

Abstract
The total synthesis of (−)-kopsinine and its unnatural enantiomer is detailed, enlisting a late-stage SmI2-mediated transannular free radical conjugate addition reaction for construction of the core bicyclo[2.2.2]octane ring system with strategic C21–C2 bond formation. Key to the approach is assemblage of the underlying skeleton by an intramolecular [4+2]/[3+2] cycloaddition cascade of a 1,3,4-oxadiazole that provided the precursor C21 functionalized pentacyclic ring system 1 in a single step in which the C3 methyl ester found in the natural product served as a key 1,3,4-oxadiazole substituent, activating it for participation in the initiating Diels–Alder reaction and stabilizing the intermediate 1,3-dipole.
1. Introduction
In efforts that have targeted key members of the Aspidosperma alkaloids, including minovine,1 (−)-aspidospermine and (+)-spegazzinine,2 (+)-N-methylaspidospermidine, (−)-vindorosine and (−)-vindoline,3 as well as their extension to the total synthesis of vinblastine4 and related natural products including vincristine5 and key analogues,6 we introduced a powerful intramolecular tandem [4 + 2]/[3 + 2] cycloaddition cascade of 1,3,4-oxadiazoles that provides the stereochemically-rich pentacyclic core of the natural products in a single step.7,8 We have subsequently disclosed the use of the common Aspidosperma-like pentacyclic intermediate 1, assembled using this key cycloaddition cascade and bearing a functionalized C5 ethyl substituent (primary alcohol), in the divergent9 total synthesis of a series of additional alkaloids. This was accomplished by direct linkage of the C21 primary alcohol oxygen to C19 (4, (+)-fendleridine)10 and C6 (3, (−)-deoxoapodine)11 or through linkage of C21 itself to C2 (5, kopsinine)12 and C3 (2, (−)-kopsifoline D)11 using the C21 functionality to conduct nucleophilic or electrophilic C–C bond forming reactions (Fig. 1). Inherent in the approach, the C3 methyl ester in the natural products served as a key 1,3,4-oxadiazole substituent, activating it for participation in the initiating Diels–Alder reaction and stabilizing the intermediate 1,3-dipole in the cycloaddition cascade. The combined efforts represented the divergent total syntheses of members of four unique classes of natural products from a common intermediate deliberately functionalized for late-stage formation of four different key strategic bonds13 embedded in each unique core structure.
Fig. 1.

Divergent total synthesis of four alkaloid families from a common cascade cycloaddition product.
In these studies and by virtue of conversion of the C5 ethyl group primary alcohol to a methyl dithiocarbonate, intermediate 1 was enlisted in the total synthesis of racemic kopsinine (5),12 featuring a diastereoselective SmI2-mediated free radical transannular conjugate addition reaction for formation of the bicyclo[2.2.2]octane core. This late stage C21–C2 bond formation not only complemented prior Diels–Alder approaches to its bicyclo[2.2.2]octane core,14 but it represented the first synthetic approach that directly provided kopsinine from the underlying pentacyclic Aspidosperma alkaloid skeleton (Fig. 1). However, our original approach suffered from a low conversion in the transformation of 1 to the key conjugate addition substrate and, unlike the efforts leading to 2–4, was conducted with racemic material. Herein, we report the extension of these studies to the total synthesis of (−)-kopsinine (5), a Kopsia alkaloid first isolated from Kopsia Longiflora Merr.,15 as well as its unnatural enantiomer ent-(+)-kopsinine that addresses these limitations of our initially reported approach.
2. Results and discussion
As detailed in preceding studies,10,11 acid-catalyzed reductive cleavage of the oxido bridge in 1 upon treatment with NaCNBH3 (20% HOAc/i-PrOH) provided the alcohol 6 (88%) as a single diastereomer, arising from hydride reduction of an intermediate N-acyliminium ion exclusively from the less hindered convex face (Scheme 1). In efforts that provided improvements in a subsequent Chugaev elimination,16 6 was first converted to the corresponding Cbz (N-carboxybenzyl) carbamate 8 via the free indoline 7.
