

Abstract
Background
Vasospasm contributes to delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage (aSAH). Glutamate concentrations increase after aSAH and correlate with vasospasm in experimental SAH. The Hp2-2 genotype is associated with higher risk of vasospasm after SAH. We tested the efficacy of S-4-CPG, a metabotropic glutamate receptor inhibitor, for treatment of vasospasm after SAH in Hp2-2 and Hp1-1 mice.
Objective
To evaluate the effect on vasospasm and neurobehavioral scores after SAH of systemic S-4-CPG, as well as its toxicity, and phosphorylation of vasodilator-stimulated phosphoprotein (VASP) in Hp 2-2 mice.
Methods
Western blot was used to assess changes in VASP phosphorylation in response to glutamate with and without S-4-CPG. A pharmacokinetics study was done to evaluate S-4-CPG penetration through the blood brain barrier (BBB) in vivo. Toxicity was assessed by administering escalating S-4-CPG doses. Efficacy of S-4-CPG assessed the effect of S-4-CPG on lumen patency of the basilar artery and animal behavior after SAH in Hp 1-1 and Hp 2-2 mice. Immunohistochemistry was used to evaluate the presence of neutrophils surrounding the basilar artery after SAH.
Results
Exposure of human brain microvascular endothelial cells to glutamate decreased phosphorylation of VASP (p-VASP), but glutamate treatment in the presence of S-4-CPG maintains p-VASP. S-4-CPG crosses the BBB and was not toxic to mice. S-4-CPG treatment significantly prevents vasospasm after SAH. S-4-CPG administered after SAH resulted in a trend towards improvement of animal behavior.
Conclusions
S-4-CPG prevents vasospasm after experimental SAH in Hp2-2 mice. S-4-CPG was not toxic and is a potential therapeutic agent for vasospasm after SAH.
Keywords: Glutamate; Haptoglobin; S-4-CPG, SAH; Subarachnoid; Vasospasm
Introduction
Arterial cerebral vasospasm is a contributing factor to symptomatic ischemia in aneurysmal subarachnoid hemorrhage (SAH). Vasospasm is part of a complex process recognized as delayed cerebral ischemia, which in addition includes loss of autoregulation of cerebral blood flow and formation of microvascular thrombi. 1-4 In humans, vasospasm occurs 4 to 21 days after SAH, and is a significant cause of morbidity and mortality.5, 6 Approximately 30% of patients with aneurysmal SAH will develop symptoms of vasospasm, but angiographic evidence of vasospasm will be present in 70% of patients. The incidence of vasospasm following aneurysmal SAH ranges from 20% to 40%.7, 8
Neutrophils and macrophages are key elements of the inflammatory response and play an important role in the development of cerebral vasospasm.9-21 After blood extravasation into the subarachnoid space, neutrophils and macrophages are recruited to clear hemoglobin and erythrocyte remnants, generating an inflammatory process.22 Upregulation of cell adhesion molecules, such as intercellular cell adhesion molecule-1 (ICAM-1, CD54) and vascular cell adhesion molecule-1 (VCAM-1, CD106), promote leukocyte infiltration after SAH.9, 22-26 It is increasingly recognized that factors produced by neutrophils and macrophages initiate and sustain the inflammatory response after SAH resulting in vasospasm and delayed cerebral ischemia. The factors produced by neutrophils that lead to vasospasm include cytokines, enzymatic proteins, and cell adhesion molecules. 27-32
Glutamate, known for its role as an excitatory aminoacid and as a neurotoxic agent, has been shown to be secreted by neutrophils during inflammation.33 Studies have shown that glutamate concentrations in the extracellular fluid in the brain increase in animals after experimental SAH 34-37 and in patients who have sustained an aneurysmal SAH.38 In a rat model of SAH, the increase in glutamate concentration is accompanied by an increase in basilar artery thickness as a measure of vasospasm.37 Other studies have identified an increase in glutamate concentrations after experimental SAH and have correlated this with acute vasoconstriction and a decrease in cerebral blood flow in rats.34, 36 Based on these data we hypothesized that vasospasm could be prevented or attenuated by blocking the action of glutamate on blood vessels pharmacologically.
Glutamate exerts its action in the central nervous system through four different types of receptors, including the kainate (KA) receptor, the α-amino-3-hydroxy-5-methyl-4-isoxazolepropioninc acid (AMPA) receptor, the N-methyl-D-aspartate (NMDA) receptor, and the metabotropic glutamate receptors (mGluR).39 The KA, AMPA, and NMDA receptors are ionotropic and they exert their action through the opening of an ion channel upon binding of their ligand, permitting the flux of ions. Metabotropic glutamate receptors, when activated by their ligand, activate intracellular metabolic pathways that regulate different cell functions ranging from cell cycle to gene expression.40 (S)-4-carboxyphenylglycine (S-4-CPG) is a selective inhibitor of metabotropic glutamate receptors 1 and 5 (mGluR1 and mGluR5), which are expressed in endothelial cells and affect endothelial cell function likely through changes in endothelial cell cytoskeleton.28, 33, 41-43 Antagonists of the mGluRs have been used in experimental models of stroke and subdural hematoma showing a neuroprotective effect.44-47
Activation of mGluR 1 and 5 by glutamate disturbs the barrier function of endothelial cells by promoting dephosphorylation of vasodilator-stimulated phosphoprotein (VASP), a protein that modulates the formation of actin filaments and maintenance of cell-cell junctions.33, 48 When dephosphorylated, VASP increases actin filament formation as well as cell retraction thus increasing the permeability of the endothelial cell barrier. On the other hand, when VASP is phosphorylated it inhibits the elongation of actin filaments and inhibits endothelial cell retraction decreasing the permeability of the endothelial cell barrier. 42, 49-51 We postulated that glutamate could promote vasospasm through dephosphorylating VASP, and that this could be prevented using S-4-CPG to inhibit activation of mGluR1 and 5 receptors in endothelial cells.
Haptoglobin (Hp) is a protein that avidly binds free hemoglobin, playing an essential role in its clearance as Hp-hemoglobin complexes by immune cells after extravasation.21, 22, 52 Hp has two alleles in humans, Hp1 and Hp2, but not in other mammalian species.22 After induction of SAH in a transgenic murine model that expresses the human Hp 2-2 gene, vasospasm is more severe than in mice expressing Hp1-1 genotype. Also, leukocyte infiltration after SAH is more severe in Hp2-2 mice.21 In addition, the Hp 2-2 genotype appears to be associated with higher risk for developing sonographic cerebral vasospasm after SAH as compared to patients with Hp1-1 genotype.53
In this study, we assessed the effect of mGluR1 and 5 inhibition with S-4-CPG on the phosphorylation status of VASP in vitro, and observed that S-4-CPG maintained the phosphorylation status of VASP after exposure of human brain microvascular endothelial cells (HBMECs) to glutamate. Based on this in vitro result we determined the safety of systemically administered S-4-CPG and measured its penetration across the blood brain barrier (BBB). Finally, we assessed the efficacy of S-4-CPG in preventing experimental cerebral vasospasm after induction of SAH in a Hp2-2 and Hp1-1 murine model.
