histone deacetylation during DNA to mRNA transcription, effectively preventing the production of frataxin protein (Bradley et al., 2000; Campuzano et al., 1996; Grabczyk and Usdin, 2000a, 2000b; Heidenfelder et al., 2003; Lodi et al., 2006; Montermini et al., 1997; Sakamoto et al., 1999, 2001; Wells, 2008). The precise cellular role of frataxin is still unclear, however its absence results in dysfunctional iron metabolism and impaired function of iron–sulfur (Fe–S) cluster proteins, including heme, electron transport chain (ETC) complexes and the Kreb’s cycle protein, aconitase, as well as dysregulation of the cellular redox state (Delatycki et al., 2000; Gakh et al., 2006; Lodi et al., 2006), ultimately leading to progressive oxidative damage to the mitochondria (Karthikeyan et al., 2003). The damage to the mitochondria is twofold: inhibition of Fe–S containing protein function by this oxidative damage impairs cellular respiration and causes further increases in production of reactive oxygen species, and these same energy producing mitochondrial Fe–S proteins are further damaged by the increased ROS, causing a vicious cycle of mitochondrial impairment (Bradley et al., 2000; Bulteau et al., 2004; Rötig et al., 1997). Similar to the pathogenesis of many other neurodegenerative disease models including those for Parkinson’s disease, Alzheimer’s disease and ischemic stroke, this mitochondrial oxidative damage causes an impairment in aerobic ATP production and a mismatch in the ratio of ATP production and the cellular ATP demands, leading to cell death in tissues and organs most dependent on oxidative phosphorylation for survival (Bulteau et al., 2004; Chantrel-Groussard et al., 2001; Gakh et al., 2006; Jauslin et al., 2002; Santos et al., 2010).
Studies in yeast by Karthikeyan et al. (2003) have shown that the depletion of frataxin homologs is related to oxidative damage and results in progressive accumulation of mitochondrial damage, a key precipitating factor in symptom development. Cells and tissues most dependent on aerobic respiration and oxidative phosphorylation for ATP production are the first to succumb to the oxidative damage, including neurons in the brain and spinal cord, cardiomyocytes and pancreatic beta cells, giving rise to the typical neurological and cardiac symptoms as well as the concurrent diabetes and glucose intolerance. As yet, it is unclear why there is variable cell death and dysfunction within these specific tissues, including why certain spinal cord tracts such as the posterior columns and spinocerebellar tracts are affected and others are not. It should also be noted that there is a phenotypic contribution to the disease process of cells in these organ systems that are dysfunctional but still viable (Santos et al., 2010).
The length of the GAA triplet repeats in the first intron of the FXN gene is an important factor in the pathogenesis of FA. Both alleles must have >100–200 GAA repeats for FA to be clinically apparent, however it is generally thought to be the shorter allele (the one with the fewest repeats) that is the determinant of neurological symptoms in terms of disease severity, age of onset and progression (Campuzao et al., 1997; Dürr et al., 1997; Isnard et al., 1997). Many of the associated neurological symptoms, including ataxia caused primarily by loss of posterior column fibers, visual and auditory loss, dysarthria and scoliosis are directly correlated to the number of GAA repeats on the shorter allele (Campuzano et al., 1997; Dürr et al., 1996; Isnard et al., 1997; Montermini et al., 1997). However, only 50% of the variation in disease progression and age of onset can be explained by the length of the GAA trinucleotide repeat expansion (Delatycki et al., 1999; Montermini et al., 1997). Other poorly defined factors may contribute to disease progression, including genetic mosaicism and mitochondrial genotype. The concurrent cardiomyopathy is independent of the ataxia and other neurological symptom progression, but it generally occurs in individuals with longer repeat sections in the smaller of the two alleles (Dürr et al., 1996; Montermini et al., 1997). Development of diabetes or insulin resistance has no known correlation to the number of repeats (Dürr et al., 1996; Montermini et al., 1997).
3. Therapeutic Strategies
Currently, there is no viable treatment option for FA patients. Treatment and therapeutic strategies in FA has been divided into four categories: palliative and symptomatic treatments, iron chelators, antioxidants and frataxin level modifiers. Palliative treatments has typically consisted of the use of wheelchairs in later stages of the disease, β-blockers, ACE-inhibitors and surgery for cardiac manifestations and physical therapy (Bradley et al., 2000; Campuzano et al., 1996; Pandolfo, 2009). Iron chelation, antioxidants, frataxin level modification and estrogen/methylene blue therapy are considered below.