Scheme 1.

Chromatographic separation of the enantiomers of 8 (α = 1.8, 20% i-PrOH/hexanes) was carried out on a semi-preparative Daicel ChiralCel OD column, providing (+)-8 and ent-(−)-8. It was the remarkable ease and scale (100 mg/injection) on which this chiral phase chromatographic separation of the enantiomers of 8 could be carried out even on a semipreparative column (Fig. 2) that precipitated our efforts to revisit and improve our reported approach to kopsinine, providing a synthesis of optically active material.
Fig. 2.

Top: Chromatographic separation (α = 1.8) of the enantiomers of 8 on a ChiralCel OD semipreparative column (2 × 20 cm, 20% i-PrOH/hexanes, 7 mL/min).
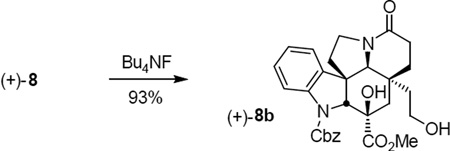
Determination of the structure and absolute stereochemistry of the natural enantiomer (+)-8 were established by a single-crystal X-ray structure determination conducted on crystals of the corresponding primary alcohol (+)-8b derived from 8 (eq 1).
 |
(1) |
Conversion of the natural enantiomer (+)-8 to the methyl dithiocarbonate (+)-9 (NaH, CS2, THF, 0 °C, 1 h followed by MeI, 25 °C, 1 h, 93%) set the stage for a Chugaev elimination (Scheme 1).16 Xanthate (+)-9 underwent clean elimination under mild thermal conditions (toluene, 100 °C, bath, 48 h or 150 °C bath, 36 h), affording good yields of (−)-10 (60%). The Cbz carbamate (vs N-benzyl) presumably activates C2-H for xanthate syn elimination, favoring formation of the more substituted and stable olefin, improving an important element of our prior reported synthesis of kopsinine where the elimination regioselectivity of the corresponding xanthate derived from 6 was less favorable.12
Silyl ether cleavage in (−)-10 (3 equiv Bu4NF, THF, 25 °C, 1 h, 98%) and subsequent conversion of the primary alcohol (−)-11 to the primary iodide (−)-12 (Et3N, CH3SO2Cl, THF, −78 °C, 1 h, then NaI, acetone, 50 °C bath temperature, 12 h, 81%) set the stage for the pivotal transannular cyclization (Scheme 1). Treatment of (−)-12 with SmI2 (Aldrich) in 10:1 THF–HMPA (0 °C, 1 h) smoothly proceeded to provide (−)-13 in excellent yield (85%) with only a trace amount of the C3 diastereomer (<5%). This generation of essentially a single diastereomer (>17:1) presumably results from a radical-mediated transannular cyclization followed by kinetic protonation of the further reduced conjugate addition ester enolate from the less hindered convex face.17 Although employed in our original synthesis with an analogous substrate bearing a N-benzyl group,12 the alternative conversion of 12 to the corresponding methyl dithiocarbonate and subsequent SmI2-promoted ring closure for formation of the bicyclo[2.2.2]octane 13 proved less productive (45%), albeit without optimization. Reductive removal of the lactam carbonyl of (−)-13 upon treatment with BH3·THF (THF, 0 °C, 1 h) provided (−)-14 and subsequent cleavage of the Cbz group (H2, 10% Pd/C, EtOAc/MeOH, 25 °C, 30 min, 82% over two steps) afforded natural (−)-kopsinine (5, [α]D −56 (c 0.15, CHCl3) vs [α]D −76.9 (c 2.09, CHCl3)15a and [α]D −69 (c 0.856, CHCl3)15c), which otherwise proved identical in all respects with reported properties of the natural product. We are unclear about the origin of the discrepancy in our optical rotation for (−)-kopsinine relative to that recorded for the natural product15 as well as those reported for additional synthetic samples ([α]D −43.1 (c 1.8, CHCl3)14d and [α]D −65.8 (c 1.13, CHCl3)14e) although we can conclusively say that it is not derived for either the enantiopurity (>99%, Fig. 3) or chemical purity (Supporting Information) of our synthetic sample. By enlisting ent-(−)-8, the unnatural enantiomer of kopsinine (ent-(+)-5) was similarly prepared ([α]D +57 (c 0.84, CHCl3).