Materials and Methods
Experimental Design
Five different experiments were performed: (1) a biochemical analysis of phosphorylation of VASP in response to glutamate, (2) a pharmacokinetics experiment to evaluate the penetration of S-4-CPG into the central nervous system, (3) a toxicology experiment to assess the adverse effects of the administration of S-4-CPG, (4) an efficacy study to test the usefulness of S-4-CPG in preventing vasospasm, and (5) an immunohistochemical experiment to assess neutrophil infiltration around the basilar artery after experimental SAH. In the biochemical analysis of the phosphorylation of VASP in response to glutamate, human brain microvascular endothelial cells (HBMEC) were incubated with glutamate in the presence or absence of S-4-CPG and the phosphorylation status of VASP was analyzed biochemically. In the pharmacokinetics experiment, S-4-CPG was administered intraperitoneally to 2 Fisher rats, brain extracellular fluid (ECF) was collected, and concentrations of S-4-CPG were measured using HPLC with tandem mass spectrometry detection (MS/MS) to assess the penetration of S-4-CPG across the BBB in vivo. In the toxicology experiment, increasing doses of S-4-CPG were administered intraperitoneally to mice and their behavior and weight gain were evaluated over a period of 90 days. In the efficacy study, the effect of inhibition of mGluR1 and mGluR5 with S-4-CPG on lumen patency of the basilar artery and animal behavior after induction of experimental SAH was evaluated in mice. SAH was induced in mice of genotype Hp1-1 and Hp2-2, the brainstem and the basilar artery were collected, fixed in formalin, and sectioned. Hp 1-1 and Hp 2-2 mice were randomized to four experimental groups to measure basilar artery lumen patency. One group underwent surgery without injection of blood into the cisterna magna (Sham Hp1-1 n=4, Hp2-2 n=5). Another group underwent injection of autologous arterial blood into the cisterna magna with no treatment (SAH-no treatment Hp1-1 n=5, Hp2-2 n=5). A third group underwent injection of autologous blood and administration of vehicle (100nM NaOH, SAH + vehicle Hp1-1 n=7, Hp2-2 n=7), and a fourth group underwent injection of autologous arterial blood into the cisterna magna with treatment with a single dose of S-4-CPG (200 mg/kg) (SAH + S-4-CPG, Hp1-1 n=6, Hp2-2 n=8). In the immunohistochemical experiment, the presence of neutrophils surrounding the basilar artery after induction of SAH was assessed.
Cell Culture and Immunoblotting
HBMEC were cultured according to manufacturer's instructions (Cell Systems). Cells used in the experiments were in their sixth passage. Previous work shows that neutrophils secrete glutamate upon extravasation and that human brain endothelial cells express mGluR1 and 5. 33 Because of this we studied the phosphorylation status of VASP in response to glutamate with and without inhibition of mGluR1 and 5 with S-4-CPG. Cells were plated on a 10 cm dish and exposed to the different experimental conditions. At the end of the experiment, cell were harvested using a cell scraper in lysis buffer (150mM NaCl, 10mM Tris, pH 7.5, 1% NP40, 1% deoxycholate, 0.1% SDS, protease inhibitor cocktail (Roche), and Halt™ Phosphatase inhibitor cocktail (Thermo)). Proteins from whole cell lysates were resolved using the NuPAGE 4–12% Bis-Tris gradient gel (Invitrogen, Carlsbad, CA). Proteins were transferred to PVDF membrane, blocked in 5% non-fat milk in TBS-Tween-20, and blotted with the antibodies for VASP (1:1000, Cell Signaling) and beta-actin (1:10,000, Abcam, Cambridge, MA). Appropriate secondary antibodies were used to detect chemiluminescence.
After the Western blot was developed, densitometry of each band was quantified using Image J software. Actin bands were used as loading controls. After background was subtracted, the densitometry of p-VASP and VASP was quantified. The product of the division of p-VASP/VASP densitometry value was represented in a scatter plot as shown in Figure 1.
Figure 1.
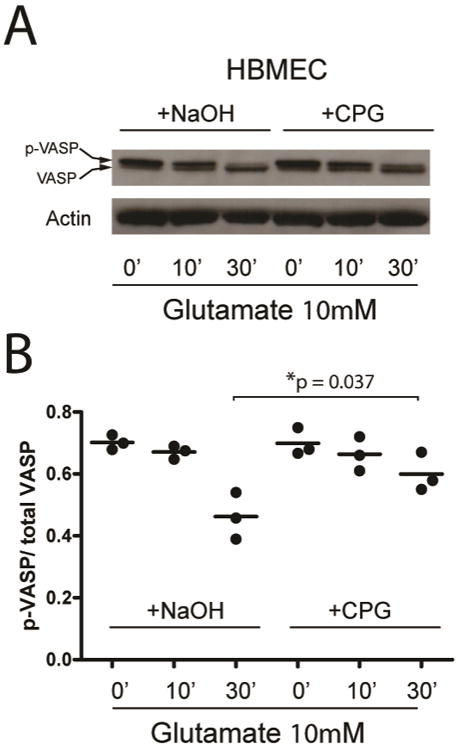
Inhibition of VASP dephosphorylation through antagonism of metabotrobic glutamate receptors by S-4-CPG in HBMEC cells. A) Western blot showing that inhibition of metabotropic glutamate receptors by S-4-CPG maintains VASP phosphorylation status. B) Quantification of the ratio of phosphorylated VASP/total VASP of three separate experiments where VASP phosphorylation status was studied after glutamate application without and with inhibition of metabotropic glutamate receptors by S-4-CPG (10μM). S-4-CPG maintains a significantly higher VASP phosphorylation status 30 minutes after application of glutamate.
Animals
The Johns Hopkins Animal Care and Use Committee approved all experimental protocols. Rats and mice were housed in standard animal facilities with free access to water and rodent chow. Fisher 344 male rats (Harlan Laboratories; Indianapolis, Indiana) were used for the pharmacokinetics studies. C57Bl/6J Hp 1-1 mice (Jackson Laboratories; Bar Harbor, Maine) and C57Bl/6J Hp 2-2 mice (Technion Institute; Haifa, Israel) weighing between 22-30 grams were used for the toxicity and efficacy studies. The development of Hp2-2 transgenic mice was previously reported.21 Briefly, the Hp2-2 allele, which is only found in humans, was generated by producing an intragenic duplication of exons 3 and 4 of the Hp 1 allele in a transgenic mouse model.
Anesthesia
Rats and mice were anesthetized using xylazine (10 mg/kg [100 mg/mL; Phoenix Pharmaceutical]) and ketamine (50 mg/kg [100mg/mL, Phoenix Pharmaceuticals]) administered intraperitoneally. Absence of tail and pedal pinch reflexes was verified before starting the surgeries.
Pharmacokinetics of S-4-CPG in the central nervous system
Microdialysis
Anesthetized Fisher 344 rats (n=2) were secured in a stereotactic frame and a guide cannula with a solid probe was placed in the right basal ganglia. The guide cannula was secured to the skull with dental cement (Geristore Syringeable value kit A2, Denmat, Santa-Maria, CA,). Skin was closed with sutures. After surgery, animals were given buprenorphine, 1 mg/kg, SC, for analgesia. One day after the implantation of the guide cannula the solid probe was replaced with a 2-mm CMA12 microdialysis catheter. The inlet tubing was attached to a CMA microsyringe and pump, and lactated ringers solution was perfused through the catheter tip at a rate of 1 μL/min. After a 60 minute period of equilibration, 112 mg/kg of S-4-CPG (Tocris Biosciences, Ellisville, MO) was administered to rats as a single intraperitoneal dose. Brain ECF dialysate samples were collected at baseline and every 60 minutes for 6 hours. All extracellular fluid samples were assessed for drug concentrations using HPLC-MS/MS. Samples were quantitated over the assay range of 10 to 500 ng/mL, with a 1:10 dilution allowing for quantitation up to 5,000 ng/mL (5 μg/mL).