3.1. Iron Chelators
There is a great deal of evidence that implicates dysregulation of iron and impaired iron homeostasis as a pathological hallmark of FA in cellular and animal models, as well as FA patients (Santos et al., 2010). As such, much effort has been expended in designing and testing drugs with iron chelating potential for use in FA (Lodi et al., 2006; Santos et al., 2010). Iron accumulation is known to increase the intracellular concentration of ROS by Fenton chemistry, and these ROS damage mitochondrial energy producing proteins in cardiac and neuronal cells (Bradley et al., 2000; Bulteau et al., 2004; Rötig et al., 1997; Rustin et al., 1999). Several iron chelators that target the mitochondria (Richardson, 2003) have been evaluated in both in vitro models (Goncalves et al., 2008; Kakhlon et al., 2008) and in clinical trials (Boddaert et al., 2007), including deferoxamine and deferiprone. Deferoxamine has not performed well in FA, as it is able to chelate iron in cell culture, but it decreases the mRNA levels of both aconitase and frataxin, making it unsuitable for use in FA (Li et al., 2008). Deferiprone has had mixed results in its assessments as well. It successfully protected the mitochondria and reduced ROS damage to mitochondrial proteins in one study (Kakhlon et al., 2008) and reduced iron buildup in the brain with a small improvement in neurological function in another study (Boddaert et al., 2007), however it also reduces the activity of aconitase (Goncalves et al., 2008).
3.2. Antioxidants
Additionally, much work has been done in evaluating the potential of antioxidants in preventing mitochondrial damage and preserving aerobic respiration. Patients with FA have increased oxidative stress and resulting DNA damage (Schulz et al., 2000), increased system-wide levels of lipid peroxidation (Emond et al., 2000) and impaired ROS defenses including manganese superoxide dismutase (Pandolfo, 2002). The first drug to reach Phase III clinical trials for FA, idebenone, is a CoQ10 analog that both shuttles electrons between damaged ETC complex proteins and attenuates intracellular ROS, utilizing both mechanisms for promoting increased oxidative phosphorylation and aerobic respiration in FA (Meier and Buyse, 2009; Rustin et al., 1999). Idebenone improved intracellular markers of ROS damage and FA symptoms in both cellular (Jauslin et al., 2002, 2007) and murine models (Seznec et al., 2004). Clinical studies of this compound were initially promising as one study showed a decrease in neurologic symptoms (Di Prospero et al., 2007), while multiple studies showed decreases in oxidative stress, lipid peroxidation and a slowing of the progression of heart disease (Rustin et al., 1999; Schulz et al., 2000). In 2011, however, idebenone failed its Phase III study because it was found not to significantly improve lifespan or adequately improve cardiac outcomes in patients (Lagedrost et al., 2011). This failure does not rule out antioxidants or ETC modifiers as potential therapeutic agents however. The Phase III trial was conducted in older individuals who already had clinical symptoms of FA, indicating that they had significant neuron and cardiomyocyte dysfunction and death, outcomes that likely cannot be overcome with pharmacotherapy. Similar compounds may be able to prevent these outcomes if given before cellular dysfunction has given way to cell death or widespread organ system dysfunction.
3.3. Frataxin level Modifiers
One of the most promising treatment strategies for FA is to increase the intracellular content of frataxin, thus preventing the cascade of protein dysfunction and ROS mediated damage that ultimately leads to the clinical syndrome. Several compounds have been used to combat the FXN gene silencing and increase frataxin levels. Erythropoietin (EPO) has been shown to significantly increase frataxin protein levels in human-derived fibroblasts (Sturm et al., 2005). This treatment modality would also likely necessitate the use of recurrent phlebotomies, since the physiologic effect of EPO increases the production of erythrocyte lines. EPO was shown to increase only frataxin protein, without a concurrent rise in frataxin mRNA levels, signifying that this effect is likely due to post-translational effects promoting the translation of frataxin mRNA into protein (Acquaviva et al., 2008). Other compounds have also shown success in increasing frataxin protein levels: the histone deacetylase inhibitors BML-210 and compound 106 (Herman et al., 2006; Rai et al., 2008) have had modest successes in lymphocytes and FA mouse models.