Fig. 3.

Chiral phase assessment of the enantiopurity of the individual enantiomers of kopsinine (5) on an analytical Chiralpak IC column (0.46 × 20 cm, 5% i-PrOH/hexanes + 0.4% Et2NH, 1 mL/min, α = 1.7).
We do note that simple chromatographic purification (SiO2) or preparative thin-layer chromatography purification of 5 even in the presence of Et3N were found to be problematic, requiring chromatographic purification of 5 on basic alumina (Al2O3) in our hands. We have also found that the quality of the natural product spectroscopic properties appears to be sensitive to potential acidic contaminants in some conventional NMR solvents (e.g., CDCl3), requiring their passage through basic alumina prior to use. Perhaps this has also contributed to the minor discrepancies in the recorded optical rotations of 5 as well.
3. Conclusions
The total synthesis of (−)-kopsinine and its unnatural enantiomer are detailed from the common intermediate 1 functionalized for late-stage formation of four different key strategic bonds embedded in four different natural product core structures. For kopsinine, this entailed development of a remarkably effective SmI2-mediated transannular free radical conjugate addition reaction for formation of the bicyclo[2.2.2]octane core central to its hexacyclic ring system with C21–C2 bond formation. The basis of the approach and central to the assemblage of the underlying skeleton of 1 is a powerful intramolecular [4 + 2]/[3 + 2] cycloaddition cascade of a 1,3,4-oxadiazole that provided the C21 functionalized pentacyclic ring system in a single step in which the C3 methyl ester found in the natural product served as a key substituent, activating the 1,3,4-oxadiazole for participation in the initiating Diels–Alder reaction and stabilizing the intermediate 1,3-dipole. Continued examination of the applications of such cycloaddition cascades in the total synthesis of natural products are in progress and will be disclosed in due course.
4. Experimental
4.1. Compound 6
A solution of 110 (953 mg, 1.62 mmol) in i-PrOH and acetic acid (16 mL/4 mL) was treated with NaCNBH3 (814 mg, 12.95 mmol, 8 equiv). The mixture was allowed to stir at 25 °C for 16 h before it was cooled to 0 °C and quenched with the addition of saturated aqueous NaHCO3. The mixture was diluted with EtOAc, the layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, gradient elution: 100% EtOAc to 5% MeOH–EtOAc) to provide 610 (841 mg, 88%) as a white foam: 1H NMR (500 MHz, CDCl3) δ 7.20–7.28 (m, 3H), 7.12–7.17 (m, 2H), 7.10 (t, J = 7.6 Hz, 1H), 6.98 (d, J = 7.3 Hz, 1H), 6.77 (t, J = 7.4 Hz, 1H), 6.58 (d, J = 7.9 Hz, 1H), 4.53 (d, J = 15.9 Hz, 1H), 3.95 (d, J = 15.9 Hz, 1H), 3.79 (s, 1H), 3.53–3.69 (m, 3H), 3.61 (s, 3H), 3.37 (d, J = 1.8 Hz, 1H), 3.29 (td, J = 11.9, 6.7 Hz, 1H), 2.20–2.35 (m, 2H), 2.04 (ddd, J = 13.5, 10.9, 4.9 Hz, 1H), 1.93 (d, J = 14.8 Hz, 1H), 1.81–1.90 (m, 2H), 1.78 (dd, J = 14.9, 1.9 Hz, 1H), 1.48 (dt, J = 13.6, 6.7 Hz, 1H), 1.36 (dd, J = 13.0, 6.6 Hz, 1H), 1.25 (dt, J = 13.3, 6.4 Hz, 1H), 0.83 (s, 9H), −0.04 (s, 3H), −0.05 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 175.9, 171.1, 151.7, 137.5, 132.5, 128.9, 128.3, 128.0, 127.4, 122.9, 119.5, 110.7, 76.5, 73.8, 65.9, 59.0, 54.7, 54.5, 52.6, 45.1, 43.0, 41.7, 35.0, 33.9, 32.0, 30.2, 25.8, 18.1, −5.4, −5.5; IR (film) υmax 3233, 2928, 1731, 1633, 1250, 725 cm−1; HRMS (ESI) m/z 591.3248 [(M+H)+, C34H46N2O5Si requires 591.3249].