In vivo assessment of probe recovery
In vivo dialysate recovery experiments were done at the end of each collection period to allow estimation of in vivo recovery and assess the integrity of the microdialysis system. At the end of extracellular fluid collection, the probes were perfused at a rate of 1 μL/min with lactated ringer solution containing 200 ng/ml of S-4-CPG to determine the in vivo probe recovery using the retrodialysis method; microdialysate samples were collected at 10-min intervals for 40 min, and the percentage relative recovery was calculated as follows:54, 55
Where Cperfusate is the drug concentration (200 ng/mL) in the perfusate, and Cdialysate is the drug concentration in the microdialysate, which was the average concentration at each collection time. The recovery was utilized to estimate the estimated ECF concentration according to the equation:54, 55
Individual pharmacokinetic parameters for the estimated ECF were calculated by standard non-compartmental analysis using the WinNonlin version 5.3 (Pharsight Corporation, Mountain View, CA). The maximum plasma concentration (Cmax) and the time of Cmax after intraperitoneal administration (Tmax) were obtained by visual inspection of the concentration-time curve. The area under the plasma concentration-time curve (AUC) was calculated using the log-linear trapezoidal rule to the end of sample collection (AUC0-5.5h) and extrapolated to infinity (AUC0-∞) by dividing the last quantifiable concentration by the terminal disposition rate constant (λz), which was determined from the slope of the terminal phase of the concentration-time profile. The half-life (T1/2) was determined by dividing 0.693 by λz.
Toxicity Study
S-4-CPG was administered to C57Bl/6J mice intraperitoneally. In a dose-escalation study, mice received a single dose of 50mg/kg (n=3), 100mg/kg (n=3), or 200mg/kg (n=3) of S-4-CPG. Mice were evaluated daily for weight and neurological function assessed using a posture, grooming, and ambulation (PGA) scale for 90 days, and euthanized for histopathological evaluation.
Surgical Technique for Experimental Subarachnoid Hemorrhage
We have previously described the surgical methodology used to induce subarachnoid hemorrhage in mice. Briefly, the atlanto-occipital membrane was exposed under the surgical microscope (Zeiss Co, Oberkochen, Germany) and pierced to allow for slow drainage of cerebrospinal fluid. Later, the right femoral artery was exposed, and autologous arterial blood drawn (∼ 60 μL) and injected into the cisterna magna. The blood was not anticoagulated. After injection, animals were positioned head down to allow for blood distribution to other subarachnoid cisterns. Dissected tissues were reapproximated and incisions were closed with staples. S-4-CPG or vehicle was administered one hour after induction of SAH.
Lumen Patency Analysis and Activity and Behavior Level Assessment
Nine sections of each basilar artery were used for analysis of patency. Digital pictures were taken of the sections stained with hematoxylin and eosin. The circumference was measured using computerized analysis by outlining the lumen between the internal lamina and the tunica media (MCID; Imaging Research, Inc., Linton, Cambridge, England). The circumference was preferred to the area, to avoid the error introduced by vessel deformation at the time of sectioning. The circumference of the basilar artery (C = 2πr) was used to calculate the cross-sectional area (Area = C2/4π) of the artery.
Mice were evaluated 24 hours after the surgery for changes in activity and behavior using the PGA score, a three point scale that qualitatively grades posture, grooming behavior, and activity levels. Buprenorphine (0.05 mg/kg [0.3 mg/mL, Abbott Laboratories]) was administered every 12 hours for analgesia. Vasospasm in this murine model of SAH began in the first hour after blood injection into the cisterna magna, peaked at 12h post-hemorrhage, and was maintained for 1-3 days. Activity and behavior of the mice were measured using a scale that evaluates posture, grooming, and ambulation, previously described. Three blinded observers graded animals and the scores of their activity level were recorded and averaged.
Histology Preparation for Lumen Patency and Neutrophil/Macrophage Infiltration Analysis
Twenty-four hours after experimental SAH, mice were anesthetized and perfused by opening of the right atrium and cannulation of the left ventricle with 50 mL of saline solution (0.9%NaCl) followed by perfusion with 50 mL of 4% paraformaldehyde in phosphate buffered saline. The brain was harvested and post-fixed in 4% paraformaldehyde for 24 hours. The brainstems with the basilar artery were removed and paraffin embedded. Transverse sections of the tissue blocks were obtained at a thickness of 10 μm mounted on glass slides and stained with hematoxylin and eosin or for markers of neutrophils/macrophages.
Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). To determine significance of differences among groups, percent lumen patency, and activity level were compared using a 1-way, nonparametric analysis of variance (ANOVA) (Kruskal-Wallis test), with the difference between each group determined by Mann-Whitney's test using Graph Pad 5.0 (Prism software). Values of P ≤ 0.05 were considered significant. For PK analysis, parameters were summarized using descriptive statistics.
Neutrophil/Macrophage Infiltration Analysis
Representative sections of the basilar artery of animals subjected to Sham surgery or SAH were stained for the presence of neutrophils/macrophages using the CD45 antibody (R&D systems, Mouse CD45 MAb Clone 30-F11, Rat IgG2B) at a dilution of 1:200. Sections were deparaffinized and antigen recovery was performed. Briefly, sections were submerged in sodium citrate solution with 0.1% Triton X-100 at 95 °C for 20 minutes. Sections were taken out of the water-bath and were left at room temperature in the sodium citrate solution for 20 minutes. Then sections were washed with phosphate buffered saline with 0.1% Triton X-100 three times for five minutes each. Sections were incubated in 0.3% H2O2, blocked in 10% normal goat serum (Vector Laboratories), and incubated with primary antibody mentioned above. The sections were washed, incubated with biotin-conjugated anti-rat IgG secondary antibody (1:500; Vector Labs), followed by 30 minutes in streptavidin-HRP (BD Biosciences), stained with 3,3-diaminobenzidine (DAB; BD Biosciences) and counterstained with hematoxylin. Negative controls were prepared by omitting the primary antibody.
Results
VASP phosphorylation status after SAH and S-4-CPG
Administration of S-4-CPG in the presence of glutamate prevents dephosphorylation of VASP in HBMECs. Upon resolution on SDS-PAGE, changes in phosphorylation of VASP result in a shift in molecular mass from 50kD to 46 kD.49 In basal conditions, phosphorylated VASP (p-VASP) is abundant. After exposing HBMECs to 10μM glutamate, we found that a shift from the p-VASP to dephosphorylated VASP starts to occur at 10 minutes, becoming significant and more evident at 30 minutes of exposure. This shift can be prevented by the addition of S-4-CPG (10 μM) in the presence of glutamate (Figure 1A). Densitometric quantification of three separate experiments confirms these findings (Figure 1B).
Pharmacokinetics of systemically administered S-4-CPG in brain extracellular fluid
S-4-CPG crosses the blood-brain barrier after systemic administration via the intraperitoneal route. After intraperitoneal administration of 112mg/kg of S-4-CPG, the mean corrected S-4-CPG ECF Cmax ranged from 3.3 to 4.7 μg/mL, which occurred at a median time of 1.5 hours. The area under the concentration curve (AUC 0-∞) ranged from 15.0 to 16.1 μg·hr/mL. The half-life ranged from 1.0 to 4.5 hr (Figure 2).
Figure 2.

HPLC-MS/MS measurement of S-4-CPG concentrations in rat cerebral extracellular fluid. After administration of S-4-CPG, rat cerebral extracellular fluid was collected by microdialysis and S-4-CPG concentrations were measured. Samples were taken every hour. Each rat is displayed (n=2).
Toxicity of S-4-CPG in mice
Administration of increasing doses of S-4-CPG did not have deleterious effects on activity nor weight gain in C57Bl6 wild-type mice. These data indicate that S-4-CPG is not toxic and is safe to use in a murine model of SAH. (Figure 3).
