

Abstract
Successful evolutionary enzyme engineering requires a high throughput screening or selection method, which considerably increases the chance of obtaining desired properties and reduces the time and cost. In this review, a series of high throughput screening and selection methods are illustrated with significant and recent examples. These high throughput strategies are also discussed with an emphasis on compatibility with phenotypic analysis during directed enzyme evolution. Lastly, certain limitations of current methods, as well as future developments, are briefly summarized.
Introduction
Directed evolution has been successfully used to engineer enzymes with a wide variety of properties such as substrate specificity, organic solvent resistance, thermostability, and optimum working pH.1−5 A successful directed evolution experiment depends on two aspects: genetic diversity and high throughput screening or selection (HTSOS) methods. Thanks to the advances in molecular biology, various methods including random mutagenesis, gene recombination, and semirational mutagenesis have been developed to introduce sufficient genetic diversity.6−8 Consequently, the primary limitation in most directed evolution experiments is the identification of the desired mutants.9 Therefore, an HTSOS method is extremely desirable in directed evolution.
Screening and selection are the two main methods of library analysis. Screening refers to evaluation of every protein for the desired property, while selection automatically eliminates nonfunctional variants.10 High throughput screening and selection methods enable the rapid identification of desirable traits from multifarious candidates. However, coupling phenotype analysis with a compatible HTSOS method is the most challenging part of developing HTSOS methods for enzyme engineering.
The purpose of this review is to familiarize readers with the strategies for coupling phenotype analysis with a compatible HTSOS method. In this review, we focus on high throughput screening and selection methods for evolutionary enzyme engineering and highlight their significant applications. General principles and specific examples are included in each method. High throughput screening methods developed for other fields such as drug discovery will not be discussed here.
High Throughput Screening Methods
Evaluation of individual protein variants is normally required in screening. It greatly reduces the chance of missing a desired mutant. However, the throughput is also reduced. By taking advantage of automation, high throughput screening methods can streamline traditional screening processes. Most importantly, methods such as fluorescence-activated cell sorting (FACS) can make the desired mutants easy to detect.
Microtiter Plates
A microtiter plate miniaturizes test tubes to multiple wells (Figure 1A). Although higher density formats, e.g., 1536-well and 9600-well, are commercially available, the 96-well plate is the most widely used format. Traditional enzyme activity assays can be performed in a microtiter plate by adding reaction components and either crude cell extracts or purified proteins manually. However, the throughput can be greatly improved with the aid of robotic systems.11 Colorimetric or fluorometric assays are the most convenient ones among numerous microtiter plate-based enzyme activity assays.12−14 For certain enzymatic reactions, the disappearance of substrates or formation of products can be easily identified by macroscopic observation or measuring UV–vis absorbance or fluorescence using a plate reader. Since these assays highly depend on the chemistry and availability of suitable native substrates, it is not surprising that they are not generally applicable. Recently, microtiter plates have been used for detecting cellulase and protease activities using a micro-bioreactor system, Biolector.15−17 Adding light excitation and emission filters to the microtiter plate on a specially designed shaking machine, Biolector is able to online monitor the light scatter and reduced nicotinamide adenine dinucleotide (NADH) fluorescence signals, which indicate the different levels of hydrolysis of an insoluble protein substrate or NADH-coupled enzyme activity. Microtiter plates are also regarded as a well-developed alternative to shake flasks that allow screening of mutants with diverse profiles of cell growth, substrate uptake, and product formation.18,19
Figure 1.

Schematic overview of high throughput screening methods. (A) Microtiter plates. A mutant DNA library was transformed into competent cells and plated onto plates. Enzyme variants were expressed inside the cells. The cells were then lysed and lysates transferred to a microtiter plate for enzymatic assay. The enzyme activity can be visualized by macroscopic observations, and enzymes with improved properties were selected for the next round of evolution. (B) Digital imaging. Digital imaging employs advanced imaging devices during screening of individual clones, which greatly enhances the throughput. (C) Product entrapment. The bar, oval, triangle, and star represent the gene, the gene product, the substrate, and the product, respectively. A fluorescent product of certain enzymatic reactions was trapped inside the cell, which made the cell screenable. (D) Cell surface display. The fluorescent product was enzymatically linked onto the cell surface. (E) In vitro compartmentalization (IVTC). The yellow oval represents the emulsion droplet. It acts as a man-made bioreactor. (F) FACS. Cells or emulsion droplets exhibiting different fluorescent signals can be sorted by FACS with high throughput.
Digital Imaging
Digital imaging (DI) allows the solid-phase screening of colonies via integrating single pixel imaging spectroscopy (Figure 1B). As it relies on simple, widely known colorimetric activity assays, DI has been applied to screen enzyme variants on problematic substrates.20,21 One classic example is to exploit DI as a screening method for directed evolution of transglycosidases. A covalent glycosyl–enzyme intermediate was involved in screening. In the absence of the acceptor cellobiose, mutants with nonhydrolytic or low hydrolytic activity gives light colored colonies using X-glycosyl as donor substrate. However, in the presence of the acceptor, mutants displaying high transferase activity resulted in an increase in the colony coloration. By using such a method, a 70-fold improvement on transglycosidase/hydrolysis activity ratio was obtained.22
FACS
Based upon the fluorescent signal of individual cells, FACS provides a method for sorting cells into two or more containers at rates of up to 30,000 cells/s23 (Figure 1F). Surface display (see the cell surface display subsection), in vitro compartmentalization (IVTC; see the IVTC subsection), green fluorescent protein (GFP)-reporter assays and product entrapment are the main applications of FACS screening approaches for enzyme activities.24
GFP and other fluorescent proteins are ideal objects for cell fluorescence analysis. By coupling target enzyme activity with the expression level of GFP, FACS has been successfully applied for screening Cre recombinase mutants with altered site specificity25 (Figure 2). In another example, GFP was used as a substrate of GroEL chaperonins with low folding efficiency. An inducible GFP was used to monitor the folding efficiency mediated by chaperonin mutants.26
Figure 2.