3.4. 17β-Estradiol and methylene blue interaction
17β-Estradiol (E2) has long been known to have neuroprotective effects in a wide variety of neurological disorders in many different model systems (Behl, 2002; Simpkins et al., 2008). Previously, we have shown that estrogens are able to attenuate reactive oxygen species, prevent the resulting lipid and protein damage, improve mitochondrial function and promote survival in human FA fibroblasts in a non-genomic ER-independent manner (Richardson et al., 2011, 2012), likely through direct antioxidant effects mediated by phenol-quinol cycling (Prokai et al., 2003; Prokai-Tatrai et al., 2008). The potency and efficacy of estrogen-derived compounds depends in FA fibroblasts on the presence of at least one phenol ring in the structure and potency is directly correlated to the number of phenol rings (Behl et al., 1997; Moosmann and Behl, 1999; Richardson et al., 2011). Estrogens are ideal candidate drugs for FA in that they contain a phenol ring which has been shown to act as an antioxidant and iron chelator. E2 also has been shown to stabilize the mitochondrial membrane potential, maintain activity of ETC complexes, maintain aerobic respiration and maintain a favorable balance of anti-apoptotic:pro-apoptotic proteins (Simpkins and Dykens, 2008; Perez et al., 2006; Prokai et al., 2003; Prokai-Tatrai et al., 2008; Wang et al., 2001; Wang et al., 2003; Jayachandran et al., 2010). It is also able to intercalate into membranes and stop the lipid peroxidation cascades, and so is well suited to protect mitochondria and other organelles from ROS-mediated damage (Simpkins et al., 1997; Simpkins et al., 2008; Simpkins et al., 2010; Yi et al., 2011).
Methylene blue (MB) has been used for many different indications in the past century, including for neuroprotection and improvement of neurological function in Alzheimer’s disease (Atamna and Kumar, 2010; Oz et al., 2011), retinal disease (Zhang et al., 2006), optic neuropathy (Rojas et al., 2009a), a Parkinson’s model (Rojas et al., 2009b; Wen et al., 2011), a stroke model (Wen et al., 2011) and recently in cytoprotection of FA fibroblasts against oxidative stress (Yu et al., 2011). The mechanism of MB protection in neurodegenerative states is thought to be due to both its antioxidant capabilities and its ability to shuttle electrons through a damaged or otherwise nonfunctional electron transport chain (Wen et al., 2011), similar to that of idebenone.
Here, we further evaluated the synergistic mechanisms between these two promising compounds (Fig. 1) in an FA fibro-blast cell model. A dose-response curve of MB was evaluated from 1 pM to 10 μM with and without a non-protective E2 dose of 10 nM (Fig. 2a) in BSO-treated cells. We found significant protection in the BSO and MB-alone treated cells between 10 nM and 1 μM, with maximal effects at 100 nM and toxicity at 10 μM. With the addition of 10 nM E2 the curve shifted significantly to the left indicating an increase in potency of the MB. There was also a significant protective effect at 1 nM MB in the presence of estrogen, an increase in efficacy from 10 nM to 1 μM and protection from toxicity at 10 μM (Fig. 2a). The addition of estrogen significantly reduced the EC50 of MB from 9.7 nM to 880pM. A dose-response curve of E2 was also evaluated from 1pM to 10 μM with and without a non-protective MB dose of 1 nM (Fig. 2a) in BSO-treated cells. There was again a significant left-shift of the E2 viability curve with 10 nM to 10 μM being protective in the presence of MB, while only 100 nMm to 10 μM was protective without it (Fig. 2b). There was also a small increase in efficacy between 100 nM and 10 μM with 1 nM MB and an ~11-fold decrease in the EC50. (For materials and methods regarding these experiments see: Richardson et al., 2011.)
Fig. 1.
Structures of compounds assessed for protection against BSO toxicity in FA fibroblasts.
Fig. 2.