Figure 3.
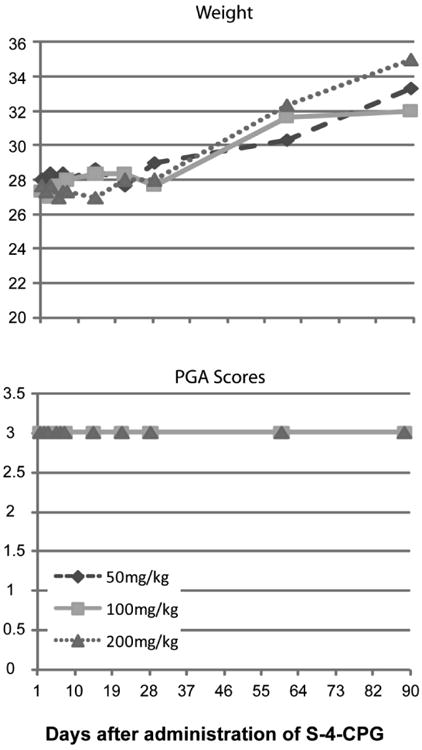
S-4-CPG administration does not have any effects on the long-term on behavior and weight gain of mice. S-4-CPG was administered intraperitoneally in different doses to C57Bl mice (n=3 per group) and weight (A) and behavior (B) were not affected.
Activity levels after SAH
Treatment with S-4-CPG resulted in a trend toward improved activity levels after SAH, which was not statistically significant (Figure 4A). Activity level, as measured by the PGA score, was significantly reduced in mice with SAH-no treatment in both Hp1-1 and Hp2-2 mice as compared to animals in the Sham group (Hp1-1 Sham = 2.5±0.2 vs. Hp1-1 SAH-no treatment = 1.8±0.1, P = 0.008; Hp2-2 Sham = 2.2±0.1 vs. Hp2-2 SAH-no treatment = 1.6±0.3, P = 0.025) (Table 1). Further statistical analysis was prevented by the sample size.
Figure 4.
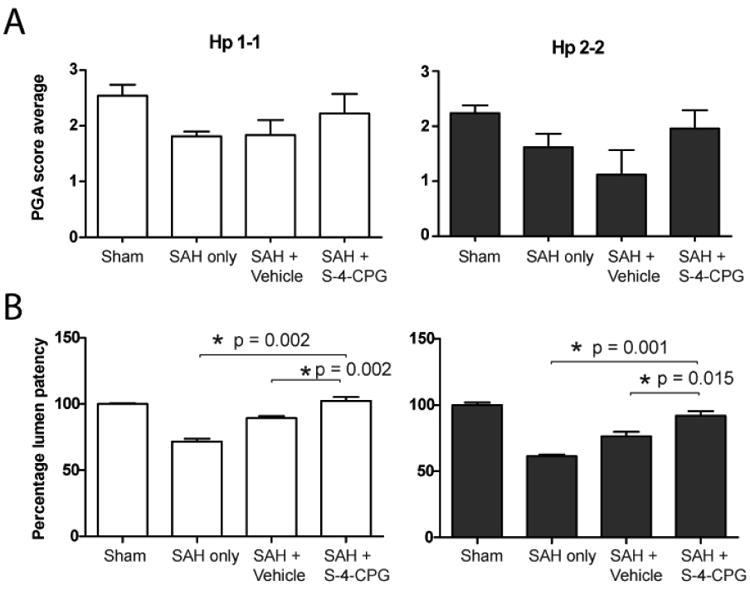
Administration of S-4-CPG (200mg/kg) improves PGA scores and percent lumen patency of animals subjected to experimental SAH. A) Using the PGA scale, we assessed the behavior of mice 24 hours after induction of experimental SAH. B) Percent lumen patency of the basilar artery measurements in Hp1-1 and Hp2-2 mice after the different experimental conditions. Data is presented as mean ± SEM.
Table 1.
PGA scores of Hp1-1 and Hp2-2 animals subjected to the different experimental procedures.
| Groups | P values | ||||
|---|---|---|---|---|---|
| Hp1-1 | PGA Scores | Sham | SAH-no treatment | SAH + Vehicle | SAH + S-4-CPG |
| Sham | 2.5±0.2 | 0.008 | 0.016 | 0.25 | |
| SAH-no treatment | 1.8±0.1 | 0.008 | 0.47 | 0.16 | |
| SAH + Vehicle | 1.8±0.3 | 0.016 | 0.47 | 0.19 | |
| SAH + S-4-CPG | 2.2±0.4 | 0.25 | 0.16 | 0.19 | |
| Groups | P values | ||||
| Hp2-2 | PGA Scores | Sham | SAH-no treatment | SAH + Vehicle | SAH + S-4-CPG |
| Sham | 2.2±0.1 | 0.025 | 0.047 | 0.38 | |
| SAH-no treatment | 1.6±0.3 | 0.025 | 0.184 | 0.2 | |
| SAH + Vehicle | 1.1±0.4 | 0.047 | 0.184 | 0.064 | |
| SAH + S-4-CPG | 2±0.3 | 0.38 | 0.2 | 0.064 | |
Lumen patency of the basilar artery after SAH
Administration of S-4-CPG (200mg/kg) after SAH significantly decreased vasospasm in both Hp1-1 and Hp2-2 animals. Lumen patency of Hp1-1 mice that underwent SAH-no treatment was 71.5% ± 2.2%, SAH + vehicle was 89.3 ± 1.5%, and SAH + S-4-CPG was 102.3 ± 2.9% as compared to Hp1-1 Sham animals. Since vasospasm is more severe in mice expressing the Hp2-2 genotype 21 we decided to test the efficacy of S-4-CPG in the treatment of vasospasm in the Hp2-2 mouse model of SAH. Lumen patency of Hp2-2 mice that underwent SAH-no treatment was 61.5% ± 1.1%, SAH + vehicle was 76.4 ± 3.4%, and SAH + S-4-CPG was 91.9 ± 3.6% as compared to Hp2-2 Sham animals (Hp1-1 SAH + vehicle vs. SAH + S-4-CPG P value = 0.002, Hp2-2 SAH + vehicle vs. SAH + S-4-CPG P value = 0.015, See Table 2). Hp2-2 animals in the SAH-no treatment group had more pronounced vasospasm than Hp1-1 animals in the group of SAH-no treatment, consistent with our previous results.21 Lumen patency of Hp1-1 animals treated with S-4-CPG after SAH was not different than that of Sham animals (Figure 4B). These results show that S-4-CPG administration after experimental SAH prevents vasospasm in both Hp1-1 and Hp2-2 genotypes.
Table 2.
Percent lumen patency of the basilar artery of Hp1-1 and Hp2-2 animals subjected to the different experimental procedures.
| Groups | P values | ||||
|---|---|---|---|---|---|
| Hp1-1 | Percent lumen patency (%) | Sham | SAH-no treatment | SAH + Vehicle | SAH + S-4-CPG |
| Sham | 100±0.6 | 0.008 | 0.003 | 0.7 | |
| SAH-no treatment | 71.5±2.2 | 0.008 | 0.001 | 0.002 | |
| SAH + Vehicle | 89.3±1.5 | 0.003 | 0.001 | 0.002 | |
| SAH + S-4-CPG | 102.3±2.9 | 0.7 | 0.002 | 0.002 | |
| Groups | P values | ||||
| Hp2-2 | Percent lumen patency(%) | Sham | SAH-no treatment | SAH + Vehicle | SAH + S-4-CPG |
| Sham | 100±1.9 | 0.003 | 0.001 | 0.047 | |
| SAH-no treatment | 61.5±1.2 | 0.003 | 0.0013 | 0.0008 | |
| SAH + Vehicle | 76.4±3.4 | 0.001 | 0.0013 | 0.015 | |
| SAH + S-4-CPG | 91.9±3.6 | 0.047 | 0.0008 | 0.015 | |
Neutrophil/Macrophage infiltration of the basilar artery after SAH
Consistent with previous results we found neutrophils and macrophages in the area of the basilar artery after induction of SAH.21 S-4-CPG administration does not reduce leukocyte infiltration (Figure 5).