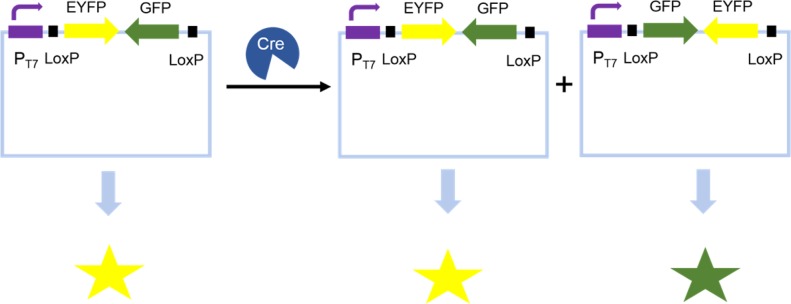
Screening Cre recombinase mutants with altered site specificity by FACS. The cells with active Cre recombinase can equally express EYFP and GFP, whereas cells with Cre recombinase which cannot recognize the loxP sites only yield yellow florescence.
For product entrapment, a fluorescent substrate is employed that can be transported both into and out of the cell and react with the enzyme of interest. For those substrates, they can be finally washed off and not be retained within the cell. Conversely, the fluorescent product cannot get out of the cell because of its size, polarity, or chemical properties. Taking advantage of the differences in physical properties between the substrate and the product, FACS enables screening of the desired phenotype through entrapment of the product inside the cells24 (Figure 1C,F). In this way, a variant of glycosyl-transferase was identified that exhibited more than 400-fold enhanced activity for the fluorescent selection substrates.27
Cell Surface Display
Among various display technologies, cell surface display is most adaptable for high throughput screening. Fused with anchoring motifs, enzymes encoded by DNA inside the cell can be expressed and displayed on the outer surface of the cell, where they directly react with substrates28 (Figure 1D). Cell surface display technologies have been developed in bacteria, yeast, and mammalian cells29−32 and have become an important application for FACS and fluorescence resonance energy transfer (FRET, see the Resonance Energy Transfer subsection) based screening methods. In particular, a new system integrating yeast surface display, enzyme-mediated bioconjugation and FACS, was reported as a general strategy for evolution of bond-forming enzymes. The enzymes displayed on yeast cell surface catalyze attachment of a diffusible substrate to the cell surface through bond formation. The surface bound substrate was subjected to fluorescence excitation and FACS screening. Impressively, this method achieved a 6,000-fold enrichment of active clones after a single round of screening.33
IVTC
As one type of compartmentalization, IVTC uses man-made compartments (e.g., water-in-oil (W/O) emulsion droplets or water-in-oil-in-water (W/O/W) double emulsion droplets) to isolate individual DNA molecules, forming independent reactors for cell-free protein synthesis and enzyme reaction34 (Figure 1E). Droplet microfluidic devices compartmentalize reactants into picoliter volumes with shorter time, higher sensitivity, and higher throughput than standard assays.35 IVTC has several advantages over in-vivo-based high throughput screening methods. First, it circumvents the regulatory network of in vivo systems, eliminating the possibility that the evolved phenotype arises from mutations not related to the target gene; second, since transformation is avoided, the library size is no longer limited to transformation efficiency of the host cell.
Though many enzymes have been proven to be incompatible with IVTC due to the different conditions between transcription–translation and screening,36 IVTC holds promise for screening enzymatic activity by combining with FACS or uniform polymer particles with diameters of 0.5–500 μm (microbeads).37−40 For example, IVTC was used to screen activity of [FeFe] hydrogenase, which is greatly inhibited by oxygen. Bound to the antibodies on the surface of microbeads, the tagged hydrogenases were subjected to oxygen exposure. Active [FeFe] hydrogenases consumed hydrogen and reduced C12-resazurin to fluorescent C12-resorufin, which adsorbed to the microbead surface, and were finally isolated by FACS.41 In another example, a single mutant of β-galactosidase was expressed in each W/O/W emulsion droplet, which allowed direct sorting of the droplets by FACS. In this way, eight mutants were identified that exhibited 300-fold higher kcat/KM values than that of the wild type enzyme.42
Resonance Energy Transfer
Resonance energy transfer (RET) is an energy transfer mechanism between two chromophores or fluorophores. The donor chromophore in an initial electronic excited state may transfer energy to the acceptor chromophore through nonradiative dipole–dipole coupling in a distance dependent manner.42 GFP, cyan fluorescent protein (CFP), yellow fluorescent protein (YFP), and red fluorescent protein (RFP) are common fluorophores used for RET.43−46 Nonradiative energy transfer between these fluorophores permits the study of protein interactions and conformations using RET.47,48
Combined with other high throughput screening methods (e.g., FACS), RET has been used for evolutionary enzyme engineering. One such example employed FRET to assay protease activity of OmpT. The substrate was composed of a fluorescent dye and a quenching partner which were linked by a scissile bond recognized by OmpT. The enzymatic cleavage led to the diffusion of the substrate’s quencher moiety, while the fluorophore remained on the cell surface. Active clones were enriched by 5000-fold after a single round of screening.49
High Throughput Selection Methods
Compared with screening methods, with which analysis of each individual enzyme variant is unavoidable, selection methods directly eliminate unwanted enzyme variants through applying certain selective pressure to the mutant library. Therefore, only positive variants are carried onto the next round of directed evolution, making the assessment of a much larger library (more than 1011) possible. This “rejective to the unwanted” feature of selection methods makes them intrinsically high throughput.
Currently, a variety of high throughput selection methods have been developed for selection of enzymes with different catalytic functions. Despite the various assay techniques involved, high throughput selection methods can be divided into two main categories, display and compartmentalization.
Display
In various display technologies, either the translated protein is physically connected to its encoding gene or the gene is restricted in a virus particle that displays the protein. The displayed protein library is readily accessible to the external environment and thus can be subjected to high throughput selection for the desired enzymatic properties. Finally, the genes linked to the selected protein variants can be easily traced and amplified.
Plasmid Display
In the plasmid display technology, a DNA binding protein was fused to the protein of interest. The fusion protein is expressed in the cell and binds to its encoding plasmid containing the recognition sequence of the DNA binding domain (DBD; Figure 3A). Then the cell is lysed, and the protein–plasmid complex can be selected. Different DBDs have been utilized to construct plasmid display systems. In one study, nuclear factor κB (NF-κB) p50 was fused to a protein of interest.50 The resulting chimera retains picomolar affinity and DNA specificity of wild type NF-κB p50. This p50-based plasmid display system was used to enrich a maltose binding protein as well as a single functional protein from large libraries. Other studies have used GAL4 DBD in the plasmid display system. Choi et al. fused model proteins, enhanced green fluorescence protein (eGFP), and glutathione S-transferase (GST), with GAL4 DBD to create GAL4 DBD/eGFP and GAL4 DBD/GST, which were efficiently isolated by plasmid display.51 These studies demonstrated the feasibility of using simple plasmid display systems to discover functional proteins from large libraries.