4.2. Compound 7
A stirred solution of 6 (251 mg, 0.425 mmol) in EtOH (8 mL, 0.053 M) was treated with excess Raney 2400 Ni (~1 g, pretreated with successive washes with EtOH) at 25 °C. After stirring at 80 °C under H2 for 30 min, the resulting mixture was filtered through a pad of Celite, rinsed with MeOH and concentrated under reduced pressure. The residue was purified by flash chromatography (SiO2, 10% MeOH–CH2Cl2) to provide 7 (196 mg, 92%) as a white solid: 1H NMR (600 MHz, CDCl3) δ 7.04 (t, J = 7.2 Hz, 1H), 7.02 (d, J = 7.2 Hz, 1H), 6.73 (t, J = 7.4 Hz, 1H), 6.54 (d, J = 7.8 Hz, 1H), 4.26 (t, J = 3.4 Hz, 1H), 3.87–3.91 (m, 1H), 3.81 (s, 3H), 3.63 (td, J = 11.4, 7.6 Hz, 1H), 3.57 (ddd, J = 12.1, 9.7, 2.2 Hz, 1H), 3.54 (s, 1H), 3.50 (td, J = 6.6, 2.8 Hz, 1H), 2.37–2.44 (m, 1H), 2.21–2.30 (m, 2H), 2.12 (ddd, J = 13.4, 7.6, 2.1 Hz, 1H), 2.06 (d, J = 15.6 Hz, 1H), 1.76–1.90 (m, 3H), 1.27 (td, J = 6.7, 3.6 Hz, 2H), 0.79 (s, 9H), −0.09 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 174.9, 170.8, 149.2, 132.2, 128.7, 123.0, 119.1, 109.5, 76.7, 68.3, 67.7, 59.0, 54.2, 52.5, 44.6, 43.7, 41.3, 34.4, 33.8, 32.5, 28.9, 25.8, 18.0, −5.4, −5.5; IR (film) υmax 3374, 2929, 1733, 1609, 1237, 669 cm−1; HRMS (ESI) m/z 501.2784 [(M+H)+, C27H40N2O5Si requires 501.2779].