Figure 5.
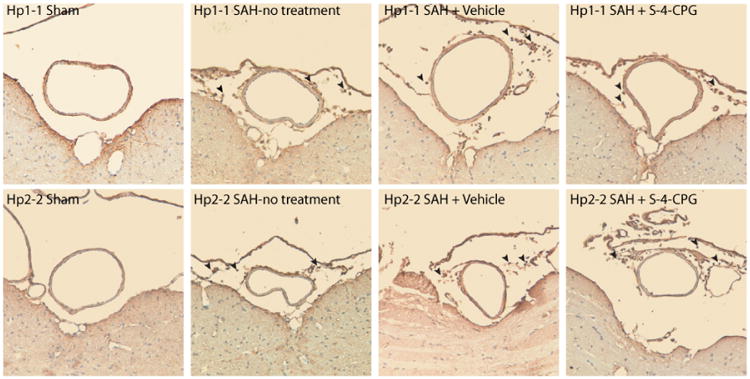
CD45 staining for neutrophils/macrophages around the basilar artery in a murine model of SAH. Representative microphotographs of the staining of the basilar artery in Hp1-1 (top panels) and Hp2-2 mice (bottom panels) subjected to SHAM surgery (left panels), SAH with no treatment (center left panels), SAH + Vehicle (center right panels), and SAH + S-4-CPG (right panels). Arrowheads point to CD45 positive cells in the subarachnoid space and around the basilar artery.
Discussion
In this study we evaluated the role of glutamate in vasospasm after SAH using S-4-CPG as a possible therapeutic agent for vasospasm after experimental SAH. Treatment of Hp2-2 and Hp1-1 mice subjected to experimental SAH with S-4-CPG resulted in a statistically significant increase in lumen patency of the basilar artery as compared to mice treated with vehicle. This was true for Hp1-1 and Hp2-2 mice. The activity and behavior of mice treated with S-4-CPG after SAH showed a trend toward improvement as compared to mice treated with vehicle, but this trend was not statistically significant. Importantly, S-4-CPG is able to cross the BBB, as assessed by HPLC- MS/MS measurement of microdialysis of the extracellular fluid of the brain of rats that received S-4-CPG systemically. These findings suggest that inhibition of metabotropic glutamate receptors may have a beneficial effect after aneurysmal SAH by preventing vasospasm.
Interestingly, we also saw a statistically significant increase in lumen patency in animals treated with the vehicle after SAH as compared to animals that only received SAH. We hypothesize that this effect is secondary to the expansion of the intravascular volume of the animal after administration of the vehicle. This may be similar to the hypervolemic-hypertensive therapy used in patients after aneurysmal SAH.59
Although the mortality of aneurysmal SAH has decreased considerably due to the changes in management of these patients in the last 25 years, there has been no change in the incidence of vasospasm.60 Ischemic complications due to vasospasm after SAH are still present in 30% of patients with SAH, although two thirds of them will have angiographic evidence of vasospasm.61 Vasospasm is not only a transient process that involves constriction of the muscular layer of intracranial arteries and arterioles; it also entails structural changes in the walls of these blood vessels and changes in the expression of receptors to potent vasoconstrictors such as 5-hydroxytryptamine and endothelin-1.62, 63
Inflammation and immune cell infiltration are important determinants of the development of vasospasm after SAH.22 Overexpression of cell adhesion molecules, such as ICAM-1, occurs after SAH and enhances leukocyte diapedesis.9, 27 After red blood cells spill into the subarachnoid space and release their hemoglobin content, immune cells infiltrate the subarachnoid space to remove Hp-hemoglobin complexes, limiting hemoglobin's toxicity.52 Inhibition of over-expression of cell adhesion molecules with anti-inflammatory agents and a decrease in vasospasm after treatment with antibodies directed against ICAM-1 support the concept that vasospasm is originated by the inflammatory process occurring after SAH.14, 24, 64
During the inflammatory response neutrophils secrete soluble factors including glutamate33, which may contribute to the development of vasospasm after SAH. In stroke, glutamate plays a pivotal role in mediating damage through neuronal and glial excitotoxicity.65, 66 Inhibition of mGluR1 using a specific inhibitor (YM-202074) in a rat stroke model showed to have a dose-dependent neuroprotective effect and decreased the infarct volume.46 Endothelial cells express mGluR1 and mGluR5 and upon binding of glutamate, they activate multiple intracellular signaling pathways, such as phospholipase C, adenylyl cyclase, and microtubule associated protein kinase pathways.67 These pathways may alter the endothelial cytoskeleton and tight junction formation.
Glutamate is the main excitatory neurotransmitter in the mammalian brain.68 Exposure of neurons and astrocytes to high concentrations of glutamate leads to cell death through a complex process called excitotoxicity.69 Glutamate, in combination with depletion of nutrients during ischemia, has been shown to lead to excitotoxic neuronal death.70 Inhibition of glutamate decreases neuronal death and improves outcomes in experimental models of stroke.71, 72 Further, glutamate concentrations increase in animal models of SAH 34-37 as well as in patients with aneurysmal SAH.38 The increase in glutamate concentration correlates with vasoconstriction and decreased cerebral blood flow in experimental SAH.34, 36, 37 These data suggest multiple potential mechanisms by which glutamate may contribute to the pathophysiology of delayed cerebral ischemia after SAH.
Further, it has been shown that glutamate disrupts the function of the endothelial barrier in a mouse model of cerebral hypoxia, in part by decreasing VASP phosphorylation.33 When VASP is phosphorylated it negatively regulates actin polymerization and contact of actin to cell-cell and cell-extracellular matrix interaction sites42 and reduces endothelial barrier permeability by allowing relaxation of the actin cytoskeleton, thus decreasing cell retraction.51 Our findings are in accordance to previous results, which show that glutamate decreases phosphorylation of VASP in human brain endothelial cells.33 In our experiments, inhibition of mGluR 1 and 5 expressed in these endothelial cells with S-4-CPG while they were exposed to glutamate maintains VASP in a phosphorylated state in vitro. In our in vivo experiments, inhibition of mGluR1 and mGluR5 after experimental SAH in Hp2-2 and Hp1-1 mice significantly improved lumen patency of the basilar artery. These data suggest that inhibition of mGluR1 and mGluR5 after SAH improves vasospasm possibly through maintaining the phosphorylation levels of VASP, thus preserving the endothelial barrier function. VASP is also expressed in a variety of tissues, including vascular smooth muscle cells.49 The phosphorylation state of VASP in vascular smooth muscle cells of coronary and pulmonary arteries is important in the regulation of vascular tone.73, 74 This has also been shown in cultured vascular smooth muscle cells.75 In these articles, the authors show that stimuli that evoke vascular smooth muscle cell contraction also correlate with decreased phosphorylation of VASP. This is in accordance with our findings, where administration of S-4-CPG to animals after SAH leads to prevention of vasospasm of the basilar artery.