Figure 3.

Schematic overview of four display techniques for high throughput selection. (A) Plasmid display. The gene (green bar) encodes the enzyme of interest (green ball) fused with a DNA binding domain (pink ball). The DNA binding domain mediates the bonding between the enzyme and the plasmid. (B) SNAP display. The gene was expressed in an emulsion droplet. The encoded enzyme was then covalently bonded to the DNA through thioester bond formation between the SNAP-tag (pink ball) and benzylguanine (black box). (C) Phage display. The gene (green bar) encodes the enzyme of interest (green ball) fused to the coat protein (pink bar) of the phage. The enzyme is then consequently displayed on the phage surface. (D) Ribosome display. The gene was in vitro transcribed and translated. The transcribed mRNA (green spiral) and expressed enzyme associate with the ribosome (red) to form a complex. This complex was then subjected to selection. The selected genes can be recovered by reverse transcription polymerase chain reaction (RT-PCR).
SNAP and SNAP Dendrimer Display
Another display technique that connects DNA with the protein is SNAP display (Figure 3B). In this system, a SNAP-tag (O6-alkylguanine-DNA-alkyltransferase) is fused to the protein of interest, while the template DNA is covalently bonded to a suicide substrate of the SNAP-tag (benzylguanine). The DNA that encodes a library of protein variants is encapsulated in water-in-oil droplets where in vitro transcription and translation occur. The expressed protein is then covalently coupled to its encoding DNA through the formation of a thioester bond between the SNAP-tag and benzylguanine. SNAP display is widely used for selecting protein binders. In one study, an improved SNAP display system was developed and used to achieve a 107-fold enrichment of a Her2 binder over nonbinding proteins after three rounds of selection. This study demonstrated SNAP display as an efficient in vitro protein engineering tool for resolving proteins with different binding affinities.52 A variant of SNAP display uses SNAP dendrimers instead of monomers to allow the display of multiple copies of a protein of interest, thus taking advantage of accumulating affinity during protein–ligand binding. In this system, the template DNA is ligated to a dendrimer-like structure that is assembled from multiple Y-shaped double stranded DNA monomers and incorporates multiple benzylguanine groups. These benzylguanine groups then serve as the docking points for SNAP-tagged proteins. Using SNAP dendrimer display in a model experiment, Kaltenbach et al. were able to achieve an enhancement of enrichment by up to 5-fold higher and recovery by up to 25-fold higher compared with SNAP monomer display.53
Phage Display
Phage display is the best described and most commonly used display techniques for in vitro selection. In typical phage display experiments, filamentous phages (such as M13) are used for their ability to infect bacterial hosts without killing them. A phagemid DNA library is constructed in vitro and transformed into competent bacterial cells. The protein to be displayed is fused to one of the coat proteins (such as pIII and pVIII) and displayed on the surface of the phage when the phage is assembled in the host cell. The DNA encoding the protein will be retained in the phage particle and recovered afterward (Figure 3C). Phage display is naturally based on binding, which makes it an effective system for selecting protein binders. Using five rounds of M13 phage display, Park et al. identified several binding peptides of Bcl-2 protein, a key regulator of apoptosis associated with human disease.54 However, one limitation of using filamentous phages for phage display is that they do not display cytoplasmic proteins effectively. To select cytoplasmic protein variants, cytoplasmic phages, such as T7 phage, have been used.55 While phage display is extremely efficient in affinity-based selections, its use for selecting other enzyme properties is still restricted. Furthermore, phage display is not suited for selection of eukaryotic proteins which usually require proper folding and posttranslational modifications. To circumvent these limitations, another similar display technique termed retrovirus display was developed.56 Instead of infecting prokaryotic bacterial cells, retroviruses infect mammalian cells, thus providing a mammalian platform for protein expression. The robustness of retrovirus display was demonstrated by showing a more than 1300-fold enrichment of active wild type tissue plasminogen activator during a single selection cycle.57
mRNA and Ribosome Display
Another high throughput display technology is mRNA display. Different from DNA display techniques which link a protein to its encoding DNA, mRNA display produces proteins that are covalently bonded to its encoding mRNA.58 Basically, the mRNA is generated by in vitro transcription and then linked to puromycin, a peptidyl acceptor antibiotic, at its 3′ end. The C-terminal end of the translated protein reacts with the puromycin to form a joint mRNA–protein molecule. The mRNA display technique has been widely used for selecting protein binders.59,60 Recently, this technique has been further developed for selection of catalytic enzymes. For example, mRNA display was used for the first time to select for novel RNA ligases from a library based on a noncatalytic zinc finger scaffold.61 Libraries of more than 1012 mutants were subjected to selection, and the evolved ligases exhibited multiple turnovers with rate enhancements of more than 2-million-fold. To overcome mRNA instability in certain environments, a variation of mRNA display, cDNA display, has been developed.62
Ribosome display is conceptually similar to mRNA display. Instead of a covalent bond between the mRNA and protein, a complex of mRNA, protein, and the ribosome was formed in ribosome display. Specifically, the 3′ terminus of mRNA is fused to a special spacer sequence that lacks a stop codon. Upon translation, the spacer remains associated with the peptidyl tRNA in the ribosome channel, thus connecting the already translated peptide and the mRNA (Figure 3D). Currently, ribosome display is mainly adapted for selection of binding proteins such as high affinity antibodies.63,64
Compartmentalization
Compartmentalization spatially constrains the gene and protein in a single compartment. According to the nature of the compartments used, compartmentalization can be divided into in vivo compartmentalization (IVVC) and in vitro compartmentalization (IVTC). IVVC uses biological compartments such as phage particles, bacterial cells, and yeast cells, while IVTC uses man-made compartments, in most cases W/O emulsion droplets or W/O/W double emulsion droplets.
IVVC
Regardless of the biological compartments used, IVVC can be roughly divided into two main categories, according to the downstream selection criteria applied. These two categories are growth complementation and reporter-based selection.