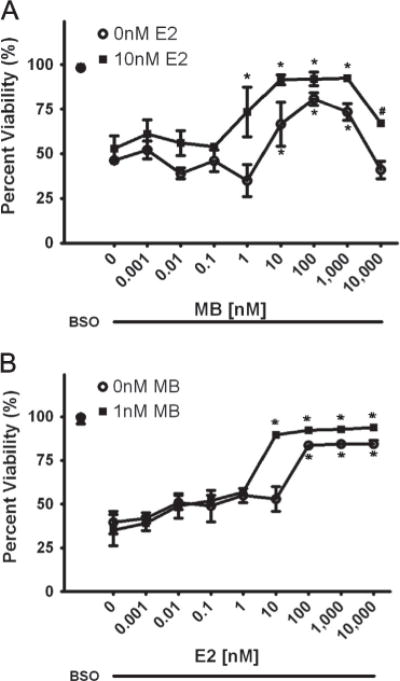
A.) Effects of MB in the presence (■) and absence (○) of 10 nM E2. BSO concentration is 1 mM and MB concentration varies from 1 pM to 10 μM. * indicated p<0.05 versus BSO alone-treated cells. # indicated p<0.01 versus 10 μM MB treated cells. B.) Effects of E2 in the presence (■2) and absence (○) of 1 nM MB. BSO concentration is 1 mM and E2 concentration varies from 1 pM to 10 μM. * indicated p<0.05 versus BSO alone-treated cells.
Depending upon the extent to which the therapeutic window for post-menopausal estrogen therapy is a reflection of the CNS toxic effects of estrogens overriding its beneficial effects (See Braunn, this Edition), our data indicated that the combination of an estrogen with MB may in part extend this therapeutic window and provide greater potential use of estrogens after the menopause.
4. Conclusions
FA is a genetic as well as mitochondrial disease with a mechanism of action of cellular and mitochondrial damage similar to Alzheimer’s disease, Parkinson’s disease and ischemic stroke (Beal, 2000; Gibson et al., 1998; Lenaz et al., 2006; Mizuno et al., 1989; Simpkins et al., 1997; Simpkins and Dykens, 2008). It is characterized by absence of functional frataxin, resulting in massive intracellular oxidative damage to the mitochondria and other organelles, lipids and proteins as well as resulting in iron dysregulation (Karthikeyan et al., 2003; Pandolfo, 1998; Santos et al., 2010). Currently there are no accepted treatments that address the underlying cause of this disorder, and with idebenone failing its Phase III clinical trial, we have not yet been able to significantly prevent cardiac damage or adequately prolong life (Lagedrost et al., 2011). Since FA can be diagnosed genetically at birth (Monros et al., 1995; Pandolfo and Montermini, 1998; Wallis et al., 1989), drugs that can penetrate into the brain and attenuate ROS, remove accumulated iron deposits, support oxidative phosphorylation or significantly increase intracellular frataxin levels, including those based on the phenolic estrogen structures, could be used to prevent or delay FA symptoms and cell death. This strategy bypasses the previously attempted methods of controlling or reversing symptoms or attempting to slightly prolong life. It will take a concerted effort to tie together all of these strategies and identify individuals who are at risk before significant and irreversible neurologic and cardiac damage has occurred.
References
- Acquaviva F, Castaldo I, Filla A, Giacchetti M, Marmolino D, Monticelli A, Pinelli M, Saccà F, Cocozza S. Recombinant human erythropoietin increases frataxin protein expression without increasing mRNA expression. Cerebellum. 2008;7:360–365. doi: 10.1007/s12311-008-0036-x. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, Webster Z, Blake J, Cooper JM, King R, Pook MA. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimer’s Dis. 2010;20(Suppl 2):S439–S452. doi: 10.3233/JAD-2010-100414. [DOI] [PubMed] [Google Scholar]
- Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- Behl C. Oestrogen as a neuroprotective hormone. Nat Rev Neurosci. 2002;3:433–442. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- Boddaert N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, Sidi D, Thalabard JC, Munnich A, Cabantchik ZI. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:521–524. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum Mol Genet. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–245. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Dürr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Cañizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Chantrel-Groussard K, Geromel V, Puccio H, Koenig M, Munnich A, Rotig A, Rustin P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum Mol Genet. 2001;10:2061–2067. doi: 10.1093/hmg/10.19.2061. [DOI] [PubMed] [Google Scholar]
- Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, MacMillan JC, Collins V, Williamson R, Forrest SM. Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet. 1999;87:168–174. doi: 10.1002/(sici)1096-8628(19991119)87:2<168::aid-ajmg8>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Delatycki MB, Williamson R, Forrest SM. Friedreich ataxia: an overview. J Med Genet. 2000;37:1–8. doi: 10.1136/jmg.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prospero NA, Baker A, Jeffries N, Fishbeck KH. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomized, placebo-controlled trial. Lancet Neurol. 2007;6:878–886. doi: 10.1016/S1474-4422(07)70220-X. [DOI] [PubMed] [Google Scholar]
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335:1169–1175. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- Emond M, Lepage G, Vanasse M, Pandolfo M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology. 2000;55:1752–1753. doi: 10.1212/wnl.55.11.1752. [DOI] [PubMed] [Google Scholar]
- Friedreich N. Uber degenerative Atrophie der spinalen Hinterstrange. Arch Pathol Anat Phys Klin Med. 1863a;26:391–419. [Google Scholar]
- Friedreich N. Uber degenerative Atrophie der spinalen Hinterstrange. Arch Pathol Anat Phys Klin Med. 1863b;26:433–459. [Google Scholar]
- Friedreich N. Uber degenerative Atrophie der spinalen Hinterstrange. Arch Pathol Anat Phys Klin Med. 1863c;27:1–26. [Google Scholar]
- Friedreich N. Über Ataxie mit besonderer Berücksichtigung der hereditären Formen. Virchow’s Arch Pathol Anat. 1876;68:145–245. [Google Scholar]
- Friedreich N. Über Ataxie mit besonderer Berücksichtigung der hereditären Formen. Virchow’s Arch Pathol Anat. 1877;70:140–142. [Google Scholar]
- Fujita R, Agid Y, Trouillas P, Seck A, Tommasi-Davenas C, Driesel AJ, Olek K, Grzeschik KH, Nakamura Y, Mandel JL, Hanauer A. Confirmation of linkage of Friedreich ataxia to chromosome 9 and identification of a new closely linked marker. Genomics. 1989;4:110–111. doi: 10.1016/0888-7543(89)90323-6. [DOI] [PubMed] [Google Scholar]
- Gakh O, Park S, Liu G, Macomber L, Imlay JA, Ferreira GC, Isaya G. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum Mol Genet. 2006;15:467–479. doi: 10.1093/hmg/ddi461. [DOI] [PubMed] [Google Scholar]
- Geoffroy G, Barbeau A, Breton G, Lemieux B, Aube M, Leger C, Bouchard JP. Clinical description and roentgenologic evaluation of patients with Friedreich’s ataxia. Can J Neurol Sci. 1976;3:279–286. doi: 10.1017/s0317167100025464. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- Goncalves S, Paupe V, Dassa EP, Rustin P. Derferiprone targets aconitase: implication for Friedreich’s ataxia treatment. BMC Neurol. 2008;8:20. doi: 10.1186/1471-2377-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E, Usdin K. Alleviating transcript insufficiency caused by Friedreich’s ataxia triplet repeats. Nucleic Acids Res. 2000a;28:4930–4937. doi: 10.1093/nar/28.24.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabczyk E, Usdin K. The GAA*TCC triplet repeat expanded in Friedreich’s ataxia impedes transcription elongation by T7 RNA polymerase in a length and supercoil dependent manner. Nucleic Acids Res. 2000b;28:2815–2822. doi: 10.1093/nar/28.14.2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanauer A, Chery M, Fujita R, Driesel AJ, Gilgenkrantz S, Mandel JL. The Friedreich ataxia gene is assigned to chromosome 9q13–q21 by mapping of tightly linked markers and shows linkage disequilibrium with D9S15. Am J Hum Genet. 1990;46:133–137. [PMC free article] [PubMed] [Google Scholar]
- Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- Heidenfelder BL, Makhov AM, Topal MD. Hairpin formation in Friedreich’s ataxia triplet repeat expansion. J Biol Chem. 2003;278:2425–2431. doi: 10.1074/jbc.M210643200. [DOI] [PubMed] [Google Scholar]
- Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- Isnard R, Kalotka H, Dürr A, Cossèe M, Schmitt M, Pousset F, Thomas D, Brice A, Koenig M, Komajda M. Correlation between left ventricular hypertrophy and GAA trinucleotide repeat length in Friedreich’s ataxia. Circulation. 1997;95:2247–2249. doi: 10.1161/01.cir.95.9.2247. [DOI] [PubMed] [Google Scholar]