In addition to its effects on the endothelium in arteries and arterioles, glutamate also regulates cerebral blood flow at the capillary level through its action on mGluR1 and mGluR5.76 Based on their results, Rancillac and collaborators propose a mechanism where activation of mGluR1 and mGluR5 expressed in glial cells and Purkinje cells are activated by extracellular glutamate, promoting the secretion of thromboxane A2 and endothelin-1 by these cells, respectively, leading to microvessel constriction.76 Other work has also shown that application of a metabotropic glutamate receptor agonist results in astrocyte-mediated secretion of 20-hydroxyeicosatetraenoic acid, which evoked microvessel constriction in hippocampal slices.77 This broadens the possible targets of metabotropic glutamate receptor antagonists, such as S-4-CPG, from the receptors present in endothelial cells to the ones present in astrocytes, both of which may regulate vessel patency.
In summary, we believe that inflammatory cells that infiltrate the perivascular space are capable of secreting multiple humoral factors, many of which have been shown to contribute to the development of vasospasm and delayed cerebral ischemia. Glutamate is included in these factors and inhibition of the activation of mGluR 1 and 5 receptors with S-4-CPG may maintain the phosphorylation status of VASP in endothelial cells and in vascular smooth muscle cells, thereby preventing vasospasm. Taken together, these results suggest an important role of glutamate in vasospasm and support the potential use of S-4-CPG for the treatment of vasospasm after SAH.
Acknowledgments
Disclosure of funding: This work was supported in part by the Analytical Pharmacology Core of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins (NIH grants P30 CA006973) and the Shared Instrument Grant (1S10RR026824-01). This publication was also made possible by Grant Number UL1 RR 025005 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
Footnotes
Conflicts of interest: The authors do not have any personal or institutional financial interest in drugs, materials, or devices described in this submission.
References
- 1.Powers WJ, Grubb RL, Jr, Baker RP, Mintun MA, Raichle ME. Regional cerebral blood flow and metabolism in reversible ischemia due to vasospasm. Determination by positron emission tomography. J Neurosurg. 1985 Apr;62(4):539–546. doi: 10.3171/jns.1985.62.4.0539. [DOI] [PubMed] [Google Scholar]
- 2.Davis SM, Andrews JT, Lichtenstein M, Rossiter SC, Kaye AH, Hopper J. Correlations between cerebral arterial velocities, blood flow, and delayed ischemia after subarachnoid hemorrhage. Stroke. 1992 Apr;23(4):492–497. doi: 10.1161/01.str.23.4.492. [DOI] [PubMed] [Google Scholar]
- 3.Yundt KD, Grubb RL, Jr, Diringer MN, Powers WJ. Autoregulatory vasodilation of parenchymal vessels is impaired during cerebral vasospasm. J Cereb Blood Flow Metab. 1998 Apr;18(4):419–424. doi: 10.1097/00004647-199804000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Pluta RM, Hansen-Schwartz J, Dreier J, et al. Cerebral vasospasm following subarachnoid hemorrhage: time for a new world of thought. Neurol Res. 2009 Mar;31(2):151–158. doi: 10.1179/174313209X393564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J Neurosurg. 1978 Feb;48(2):173–178. doi: 10.3171/jns.1978.48.2.0173. [DOI] [PubMed] [Google Scholar]
- 6.Suarez JI, Tarr RW, Selman WR. Aneurysmal subarachnoid hemorrhage. N Engl J Med. 2006 Jan 26;354(4):387–396. doi: 10.1056/NEJMra052732. [DOI] [PubMed] [Google Scholar]
- 7.Pasqualin A. Epidemiology and pathophysiology of cerebral vasospasm following subarachnoid hemorrhage. J Neurosurg Sci. 1998 Mar;42(1 Suppl 1):15–21. [PubMed] [Google Scholar]
- 8.Haley EC, Jr, Kassell NF, Torner JC, Truskowski LL, Germanson TP. A randomized trial of two doses of nicardipine in aneurysmal subarachnoid hemorrhage. A report of the Cooperative Aneurysm Study. J Neurosurg. 1994 May;80(5):788–796. doi: 10.3171/jns.1994.80.5.0788. [DOI] [PubMed] [Google Scholar]
- 9.Sills AK, Jr, Clatterbuck RE, Thompson RC, Cohen PL, Tamargo RJ. Endothelial cell expression of intercellular adhesion molecule 1 in experimental posthemorrhagic vasospasm. Neurosurgery. 1997 Aug;41(2):453–460. doi: 10.1097/00006123-199708000-00025. discussion 460-451. [DOI] [PubMed] [Google Scholar]
- 10.Thai QA, Oshiro EM, Tamargo RJ. Inhibition of experimental vasospasm in rats with the periadventitial administration of ibuprofen using controlled-release polymers. Stroke. 1999 Jan;30(1):140–147. doi: 10.1161/01.str.30.1.140. [DOI] [PubMed] [Google Scholar]
- 11.Clatterbuck RE, Oshiro EM, Hoffman PA, Dietsch GN, Pardoll DM, Tamargo RJ. Inhibition of vasospasm with lymphocyte function-associated antigen-1 monoclonal antibody in a femoral artery model in rats. J Neurosurg. 2002 Sep;97(3):676–682. doi: 10.3171/jns.2002.97.3.0676. [DOI] [PubMed] [Google Scholar]
- 12.Clatterbuck RE, Gailloud P, Ogata L, et al. Prevention of cerebral vasospasm by a humanized anti-CD11/CD18 monoclonal antibody administered after experimental subarachnoid hemorrhage in nonhuman primates. J Neurosurg. 2003 Aug;99(2):376–382. doi: 10.3171/jns.2003.99.2.0376. [DOI] [PubMed] [Google Scholar]
- 13.Frazier JL, Pradilla G, Wang PP, Tamargo RJ. Inhibition of cerebral vasospasm by intracranial delivery of ibuprofen from a controlled-release polymer in a rabbit model of subarachnoid hemorrhage. J Neurosurg. 2004 Jul;101(1):93–98. doi: 10.3171/jns.2004.101.1.0093. [DOI] [PubMed] [Google Scholar]
- 14.Pradilla G, Thai QA, Legnani FG, et al. Local delivery of ibuprofen via controlled-release polymers prevents angiographic vasospasm in a monkey model of subarachnoid hemorrhage. Neurosurgery. 2005 Jul;57(1 Suppl):184–190. doi: 10.1227/01.neu.0000163604.52273.28. discussion 184-190. [DOI] [PubMed] [Google Scholar]
- 15.Recinos PF, Pradilla G, Thai QA, Perez M, Hdeib AM, Tamargo RJ. Controlled release of lipopolysaccharide in the subarachnoid space of rabbits induces chronic vasospasm in the absence of blood. Surg Neurol. 2006 Nov;66(5):463–469. doi: 10.1016/j.surneu.2006.04.010. discussion 469. [DOI] [PubMed] [Google Scholar]
- 16.Pradilla G, Chaichana KL, Hoang S, Huang J, Tamargo RJ. Inflammation and cerebral vasospasm after subarachnoid hemorrhage. Neurosurg Clin N Am. 2010 Apr;21(2):365–379. doi: 10.1016/j.nec.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Provencio JJ, Fu X, Siu A, Rasmussen PA, Hazen SL, Ransohoff RM. CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit Care. 2010 Apr;12(2):244–251. doi: 10.1007/s12028-009-9308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mocco J, Mack WJ, Kim GH, et al. Rise in serum soluble intercellular adhesion molecule-1 levels with vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2002 Sep;97(3):537–541. doi: 10.3171/jns.2002.97.3.0537. [DOI] [PubMed] [Google Scholar]