Growth Complementation
Selection of evolutionally engineered enzymes using the growth complementation techniques is exclusively conducted in living cells. The examined enzyme property is coupled with the fitness of the host cell in such a way that only the cells containing the desired enzyme variants can survive under the selective pressure (Figure 4A). Specific growth complementation selection methods depend on the nature of the evolved enzyme. For example, a dimeric chorismate mutase has been topologically redesigned into a monomeric protein with near native activity.65 Since chorismate mutase is involved in the phenylalanine and tyrosine production pathway, it was naturally reasonable to use growth complementation in this case. A more stringent selection was developed for evolving highly active chorismate mutase using an N-terminal degradation tag.66 Other enzymes such as terminal alkane hydroxylases and β-glucosidases have been evolved into catalytically specific or thermostable variants by selecting host cells on different carbon sources.67,68 By using growth complementation, even relatively difficult enzyme properties such as enantioselectivity can be efficiently selected. In one study, a dual selection system was used to select for a lipase with desired enantioselectivity.69 First, a lipase mutant library in an aspartate auxotroph Escherichia coli (E. coli) was plated on minimal medium that was supplemented with the aspartate ester of the desired enantiomer (S)-(+)-1,2-O-isopropylidene-sn-glycerol. As a result, only lipase that hydrolyzed this substrate could be selected. Second, variants with low enantioselectivity were eliminated by adding a covalently binding phosphonate ester of the opposite (R)-(−)-1,2-O-isopropylidene-sn-glycerol. After three rounds of selection with increasing phosphonate ester concentration, a mutant with an improved enantioselectivity toward the (S)-(+)-enantiomer was selected.
Figure 4.
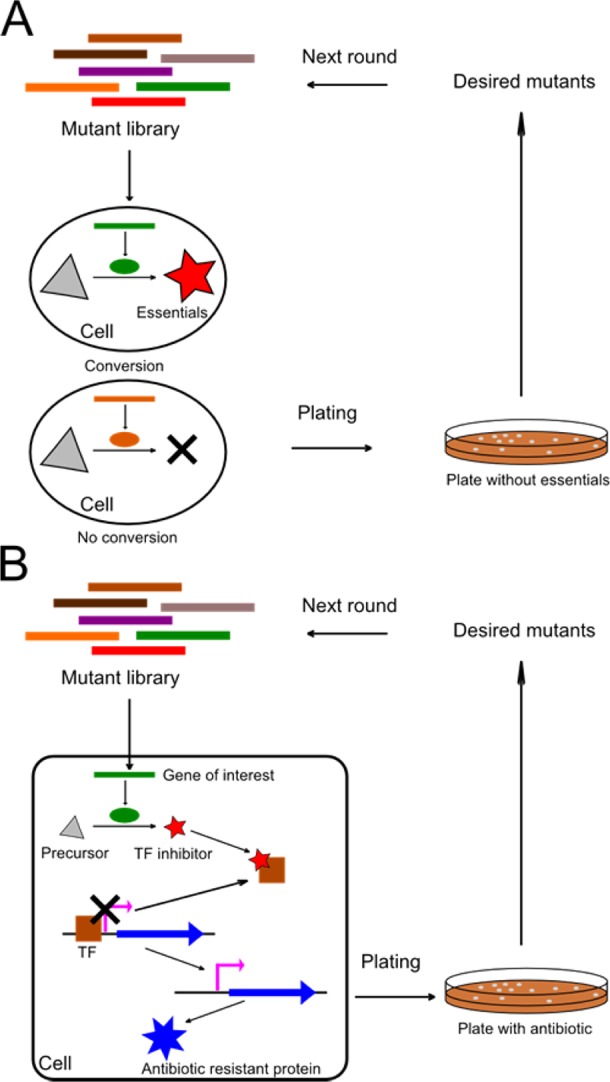
Schematic overview of growth complementation and reporter-based selection. (A) Growth complementation. A library of mutant genes was transformed into host cells. The active mutant protein (green oval) converts a precursor molecule (gray triangle) into a compound essential to cell survival (red star), while the nonactive mutant protein (brown oval) does not. After plating the cells onto plates without the compound essential to cell survival, only cells with active mutant proteins will survive and be selected. The active genes are then recovered and subjected to the next round of mutagenesis. (B) Reporter-based selection. Here a transcriptional regulator-based strategy is presented. A library of mutant genes was transformed into host cells. The active mutant protein (green oval) converted a precursor molecule (gray triangle) into a transcription factor (TF) inhibitor (red star), which binds and inhibits the TF (brown rectangle) of the reporter gene (blue arrow). The TF then dissociates, and the reporter gene is expressed. Here, the reporter gene encodes an antibiotic resistant protein as an example. After plating the cells onto plates with the antibiotic, only cells with active mutant proteins will survive and be selected. The active genes are then recovered and subjected to the next round of mutagenesis.
Reporter-Based Selection
A reporter can be used for either screening or selection. In a reporter-based selection, the activity of a specific reporter confers a survival phenotype of the host cell. For example, when the reporter gene encodes an antibiotic resistant protein, it is considered a reporter-based selection (Figure 4B). The reporter activity is designed to be controlled by the activity of the enzyme of interest. As a result, desired enzyme variants can be selected by selecting cells exhibiting reporter activity. Since enzyme activity is indirectly linked to cell growth, reporter-based selection allows for selection of more diverse enzyme functions as compared to growth complementation. Based on how the reporter gene is turned on by the enzyme of interest, reporter-based selection can be divided into transcriptional-regulator-based strategies and riboswitch/ribozyme-based strategies, as reviewed elsewhere.70 For example, Firestine et al. developed an AraC-based three hybrid system using the arabinose operon activator AraC for screening or selection.71 Instead of linking the enzyme function to transcription regulators, riboswitch/ribozyme-based strategies use a small molecule-regulated riboswitch/ribozyme. Upon binding of a specific small molecule to the riboswitch/ribozyme, the reporter gene is turned on and can be selected. The regulating small molecule is often the product of an enzymatic reaction. Michener and Smolke used a synthetic riboswitch coupled with FACS to aid in the evolution of a caffeine demethylase, increasing the enzyme activity in vivo by 33-fold and the product selectivity by 22-fold.72 The utilities of riboswitches/ribozymes for reporter-based strategies were further broadened by adapting riboswitches to recognize new small molecules through aptamer selection.73
Other than transcription regulator and riboswitch/ribozyme-based reporter systems, Esvelt et al. developed a continuous biomolecule directed evolution system termed phage-assisted continuous evolution (PACE).74 Essentially, PACE combines a reporter-based selection strategy with directed evolution, assisted by an automated phage infection cycle. The interrogated enzyme property is directly linked to the RNA polymerase activity that drives the expression of phage coat protein pIII, which is essential for making the assembled phage particle infectious. As a result, only genes of active enzymes will be selected and passed on to the next round of directed evolution. Using the PACE system, various enzyme properties such as polymerase activity, protein–peptide binding, and recombinase activity can be selected and evolved in an ultrahigh throughput manner. As compared to traditional selection methods that require human intervention between each round of directed evolution, PACE further increases the throughput by reducing the human effort and time cost.