- Jauslin ML, Vertuani S, Durini E, Buzzoni L, Ciliberti N, Verdecchia S, Palozza P, Meier T, Manfredini S. Protective effects of Fe-Aox29, a novel antioxidant derived from a molecular combination of Idebenone and vitamin E, in immortalized fibroblasts and fibroblasts from patients with Friedreich ataxia. Mol Cell Biochem. 2007;302:79–85. doi: 10.1007/s11010-007-9429-2. [DOI] [PubMed] [Google Scholar]
- Jauslin ML, Wirth T, Meier T, Shoumacher F. A cellular model for Friedreich Ataxia reveals small-molecule glutathione peroxidase mimetics as novel treatment strategy. Hum Mol Genet. 2002;11:3055–3063. doi: 10.1093/hmg/11.24.3055. [DOI] [PubMed] [Google Scholar]
- Jayachandran M, Preston CC, Hunter LW, Jahangir A, Owen WG, Korach KS, Miller VM. Loss of estrogen receptor beta decreases mitochondrial energetic potential and increases thrombogenicity of platelets in aged female mice. Age (Dordr) 2010;32:109–121. doi: 10.1007/s11357-009-9119-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhlon O, Manning H, Breuer W, Melamed-Book N, Lu C, Cortopassi G, Munnich A, Cabantchik ZI. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112:5219–5227. doi: 10.1182/blood-2008-06-161919. [DOI] [PubMed] [Google Scholar]
- Karthikeyan G, Santos JH, Graziewicz MA, Copeland WC, Isaya G, Van Houten B, Resnick MA. Reduction in frataxin causes progressive accumulation of mitochondrial damage. Hum Mol Genet. 2003;12:3331–3342. doi: 10.1093/hmg/ddg349. [DOI] [PubMed] [Google Scholar]
- Labuda M, Labuda D, Miranda C, Poirier J, Soong BW, Barucha NE, Pandolfo M. Unique origin and specific ethnic distribution of the Friedreich ataxia GAA expansion. Neurology. 2000;54:2322–2324. doi: 10.1212/wnl.54.12.2322. [DOI] [PubMed] [Google Scholar]
- Lagedrost SJ, Sutton MS, Cohen MS, Satou GM, Kaufman BD, Perlman SL, Rummey C, Meier T, Lynch DR. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA) Am Heart J. 2011;161:639–645. doi: 10.1016/j.ahj.2010.10.038. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Baracca A, Fato R, Genova ML, Solaini G. Mitochondrial complex I: structure, function, and implications in neurodegeneration. Ital J Biochem. 2006;55:232–253. [PubMed] [Google Scholar]
- Leone M, Brignolio F, Rosso MG, Curtoni ES, Moroni A, Tribolo A, Schiffer D. Friedreich’s ataxia: a descriptive epidemiological study in an Italian population. Clin Genet. 1990;38:161–169. doi: 10.1111/j.1399-0004.1990.tb03566.x. [DOI] [PubMed] [Google Scholar]
- Li K, Besse EK, Ha D, Kovtunovych G, Rouault TA. Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum Mol Genet. 2008;17:2265–2273. doi: 10.1093/hmg/ddn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi A, Tonon C, Calabrese V, Schapira AHV. Friedreich’s Ataxia: from disease mechanisms to therapeutic interventions. Antioxid and Redox Signal. 2006;8:438–443. doi: 10.1089/ars.2006.8.438. [DOI] [PubMed] [Google Scholar]
- Meier T, Buyse G. Idebenone: an emerging therapy for Friedreich ataxia. J Neurol. 2009;256:25–30. doi: 10.1007/s00415-009-1005-0. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ohta S, Tanaka M, Takamiva S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res Commun. 1989;163:1450–1455. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- Monros E, Smeyers P, Ramos MA, Prieto F, Paulo F. Prenatal diagnosis of Friedreich ataxia: improved accuracy by using new genetic flanking markers. Prenat Diagn. 1995;15:551–554. doi: 10.1002/pd.1970150608. [DOI] [PubMed] [Google Scholar]
- Montermini L, Andermann E, Labuda M, Richter A, Pandolfo M, Cavalcanti F, Pianese L, Iodice L, Farina G, Monticelli A, Turano M, Filla A, De Michele G, Cocozza S. The Friedreich ataxia GAA triplet repeat: permutation and normal alleles. Hum Mol Genet. 1997;6:1261–1266. doi: 10.1093/hmg/6.8.1261. [DOI] [PubMed] [Google Scholar]