- 19.Tam AK, Ilodigwe D, Mocco J, et al. Impact of systemic inflammatory response syndrome on vasospasm, cerebral infarction, and outcome after subarachnoid hemorrhage: exploratory analysis of CONSCIOUS-1 database. Neurocrit Care. 2010 Oct;13(2):182–189. doi: 10.1007/s12028-010-9402-x. [DOI] [PubMed] [Google Scholar]
- 20.Dumont AS, Dumont RJ, Chow MM, et al. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery. 2003 Jul;53(1):123–133. doi: 10.1227/01.neu.0000068863.37133.9e. discussion 133-125. [DOI] [PubMed] [Google Scholar]
- 21.Chaichana KL, Levy AP, Miller-Lotan R, Shakur S, Tamargo RJ. Haptoglobin 2-2 genotype determines chronic vasospasm after experimental subarachnoid hemorrhage. Stroke. 2007 Dec;38(12):3266–3271. doi: 10.1161/STROKEAHA.107.490003. [DOI] [PubMed] [Google Scholar]
- 22.Chaichana KL, Pradilla G, Huang J, Tamargo RJ. Role of inflammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg. 2010 Jan;73(1):22–41. doi: 10.1016/j.surneu.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 23.Gallia GL, Tamargo RJ. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol Res. 2006 Oct;28(7):750–758. doi: 10.1179/016164106X152025. [DOI] [PubMed] [Google Scholar]
- 24.Oshiro EM, Hoffman PA, Dietsch GN, Watts MC, Pardoll DM, Tamargo RJ. Inhibition of experimental vasospasm with anti-intercellular adhesion molecule-1 monoclonal antibody in rats. Stroke. 1997 Oct;28(10):2031–2037. doi: 10.1161/01.str.28.10.2031. discussion 2037-2038. [DOI] [PubMed] [Google Scholar]
- 25.Lin CL, Kwan AL, Dumont AS, et al. Attenuation of experimental subarachnoid hemorrhage-induced increases in circulating intercellular adhesion molecule-1 and cerebral vasospasm by the endothelin-converting enzyme inhibitor CGS 26303. J Neurosurg. 2007 Mar;106(3):442–448. doi: 10.3171/jns.2007.106.3.442. [DOI] [PubMed] [Google Scholar]
- 26.Rothoerl RD, Schebesch KM, Kubitza M, Woertgen C, Brawanski A, Pina AL. ICAM-1 and VCAM-1 expression following aneurysmal subarachnoid hemorrhage and their possible role in the pathophysiology of subsequent ischemic deficits. Cerebrovasc Dis. 2006;22(2-3):143–149. doi: 10.1159/000093243. [DOI] [PubMed] [Google Scholar]
- 27.Aihara Y, Kasuya H, Onda H, Hori T, Takeda J. Quantitative analysis of gene expressions related to inflammation in canine spastic artery after subarachnoid hemorrhage. Stroke. 2001 Jan;32(1):212–217. doi: 10.1161/01.str.32.1.212. [DOI] [PubMed] [Google Scholar]
- 28.Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5′-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med. 1998 Oct 19;188(8):1433–1443. doi: 10.1084/jem.188.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fassbender K, Hodapp B, Rossol S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry. 2001 Apr;70(4):534–537. doi: 10.1136/jnnp.70.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaetani P, Tartara F, Pignatti P, Tancioni F, Rodriguez y Baena R, De Benedetti F. Cisternal CSF levels of cytokines after subarachnoid hemorrhage. Neurol Res. 1998 Jun;20(4):337–342. doi: 10.1080/01616412.1998.11740528. [DOI] [PubMed] [Google Scholar]
- 31.Hendryk S, Jarzab B, Josko J. Increase of the IL-1 beta and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Neuro Endocrinol Lett. 2004 Feb-Apr;25(1-2):141–147. [PubMed] [Google Scholar]
- 32.Schoch B, Regel JP, Wichert M, Gasser T, Volbracht L, Stolke D. Analysis of intrathecal interleukin-6 as a potential predictive factor for vasospasm in subarachnoid hemorrhage. Neurosurgery. 2007 May;60(5):828–836. doi: 10.1227/01.NEU.0000255440.21495.80. discussion 828-836. [DOI] [PubMed] [Google Scholar]
- 33.Collard CD, Park KA, Montalto MC, et al. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem. 2002 Apr 26;277(17):14801–14811. doi: 10.1074/jbc.M110557200. [DOI] [PubMed] [Google Scholar]
- 34.Westermaier T, Jauss A, Eriskat J, Kunze E, Roosen K. The temporal profile of cerebral blood flow and tissue metabolites indicates sustained metabolic depression after experimental subarachnoid hemorrhage in rats. Neurosurgery. 2011 Jan;68(1):223–229. doi: 10.1227/NEU.0b013e3181fe23c1. discussion 229-230. [DOI] [PubMed] [Google Scholar]
- 35.Merritt JE, Williams PB. Vasospasm contributes to monosodium glutamate-induced headache. Headache. 1990 Sep;30(9):575–580. doi: 10.1111/j.1526-4610.1990.hed3009575.x. [DOI] [PubMed] [Google Scholar]
- 36.Bederson JB, Levy AL, Ding WH, et al. Acute vasoconstriction after subarachnoid hemorrhage. Neurosurgery. 1998 Feb;42(2):352–360. doi: 10.1097/00006123-199802000-00091. discussion 360-352. [DOI] [PubMed] [Google Scholar]
- 37.Wu CT, Wen LL, Wong CS, et al. Temporal changes in glutamate, glutamate transporters, basilar arteries wall thickness, and neuronal variability in an experimental rat model of subarachnoid hemorrhage. Anesth Analg. 2011 Mar;112(3):666–673. doi: 10.1213/ANE.0b013e318207c51f. [DOI] [PubMed] [Google Scholar]
- 38.Saveland H, Nilsson OG, Boris-Moller F, Wieloch T, Brandt L. Intracerebral microdialysis of glutamate and aspartate in two vascular territories after aneurysmal subarachnoid hemorrhage. Neurosurgery. 1996 Jan;38(1):12–19. doi: 10.1007/BF01921273. discussion 19-20. [DOI] [PubMed] [Google Scholar]
- 39.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 40.Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994 Nov;13(5):1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 41.Brabet I, Mary S, Bockaert J, Pin JP. Phenylglycine derivatives discriminate between mGluR1- and mGluR5-mediated responses. Neuropharmacology. 1995 Aug;34(8):895–903. doi: 10.1016/0028-3908(95)00079-l. [DOI] [PubMed] [Google Scholar]
- 42.Harbeck B, Huttelmaier S, Schluter K, Jockusch BM, Illenberger S. Phosphorylation of the vasodilator-stimulated phosphoprotein regulates its interaction with actin. J Biol Chem. 2000 Oct 6;275(40):30817–30825. doi: 10.1074/jbc.M005066200. [DOI] [PubMed] [Google Scholar]
- 43.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000 Apr;20(2):131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Vry J, Horvath E, Schreiber R. Neuroprotective and behavioral effects of the selective metabotropic glutamate mGlu(1) receptor antagonist BAY 36-7620. Eur J Pharmacol. 2001 Oct 5;428(2):203–214. doi: 10.1016/s0014-2999(01)01296-1. [DOI] [PubMed] [Google Scholar]
- 45.Szydlowska K, Kaminska B, Baude A, Parsons CG, Danysz W. Neuroprotective activity of selective mGlu1 and mGlu5 antagonists in vitro and in vivo. Eur J Pharmacol. 2007 Jan 5;554(1):18–29. doi: 10.1016/j.ejphar.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 46.Kohara A, Takahashi M, Yatsugi S, et al. Neuroprotective effects of the selective type 1 metabotropic glutamate receptor antagonist YM-202074 in rat stroke models. Brain Res. 2008 Jan 29;1191:168–179. doi: 10.1016/j.brainres.2007.11.035. [DOI] [PubMed] [Google Scholar]