IVTC
In addition to screening, IVTC can also be used in selection. Nonetheless, the adaptation of IVTC to selection has not been as widely adopted as in screening. It seems that only enzymes that directly or indirectly act on DNA can be selected using IVTC. For example, Tay et al. were able to select bacteriophage λ integrases with significantly enhanced recombination activity on a noncognate target DNA sequence.75 In this study, desired recombination product containing the encoding gene was recovered from the DNA pool by selective PCR using specifically designed primers. This in vitro selection by PCR mimics the survival-based in vivo selection process in growth complementation. The application of IVTC to selection can potentially increase the throughput by circumventing the transformation step involved in in vivo selection methods. However, selective PCR in this work made simultaneous differentiation of enzyme activity levels impossible. As a result, each selected clone needs to be further screened in order to evaluate the absolute enzyme activity and identify the best performing enzyme variant, which reduces the throughput of the entire selection process. Further study is needed for exploring IVTC as a high throughput selection tool.
Conclusion and Prospects
Fast development of HTSOS methods in the past decade has greatly advanced research in evolutionary enzyme engineering. A comprehensive overview of HTSOS methods was summarized in Table 1. Thanks to these methods, large libraries created by various diversity-generating strategies can now be comprehensively screened or selected. As a result, the chance of obtaining an enzyme variant with the desired properties is greatly enhanced. Despite the great successes, certain limitations of current screening or selection methods need to be further addressed. For example, traditional screening methods such as microtiter plates have broad utility in almost every facet of phenotype improvement, but at the cost of relatively low throughput (usually <105). Screening coupled with FACS greatly increases the throughput, yet not every enzyme property is amenable to FACS. A similar problem exists with high throughput selection methods. For instance, display techniques are mostly used for selecting binding proteins, while IVVC leads to a loss of genetic diversity during transformation of the DNA library. Currently, successful stories overcoming some of the limitations are still limited to individual cases.75 How to adapt high throughput screening or selection methods to a broader range of enzyme functions is an important question to ask in future studies.
Table 1. Summary of HTSOS Methods.
| methods | screening/library size | advantages | disadvantages | applications |
|---|---|---|---|---|
| high throughput screening methods | ||||
| microtiter plates | <104/day | applicable to many assays | laborious | enzyme reactions leading to change in color, fluorescence, pH, cell growth, etc. |
| DI | limited by transformation efficiency | high sensitivity | not generally applicable | restricted to colorimetric activity assays |
| FACS | up to 3 × 104/s23 | high sensitivity and extremely high throughput | target enzyme activity has to be coupled with the expression level of fluorescent proteins | enzyme reactions leading to change in fluorescence |
| cell surface display | limited by transformation efficiency | avoids possible cell lysis during enzyme reaction | protein expressed as fusion proteins | screening for bond-forming enzymes |
| IVTC | limited by the throughput of the detection method (e.g., FACS) but not transformation efficiency | high sensitivity and efficiency | lack of posttranslational modifications; not suitable for screening enzymes with incompatible conditions between transcription-translation and screening | screening enzymatic activities combined with FACS |
| RET | limited by transformation efficiency | high sensitivity and efficiency | target enzyme activity has to be coupled with energy transfer between two fluorophores | screening protease activity |
| high throughput selection methods | ||||
| plasmid display | limited by transformation efficiency | avoids potential problems of protein secretion, in vitro translation, and/or RNA instability | protein expressed as fusion proteins | selecting protein binders |
| SNAP display | limited to 109/(1 mL of emulsion)52 | resolves proteins with different binding affinities; display multiple copies of a protein; selection under harsh conditions | weak binders can be lost in SNAP monomer display; lack of posttranslational modifications | selecting protein binders |
| phage display | limited by transformation efficiency | multiple copies of proteins can be displayed | not applicable to selecting diverse enzyme properties; not suitable for selecting eukaryotic proteins; protein expressed as fusion proteins | extremely efficient in affinity-based selections |
| retrovirus display | limited by transformation efficiency | proper folding of eukaryotic proteins; allows posttranslational modifications | protein expressed as fusion proteins | selecting eukaryotic proteins |
| mRNA display and ribosome display | not limited by transformation efficiency | overcome the incompatible conditions between in vitro transcription and translation | mRNA instability; lack of posttranslational modifications; protein expressed as fusion proteins | selecting protein binders, high affinity antibodies, and catalytic enzymes |
| growth complementation | limited by transformation efficiency | potential to select enzymes with diverse properties | selection method needs to be individualized | selecting enzyme properties that can be coupled to host fitness |
| reporter-based selection | limited by transformation efficiency | potential to select enzymes with diverse properties | reporter needs to be constructed individually according to properties of selected enzyme | selecting enzymes that produce transcription regulation molecules |
| IVTC | not limited by transformation efficiency | high sensitivity toward variants without target enzyme activity | lack of posttranslational modifications; further screening is needed for identification variants with high enzyme activity75 | restricted to selecting enzymes that act on DNA |
Despite the limitations, development of new technologies and the interplay between different techniques are enabling a further improvement of the throughput. For example, IVTC and FACS can be combined together to preserve the genetic diversity and reduce the screening effort, at the same time. Another way to increase the throughput is by reducing human intervention during the entire directed evolution process, such as that demonstrated in PACE. A minimal human intervention may be achieved by incorporating robotic system- or microfluidic chip-based large-scale automation into the evolution process. Finally, as automation becomes more and more comprehensive, many design and practical issues such as data management and logistics may rise as new challenges in the field of evolutionary enzyme engineering.