- Moosmann B, Behl C. The antioxidant neuroprotective effects of estrogens and phenolic compounds are independent from their estrogenic properties. Proc Natl Acad Sci USA. 1999;96:8867–8872. doi: 10.1073/pnas.96.16.8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz M, Lorke DE, Hasan M, Petroianu GA. Cellular and molecular actions of methylene blue in the nervous system. Med Res Rev. 2011;31:93–117. doi: 10.1002/med.20177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfo M. Molecular genetics and pathogenesis of Friedreich ataxia. Neuromucul Disord. 1998;8:409–415. doi: 10.1016/s0960-8966(98)00039-x. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Frataxin deficiency and mitochondrial dysfunction. Mitochondrion. 2002;2:87–93. doi: 10.1016/s1567-7249(02)00039-9. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Friedreich ataxia: the clinical picture. J Neurol. 2009;256(Suppl 1):3–8. doi: 10.1007/s00415-009-1002-3. [DOI] [PubMed] [Google Scholar]
- Pandolfo M, Montermini L. Prenatal diagnosis of Friedreich ataxia. Prenat Diagn. 1998;18:831–833. [PubMed] [Google Scholar]
- Perez E, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of estratriene analogs: structure-activity relationships and molecular optimization. Drug Dev Res. 2006;66:78–92. [Google Scholar]
- Prokai L, Prokai-Tatrai K, Perjesi P, Zharikova AD, Perez EJ, Liu R, Simpkins JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci U S A. 2003;100:11741–11746. doi: 10.1073/pnas.2032621100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai-Tatrai K, Perjesi P, Rivera-Portalatin NM, Simpkins JW, Prokai L. Mechanistic investigation on the antioxidant action of a neuroprotective estrogen derivative. Steroids. 2008;73:280288. doi: 10.1016/j.steroids.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai M, Soragni E, Jenssen K, Burnett R, Herman D, Coppola G, Geschwind DH, Gottesfeld JM, Pandolfo M. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS ONE. 2008;3:e1958. doi: 10.1371/journal.pone.0001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DR. Friedreich’s ataxia: iron chelators that target the mitochondrion as a therapeutic strategy? Expert Opin Invest. Drugs. 2003;12:235–245. doi: 10.1517/13543784.12.2.235. [DOI] [PubMed] [Google Scholar]
- Richardson TE, Yang SH, Wen Y, Simpkins JW. Estrogen protection in Friedreich’s ataxia skin fibroblasts. Endocrinology. 2011;152:2742–2749. doi: 10.1210/en.2011-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson TE, Yu AE, Wen Y, Yang SH, Simpkins JW. Estrogen prevents oxidative damage to the mitochondria in Friedreich’s ataxia skin fibroblasts. PLoS ONE. 2012;7:e34600. doi: 10.1371/journal.pone.0034600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, John JM, Lee J, Gonzalez-Lima F. Methylene blue provides behavioral and metabolic neuroprotection against optic neuropathy. Neurotox. Res. 2009a;15:260–273. doi: 10.1007/s12640-009-9027-z. [DOI] [PubMed] [Google Scholar]
- Rojas JC, Simola N, Kermath BA, Kane JR, Schallert T, Gonzalez-Lima F. Striatal neuorprotection with methylene blue. Neuroscience. 2009b;163:877–889. doi: 10.1016/j.neuroscience.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rötig A, De Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulfur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, Sidi D, Munnich A, Rotic A. Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: a preliminary study. Lancet. 1999;354:477–479. doi: 10.1016/S0140-6736(99)01341-0. [DOI] [PubMed] [Google Scholar]
- Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD, Wells RD. Sticky DNA: self-association properties of long GAA. TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol Cell. 1999;3:465–475. doi: 10.1016/s1097-2765(00)80474-8. [DOI] [PubMed] [Google Scholar]
- Sakamoto N, Ohshima K, Montermini L, Pandolfo M, Wells RD. Sticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcription. J Biol Chem. 2001;276:27171–27177. doi: 10.1074/jbc.M101879200. [DOI] [PubMed] [Google Scholar]