- 47.Murotomi K, Takagi N, Takayanagi G, Ono M, Takeo S, Tanonaka K. mGluR1 antagonist decreases tyrosine phosphorylation of NMDA receptor and attenuates infarct size after transient focal cerebral ischemia. J Neurochem. 2008 Jun;105(5):1625–1634. doi: 10.1111/j.1471-4159.2008.05260.x. [DOI] [PubMed] [Google Scholar]
- 48.Pula G, Krause M. Role of Ena/VASP proteins in homeostasis and disease. Handb Exp Pharmacol. 2008;(186):39–65. doi: 10.1007/978-3-540-72843-6_3. [DOI] [PubMed] [Google Scholar]
- 49.Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch BM, Walter U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 1992 Jun;11(6):2063–2070. doi: 10.1002/j.1460-2075.1992.tb05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sporbert A, Mertsch K, Smolenski A, et al. Phosphorylation of vasodilator-stimulated phosphoprotein: a consequence of nitric oxide- and cGMP-mediated signal transduction in brain capillary endothelial cells and astrocytes. Brain Res Mol Brain Res. 1999 Apr 20;67(2):258–266. doi: 10.1016/s0169-328x(99)00067-4. [DOI] [PubMed] [Google Scholar]
- 51.Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J. 2002 Apr;16(6):583–585. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 52.Ascenzi P, Bocedi A, Visca P, et al. Hemoglobin and heme scavenging. IUBMB Life. 2005 Nov;57(11):749–759. doi: 10.1080/15216540500380871. [DOI] [PubMed] [Google Scholar]
- 53.Borsody M, Burke A, Coplin W, Miller-Lotan R, Levy A. Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology. 2006 Mar 14;66(5):634–640. doi: 10.1212/01.wnl.0000200781.62172.1d. [DOI] [PubMed] [Google Scholar]
- 54.Le Quellec A, Dupin S, Genissel P, Saivin S, Marchand B, Houin G. Microdialysis probes calibration: gradient and tissue dependent changes in no net flux and reverse dialysis methods. J Pharmacol Toxicol Methods. 1995 Feb;33(1):11–16. doi: 10.1016/1056-8719(94)00049-a. [DOI] [PubMed] [Google Scholar]
- 55.Bungay PM, Morrison PF, Dedrick RL. Steady-state theory for quantitative microdialysis of solutes and water in vivo and in vitro. Life Sci. 1990;46(2):105–119. doi: 10.1016/0024-3205(90)90043-q. [DOI] [PubMed] [Google Scholar]
- 56.Froehler MT, Kooshkabadi A, Miller-Lotan R, et al. Vasospasm after subarachnoid hemorrhage in haptoglobin 2-2 mice can be prevented with a glutathione peroxidase mimetic. J Clin Neurosci. 2010 Sep;17(9):1169–1172. doi: 10.1016/j.jocn.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 57.Momin EN, Schwab KE, Chaichana KL, Miller-Lotan R, Levy AP, Tamargo RJ. Controlled delivery of nitric oxide inhibits leukocyte migration and prevents vasospasm in haptoglobin 2-2 mice after subarachnoid hemorrhage. Neurosurgery. 2009 Nov;65(5):937–945. doi: 10.1227/01.NEU.0000356974.14230.B8. discussion 945. [DOI] [PubMed] [Google Scholar]
- 58.Lin CL, Calisaneller T, Ukita N, Dumont AS, Kassell NF, Lee KS. A murine model of subarachnoid hemorrhage-induced cerebral vasospasm. J Neurosci Methods. 2003 Feb 15;123(1):89–97. doi: 10.1016/s0165-0270(02)00344-8. [DOI] [PubMed] [Google Scholar]
- 59.Diringer MN. Management of aneurysmal subarachnoid hemorrhage. Crit Care Med. 2009 Feb;37(2):432–440. doi: 10.1097/CCM.0b013e318195865a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lovelock CE, Rinkel GJ, Rothwell PM. Time trends in outcome of subarachnoid hemorrhage: Population-based study and systematic review. Neurology. 2010 May 11;74(19):1494–1501. doi: 10.1212/WNL.0b013e3181dd42b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dorsch NW. Cerebral arterial spasm--a clinical review. Br J Neurosurg. 1995;9(3):403–412. doi: 10.1080/02688699550041403. [DOI] [PubMed] [Google Scholar]
- 62.Vollrath BA, Weir BK, Macdonald RL, Cook DA. Intracellular mechanisms involved in the responses of cerebrovascular smooth-muscle cells to hemoglobin. J Neurosurg. 1994 Feb;80(2):261–268. doi: 10.3171/jns.1994.80.2.0261. [DOI] [PubMed] [Google Scholar]
- 63.Hansen-Schwartz J, Ansar S, Edvinsson L. Cerebral vasoconstriction after subarachnoid hemorrhage--role of changes in vascular receptor phenotype. Front Biosci. 2008;13:2160–2164. doi: 10.2741/2831. [DOI] [PubMed] [Google Scholar]
- 64.Kapiotis S, Sengoelge G, Sperr WR, et al. Ibuprofen inhibits pyrogen-dependent expression of VCAM-1 and ICAM-1 on human endothelial cells. Life Sci. 1996;58(23):2167–2181. doi: 10.1016/0024-3205(96)00210-x. [DOI] [PubMed] [Google Scholar]
- 65.Hazell AS. Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem Int. 2007 Jun;50(7-8):941–953. doi: 10.1016/j.neuint.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 66.Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006 Jan 15;53(2):212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- 67.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 69.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008 Nov;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999 Sep;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 71.McCulloch J. Excitatory amino acid antagonists and their potential for the treatment of ischaemic brain damage in man. Br J Clin Pharmacol. 1992 Aug;34(2):106–114. doi: 10.1111/j.1365-2125.1992.tb04118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schurr A. Neuroprotection against ischemic/hypoxic brain damage: blockers of ionotropic glutamate receptor and voltage sensitive calcium channels. Curr Drug Targets. 2004 Oct;5(7):603–618. doi: 10.2174/1389450043345209. [DOI] [PubMed] [Google Scholar]
- 73.Chettimada S, Rawat DK, Dey N, et al. Glc-6-PD and PKG contribute to hypoxia-induced decrease in smooth muscle cell contractile phenotype proteins in pulmonary artery. Am J Physiol Lung Cell Mol Physiol. 2012 Jul 1;303(1):L64–74. doi: 10.1152/ajplung.00002.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ata H, Rawat DK, Lincoln T, Gupte SA. Mechanism of glucose-6-phosphate dehydrogenase-mediated regulation of coronary artery contractility. Am J Physiol Heart Circ Physiol. 2011 Jun;300(6):H2054–2063. doi: 10.1152/ajpheart.01155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim HR, Graceffa P, Ferron F, et al. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol. 2010 Mar;298(3):C559–571. doi: 10.1152/ajpcell.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rancillac A, Rossier J, Guille M, et al. Glutamatergic Control of Microvascular Tone by Distinct GABA Neurons in the Cerebellum. J Neurosci. 2006 Jun 28;26(26):6997–7006. doi: 10.1523/JNEUROSCI.5515-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004 Sep 9;431(7005):195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]