Acknowledgments
We thank the National Institutes of Health (Grant GM077596) and the National Science Foundation as part of the Center for Enabling New Technologies through Catalysis (CENTC), Grant CHE-0650456, for financial support in our directed evolution projects. In addition, we thank Dr. Ryan E. Cobb (Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana–Champaign, Urbana, IL, USA) for helpful suggestions.
Glossary
Abbreviations
- CFP
cyan fluorescent protein
- DBD
DNA binding domain
- DI
digital imaging
- eGFP
enhanced green fluorescence protein
- FACS
fluorescence-activated cell sorting
- FRET
fluorescence resonance energy transfer
- GFP
green fluorescent protein
- HTSOS
high throughput screening or selection
- IVTC
in vitro compartmentalization
- IVVC
in vivo compartmentalization
- PACE
phage-assisted continuous evolution
- RET
resonance energy transfer
- RFP
red fluorescent protein
- W/O
water-in-oil
- W/O/W
water-in-oil-in-water
- YFP
yellow fluorescent protein
Author Contributions
∥ H.X. and Z.B. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Goldsmith M.; Tawfik D. S. Directed enzyme evolution: beyond the low-hanging fruit. Curr. Opin. Struct. Biol. 2012, 224406–412. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Ang E. L.; Zhao H., Directed evolution of enzymes for industrial biocatalysis. In Industrial Biocatalysis; Grunwald P., Ed.; Pan Stanford: Singapore, 2014; pp 73–110. [Google Scholar]
- Cobb R. E.; Si T.; Zhao H. Directed evolution: An evolving and enabling synthetic biology tool. Curr. Opin. Chem. Biol. 2012, 163–4285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb R. E.; Chao R.; Zhao H. Directed evolution: past, present, and future. AIChE J. 2013, 5951432–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Singh S. Directed evolution: Tailoring biocatalysts for industrial applications. Crit. Rev. Biotechnol. 2013, 334365–378. [DOI] [PubMed] [Google Scholar]
- Nair N. U.; Zhao H.. Improving proteins by directed evolution. In Handbooks for Metabolic Engineering; Smolke C., Ed.; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2009; pp 2.1–2.37. [Google Scholar]
- Labrou N. E. Random mutagenesis methods for in vitro directed enzyme evolution. Curr. Protein Pept. Sci. 2010, 11191–100. [DOI] [PubMed] [Google Scholar]
- Ruff A. J.; Dennig A.; Schwaneberg U. To get what we aim for—Progress in diversity generation methods. FEBS J. 2013, 280132961–2978. [DOI] [PubMed] [Google Scholar]
- Yuan L.; Kurek I.; English J.; Keenan R. Laboratory-directed protein evolution. Microbiol. Mol. Biol. Rev. 2005, 693373–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis H.; Kelly R. M.; Dijkhuizen L. Directed evolution of enzymes: Library screening strategies. IUBMB Life 2009, 613222–228. [DOI] [PubMed] [Google Scholar]
- Watt A. P.; Morrison D.; Locker K. L.; Evans D. C. Higher throughput bioanalysis by automation of a protein precipitation assay using a 96-well format with detection by LC-MS/MS. Anal. Chem. 2000, 725979–984. [DOI] [PubMed] [Google Scholar]
- Behrendorff J. B. Y. H.; Vickers C. E.; Chrysanthopoulos P.; Nielsen L. K. 2,2-Diphenyl-1-picrylhydrazyl as a screening tool for recombinant monoterpene biosynthesis. Microb. Cell Fact. 2013, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack M.; Burger M.; Pietschmann P.; Hock B. A high-throughput microtiter plate-based screening method for the detection of full-length recombinant proteins. Protein Expression Purif. 2008, 61192–98. [DOI] [PubMed] [Google Scholar]
- He Y.-C.; Ma C.-L.; Xu J.-H.; Zhou L. A high-throughput screening strategy for nitrile-hydrolyzing enzymes based on ferric hydroxamate spectrophotometry. Appl. Environ. Microbiol. 2011, 893817–823. [DOI] [PubMed] [Google Scholar]
- Huber R.; Wulfhorst H.; Maksym L.; Stehr R.; Poehnlein M.; Jaeger G.; Spiess A. C.; Buechs J. Screening for enzyme activity in turbid suspensions with scattered light. Biotechnol. Prog. 2011, 272555–561. [DOI] [PubMed] [Google Scholar]
- Jaeger G.; Wulfhorst H.; Zeithammel E. U.; Elinidou E.; Spiess A. C.; Buechs J. Screening of cellulases for biofuel production: Online monitoring of the enzymatic hydrolysis of insoluble cellulose using high-throughput scattered light detection. Biotechnol. J. 2011, 6174–85. [DOI] [PubMed] [Google Scholar]
- Samorski M.; Muller-Newen G.; Buchs J. Quasi-continuous combined scattered light and fluorescence measurements: A novel measurement technique for shaken microtiter plates. Biotechnol. Bioeng. 2005, 92161–68. [DOI] [PubMed] [Google Scholar]
- Duetz W. A. Microtiter plates as mini-bioreactors: miniaturization of fermentation methods. Trends Biotechnol. 2007, 1510469–475. [DOI] [PubMed] [Google Scholar]
- Weis R.; Luiten R.; Skranc W.; Schwab H.; Wubbolts M.; Glieder A. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 2004, 52179–189. [DOI] [PubMed] [Google Scholar]
- Delagrave S.; Murphy D. J.; Pruss J. L.; Maffia A. M. 3rd; Marrs B. L.; Bylina E. J.; Coleman W. J.; Grek C. L.; Dilworth M. R.; Yang M. M.; Youvan D. C. Application of a very high-throughput digital imaging screen to evolve the enzyme galactose oxidase. Protein Eng. 2001, 144261–267. [DOI] [PubMed] [Google Scholar]
- Bylina E. J.; Grek C. L.; Coleman W. J.; Youvan D. C.. Directed evolution and solid phase enzyme screening. In Advances in nucleic acid and protein analyses, manipulation, and sequencing, Limbach P. A.; Owicki J. C.; Raghavachari R.; Tan W., Eds.; SPIE: Bellingham, WA, USA, 2000; Vol. 1, pp 186–191. [Google Scholar]