- Santos R, Lefevre S, Silwa D, Seguin A, Camadro JM, Lesuisse E. Friedreich ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid. Redox Signal. 2010;13:651–690. doi: 10.1089/ars.2009.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Boesch S, Bürk K, Dürr A, Giunti P, Mariotti C, Pousset F, Schöls L, Vankan P, Pandolfo M. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Rev Neurol. 2009;5:222–234. doi: 10.1038/nrneurol.2009.26. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Dehmer T, Schöls L, Mende H, Hardt C, Vorgerd M, Bürk K, Matson W, Dichgans J, Beal MF, Bogdanov MB. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000;55:1719–1721. doi: 10.1212/wnl.55.11.1719. [DOI] [PubMed] [Google Scholar]
- Seznec H, Simon D, Monassier L, Criqui-Filipe P, Gansmuller A, Rustin P, Koenig M, Puccio H. Idebenone delays the onset of cardiac function alteration without correction of Fe-S enzymes deficit in a mouse model for Friedreich ataxia. Hum Mol Genet. 2004;13:1017–1024. doi: 10.1093/hmg/ddh114. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Brain Res Rev. 2008;57:421–430. doi: 10.1016/j.brainresrev.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Green PS, Gridley KE, Singh M, de Fiebre NC, Rajakumar G. Role of estrogen replacement therapy in memory enhancement and the prevention of neuronal loss associated with Alzheimer’s disease. Am J Medicine. 1997;103:195–255. doi: 10.1016/s0002-9343(97)00260-x. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Yang SH, Sarkar SN, Pearce V. Estrogen actions on mitochondria–physiological and pathological implications. Mol Cell Endocrinol. 2008;290:51–59. doi: 10.1016/j.mce.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochim Biophys Acta. 2010;1800:1113–1120. doi: 10.1016/j.bbagen.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm B, Stupphann D, Kaun C, Boesch S, Schranzhofer M, Wojta J, Goldenberg H, Scheiber-Mojdehkar B. Recombinant human erythropoietin: effects on frataxin expression in vitro. Eur J Clin Invest. 2005;35:711–717. doi: 10.1111/j.1365-2362.2005.01568.x. [DOI] [PubMed] [Google Scholar]
- Wallis J, Shaw J, Wilkes D, Farrall M, Williamson R, Chamberlain S, Skare JC, Milunsky A. Prenatal diagnosis of Friedreich ataxia. Am J Med Genet. 1989;34:458–461. doi: 10.1002/ajmg.1320340327. [DOI] [PubMed] [Google Scholar]
- Wang J, Green PS, Simpkins JW. Estradiol protects against ATP depletion, mitochondrial membrane potential decline and the generation of reactive oxygen species induced by 3-nitroproprionic acid in SK-N-SH human neuroblastoma cells. J Neurochem. 2001;77:804–811. doi: 10.1046/j.1471-4159.2001.00271.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Simpkins JW, Dykens JA, Cammarata PR. Oxidative damage to human lense epithelial cells in culture: estrogen protection of mitochondrial potential, ATP, and cell viability. Invest Ophthalmol Vis Sci. 2003;44:2067–2075. doi: 10.1167/iovs.02-0841. [DOI] [PubMed] [Google Scholar]
- Wells RD. DNA triplexes and Friedreich ataxia. FASEB J. 2008;22:1625–1634. doi: 10.1096/fj.07-097857. [DOI] [PubMed] [Google Scholar]
- Wen Y, Li W, Poteet EC, Xie L, Tan C, Yan LJ, Ju X, Liu R, Qian H, Marvin MA, Goldberg MS, She H, Mao Z, Simpkins JW, Yang SH. Alternative mitochondrial electron transfer as a strategy for neuroprotection. J Biol Chem. 2011;286:16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi KD, Perez E, Yang S, Liu R, Covey DF, Simpkins JW. The assessment of non-feminzing estrogens for use in neuroprotection. Brain Res. 2011;1379:61–70. doi: 10.1016/j.brainres.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AE, Poteet E, Ryou MG, Li W, Wen Y, Simpkins JW, Yang SH. Methylene Blue Enhances Mitochondrial Function and Provides Protection Against Oxidative Stress in a Cellular Model of Friedreich’s Ataxia. Washington, DC: Society for Neuroscience; 2011. (Program No.361.23.2011 Neuroscience MeetingPlanner). Online. [Google Scholar]
- Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006;9:47–57. doi: 10.1007/BF03033307. [DOI] [PubMed] [Google Scholar]