- Kone F. M. T.; Le Bechec M.; Sine J.-P.; Dion M.; Tellier C. Digital screening methodology for the directed evolution of transglycosidases. Protein Eng., Des. Sel. 2009, 22137–44. [DOI] [PubMed] [Google Scholar]
- Becker S.; Schmoldt H. U.; Adams T. M.; Wilhelm S.; Kolmar H. Ultra-high-throughput screening based on cell-surface display and fluorescence-activated cell sorting for the identification of novel biocatalysts. Curr. Opin. Biotechnol. 2004, 154323–329. [DOI] [PubMed] [Google Scholar]
- Yang G.; Withers S. G. Ultrahigh-throughput FACS-based screening for directed enzyme evolution. ChemBioChem 2009, 10172704–2715. [DOI] [PubMed] [Google Scholar]
- Santoro S. W.; Schultz P. G. Directed evolution of the site specificity of Cre recombinase. Proc. Natl. Acad. Sci. U. S. A. 2002, 9974185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. D.; Herman C.; Tipton K. A.; Gross C. A.; Weissman J. S. Directed evolution of substrate-optimized GroEL/S chaperonins. Cell 2002, 11171027–1039. [DOI] [PubMed] [Google Scholar]
- Aharoni A.; Thieme K.; Chiu C. P.; Buchini S.; Lairson L. L.; Chen H.; Strynadka N. C.; Wakarchuk W. W.; Withers S. G. High-throughput screening methodology for the directed evolution of glycosyltransferases. Nat. Methods 2006, 38609–614. [DOI] [PubMed] [Google Scholar]
- Lee S. Y.; Choi J. H.; Xu Z. Microbial cell-surface display. Trends Biotechnol. 2003, 21145–52. [DOI] [PubMed] [Google Scholar]
- Kim Y. S.; Jung H. C.; Pan J. G. Bacterial cell surface display of an enzyme library for selective screening of improved cellulase variants. Appl. Environ. Microbiol. 2000, 662788–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. C.; Wu C. H.; Chang C. Y.; Lu W. C.; Tseng Q. Z.; Prijovich Z. M.; Schechinger W.; Liaw Y. C.; Leu Y. L.; Roffler S. R. Directed evolution of a lysosomal enzyme with enhanced activity at neutral pH by mammalian cell-surface display. Chem. Biol. 2008, 15121277–1286. [DOI] [PubMed] [Google Scholar]
- Boder E. T.; Wittrup K. D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 156553–557. [DOI] [PubMed] [Google Scholar]
- Yi L.; Gebhard M. C.; Li Q.; Taft J. M.; Georgiou G.; Iverson B. L. Engineering of TEV protease variants by yeast ER sequestration screening (YESS) of combinatorial libraries. Proc. Natl. Acad. Sci. U. S. A. 2013, 110187229–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.; Dorr B. M.; Liu D. R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. U. S. A. 2011, 1082811399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherine C.; Lee K.-H.; Oh S.-J.; Kim D.-M. Cell-free platforms for flexible expression and screening of enzymes. Biotechnol. Adv. 2013, 316797–803. [DOI] [PubMed] [Google Scholar]
- Guo M. T.; Rotem A.; Heyman J. A.; Weitz D. A. Droplet microfluidics for high-throughput biological assays. Lab Chip 2012, 12122146–2155. [DOI] [PubMed] [Google Scholar]
- Miller O. J.; Bernath K.; Agresti J. J.; Amitai G.; Kelly B. T.; Mastrobattista E.; Taly V.; Magdassi S.; Tawfik D. S.; Griffiths A. D. Directed evolution by in vitro compartmentalization. Nat. Methods 2006, 37561–570. [DOI] [PubMed] [Google Scholar]
- Griffiths A. D.; Tawfik D. S. Directed evolution of an extremely fast phosphotriesterase by in vitro compartmentalization. EMBO J. 2003, 22124–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrobattista E.; Taly V.; Chanudet E.; Treacy P.; Kelly B. T.; Griffiths A. D. High-throughput screening of enzyme libraries: In vitro evolution of a beta-galactosidase by fluorescence-activated sorting of double emulsions. Chem. Biol. 2005, 12121291–1300. [DOI] [PubMed] [Google Scholar]
- Shim J. U.; Olguin L. F.; Whyte G.; Scott D.; Babtie A.; Abell C.; Huck W. T.; Hollfelder F. Simultaneous determination of gene expression and enzymatic activity in individual bacterial cells in microdroplet compartments. J. Am. Chem. Soc. 2009, 1314215251–15256. [DOI] [PubMed] [Google Scholar]
- Wang B. L.; Ghaderi A.; Zhou H.; Agresti J.; Weitz D. A.; Fink G. R.; Stephanopoulos G. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nat. Biotechnol. 2014, 325473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton J. A.; Swartz J. R. Development of an in vitro compartmentalization screen for high-throughput directed evolution of [FeFe] hydrogenases. PLoS One 2010, 512e15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans B. N.; Adams S. R.; Ellisman M. H.; Tsien R. Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 3125771217–224. [DOI] [PubMed] [Google Scholar]
- Nguyen A. W.; Daugherty P. S. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 2005, 233355–360. [DOI] [PubMed] [Google Scholar]
- Inglese J.; Johnson R. L.; Simeonov A.; Xia M.; Zheng W.; Austin C. P.; Auld D. S. High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 2007, 38466–479. [DOI] [PubMed] [Google Scholar]
- Lam A. J.; St-Pierre F.; Gong Y.; Marshall J. D.; Cranfill P. J.; Baird M. A.; McKeown M. R.; Wiedenmann J.; Davidson M. W.; Schnitzer M. J.; Tsien R. Y.; Lin M. Z. Improving FRET dynamic range with bright green and red fluorescent proteins. Nat. Methods 2012, 9101005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammon S. T.; Villalobos V. A.; Roshal M.; Samrakandi M.; Piwnica-Worms D. Rational design of novel red-shifted BRET pairs: platforms for real-time single-chain protease biosensors. Biotechnol. Prog. 2009, 252559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenworthy A. K. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods 2001, 243289–296. [DOI] [PubMed] [Google Scholar]
- Miyawaki A.; Tsien R. Y. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000, 327, 472–500. [DOI] [PubMed] [Google Scholar]
- Olsen M. J.; Stephens D.; Griffiths D.; Daugherty P.; Georgiou G.; Iverson B. L. Function-based isolation of novel enzymes from a large library. Nat. Biotechnol. 2000, 18101071–1074. [DOI] [PubMed] [Google Scholar]
- Speight R. E.; Hart D. J.; Sutherland J. D.; Blackburn J. M. A new plasmid display technology for the in vitro selection of functional phenotype-genotype linked proteins. Chem. Biol. 2001, 810951–965. [DOI] [PubMed] [Google Scholar]
- Choi Y. S.; Pack S. P.; Yoo Y. J. Development of a plasmid display system using GAL4 DNA binding domain for the in vitro screening of functional proteins. Biotechnol. Lett. 2005, 27211707–1711. [DOI] [PubMed] [Google Scholar]
- Houlihan G.; Gatti-Lafranconi P.; Kaltenbach M.; Lowe D.; Hollfelder F. An experimental framework for improved selection of binding proteins using SNAP display. J. Immunol. Methods 2014, 405, 47–56. [DOI] [PubMed] [Google Scholar]
- Kaltenbach M.; Stein V.; Hollfelder F. SNAP dendrimers: Multivalent protein display on dendrimer-like DNA for directed evolution. ChemBioChem 2011, 12142208–2216. [DOI] [PubMed] [Google Scholar]
- Park H. Y.; Kim J.; Cho J. H.; Moon J. Y.; Lee S. J.; Yoon M. Y. Phage display screen for peptides that bind Bcl-2 protein. J. Biomol. Screening 2011, 16182–89. [DOI] [PubMed] [Google Scholar]
- Dai M.; Temirov J.; Pesavento E.; Kiss C.; Velappan N.; Pavlik P.; Werner J. H.; Bradbury A. R. Using T7 phage display to select GFP-based binders. Protein Eng., Des. Sel. 2008, 217413–424. [DOI] [PubMed] [Google Scholar]
- Urban J. H.; Merten C. A. Retroviral display in gene therapy, protein engineering, and vaccine development. ACS Chem. Biol. 2011, 6161–74. [DOI] [PubMed] [Google Scholar]
- Granieri L.; Baret J. C.; Griffiths A. D.; Merten C. A. High-throughput screening of enzymes by retroviral display using droplet-based microfluidics. Chem. Biol. 2010, 173229–235. [DOI] [PubMed] [Google Scholar]
- Keefe A. D., Protein selection using mRNA display. Curr. Protoc. Mol. Biol. 2001, Chapter 24, Unit 24.5, DOI: 10.1002/0471142727.mb2405s53. [DOI] [PubMed] [Google Scholar]
- Takahashi T. T.; Roberts R. W. In vitro selection of protein and peptide libraries using mRNA display. Methods Mol. Biol. 2009, 535, 293–314. [DOI] [PubMed] [Google Scholar]
- Seelig B. mRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat. Protoc. 2011, 64540–552. [DOI] [PubMed] [Google Scholar]
- Seelig B.; Szostak J. W. Selection and evolution of enzymes from a partially randomized non-catalytic scaffold. Nature 2007, 4487155828–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz M.; Gu K.; Al-Gawari A.; Lohse P. A. cDNA–protein fusions: Covalent protein–gene conjugates for the in vitro selection of peptides and proteins. ChemBioChem 2001, 29666–672. [DOI] [PubMed] [Google Scholar]
- Yanagida H.; Matsuura T.; Yomo T. Ribosome display for rapid protein evolution by consecutive rounds of mutation and selection. Methods Mol. Biol. 2010, 634, 257–267. [DOI] [PubMed] [Google Scholar]
- Zahnd C.; Amstutz P.; Pluckthun A. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 2007, 43269–279. [DOI] [PubMed] [Google Scholar]
- MacBeath G.; Kast P.; Hilvert D. Redesigning enzyme topology by directed evolution. Science 1998, 27953581958–1961. [DOI] [PubMed] [Google Scholar]
- Butz M.; Neuenschwander M.; Kast P.; Hilvert D. An N-terminal protein degradation tag enables robust selection of highly active enzymes. Biochemistry 2011, 50408594–8602. [DOI] [PubMed] [Google Scholar]
- Liu W.; Hong J.; Bevan D. R.; Zhang Y. H. Fast identification of thermostable beta-glucosidase mutants on cellobiose by a novel combinatorial selection/screening approach. Biotechnol. Bioeng. 2009, 10361087–1094. [DOI] [PubMed] [Google Scholar]
- Koch D. J.; Chen M. M.; van Beilen J. B.; Arnold F. H. In vivo evolution of butane oxidation by terminal alkane hydroxylases AlkB and CYP153A6. Appl. Environ. Microbiol. 2009, 752337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma Y. L.; Droge M. J.; van der Sloot A. M.; Pijning T.; Cool R. H.; Dijkstra B. W.; Quax W. J. A novel genetic selection system for improved enantioselectivity of Bacillus subtilis lipase A. ChemBioChem 2008, 971110–1115. [DOI] [PubMed] [Google Scholar]
- Van Rossum T.; Kengen S. W. M.; van der Oost J. Reporter-based screening and selection of enzymes. FEBS J. 2013, 280132979–2996. [DOI] [PubMed] [Google Scholar]
- Firestine S. M.; Salinas F.; Nixon A. E.; Baker S. J.; Benkovic S. J. Using an AraC-based three-hybrid system to detect biocatalysts in vivo. Nat. Biotechnol. 2000, 185544–547. [DOI] [PubMed] [Google Scholar]
- Michener J. K.; Smolke C. D. High-throughput enzyme evolution in Saccharomyces cerevisiae using a synthetic RNA switch. Metab. Eng. 2012, 144306–316. [DOI] [PubMed] [Google Scholar]
- Dixon N.; Duncan J. N.; Geerlings T.; Dunstan M. S.; McCarthy J. E.; Leys D.; Micklefield J. Reengineering orthogonally selective riboswitches. Proc. Natl. Acad. Sci. U. S. A. 2010, 10772830–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt K. M.; Carlson J. C.; Liu D. R. A system for the continuous directed evolution of biomolecules. Nature 2011, 4727344499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay Y.; Ho C.; Droge P.; Ghadessy F. J. Selection of bacteriophage lambda integrases with altered recombination specificity by in vitro compartmentalization. Nucleic Acids Res. 2010, 384e25. [DOI] [PMC free article] [PubMed] [Google Scholar]