4.3. Compound 8
A solution of 7 (1.18 g, 2.36 mmol) in CH2Cl2 (80 mL, 0.029 M) was treated with K2CO3 (1.63 g, 11.79 mmol) and benzyl chloroformate (0.84 mL, 5.89 mmol). After stirring for 4 h at 25 °C, the resulting mixture was quenched with the addition of saturated aqueous NaHCO3 and diluted with H2O and CH2Cl2 (30 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, 5% MeOH–CH2Cl2) to provide 8 (1.449 g, 97%) as a white foam: 1H NMR (500 MHz, CDCl3, 50 °C) δ 7.64 (bs, 1H), 7.31–7.44 (m, 5H), 7.23 (t, J = 7.9 Hz, 1H), 7.18 (d, J = 7.6 Hz, 1H), 5.36 (d, J = 12.0 Hz, 1H), 5.12 (d, J = 12.1 Hz, 1H), 4.17 (s, 1H), 4.05 (s, 1H), 3.83 (t, J = 10.4 Hz, 1H), 3.49–3.64 (m, 3H), 3.54 (s, 3H), 2.17–2.35 (m, 3H), 2.09 (dd, J = 13.1, 6.4 Hz, 1H), 1.96 (q, J = 11.5 Hz, 1H), 1.81 (d, J = 12.9 Hz, 1H), 1.68 (d, J = 14.7 Hz, 1H), 1.58 (d, J = 14.6 Hz, 1H), 1.52 (q, J = 6.7 Hz, 1H), 1.20–1.27 (m, 1H), 0.82 (s, 9H), −0.05 (s, 3H), −0.06 (s, 3H); 13C NMR (125 MHz, CDCl3, 50 °C) δ 174.8, 170.7, 154.0, 141.6, 135.6, 133.2, 128.9, 128.7, 128.54, 128.50, 123.6, 123.1, 116.1, 75.3, 70.9, 68.1, 64.6, 58.9, 53.4, 53.1, 44.6, 43.1, 40.6, 37.5, 34.4, 31.8, 30.4, 25.8, 18.0, −5.47, −5.51; IR (film) υmax 3294, 2950, 1706, 1636, 1398, 1257, 835, 749 cm−1; HRMS (ESI) m/z 635.3147 [(M+H)+, C35H46N2O7Si requires 635.3143].
The 1H and 13C NMR spectra in CDCl3 at 25 °C exhibits broaden peaks due to Cbz rotamers. The enantiomers of 8 (100 mg/injection) were separated (α = 1.8, Fig. 2) on a semipreparative ChiralCel OD column (2 × 25 cm, 20% i-PrOH–hexanes, 7 mL/min flow rate) providing natural (+)-8 (tR: 32.3 min) and ent-(−)-8 (tR: 58.4 min). For natural enantiomer (+)-8: [α]D20 +32 (c 1.0, CHCl3), unnatural enantiomer (−)-8: [α]D20 −32 (c 1.0, CHCl3).
4.4. Compound (+)-8b
A cooled (0 °C) solution of (+)-8 (38.0 mg, 0.060 mmol) in THF (5 mL, 0.012 M) was treated with Bu4NF (0.18 mL, 1.0 M in THF, 0.18 mmol, 3 equiv). After stirring for 2 h at 25 °C, the resulting mixture was quenched with the addition of saturated aqueous NH4Cl and diluted with EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, 10% MeOH–EtOAc) providing (+)-8b (29.0 mg, 93%) as a white solid: [α]D20 +18.6 (c 0.5, CHCl3); 1H NMR (600 MHz, DMSO-d6) δ 7.52 (br s, 1H), 7.44 (d, J = 7.4 Hz, 2H), 7.36–7.42 (m, 3H), 7.33 (t, J = 7.3 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 7.04 (t, J = 7.4 Hz, 1H), 5.78 (s, 1H), 5.32 (d, J = 12.6 Hz, 1H), 5.06 (br s, 1H), 4.34 (br s, 1H), 4.17 (s, 1H), 4.06 (s, 1H), 3.59–3.66 (m, 1H), 3.47 (s, 3H), 3.30–3.44 (m, 1H), 3.22 (td, J = 9.8, 5.5 Hz, 1H), 2.96–3.04 (m, 1H), 2.15 (td, J = 14.3, 4.1 Hz, 1H), 1.91–2.08 (m, 3H), 1.64–1.71 (m, 1H), 1.57–1.64 (m, 1H), 1.48–1.54 (m, 1H), 1.48 (d, J = 15.2 Hz, 1H), 1.35 (d, J = 14.7 Hz, 1H), 1.31 (q, J = 7.5 Hz, 1H); 13C NMR (150 MHz, DMSO-d6) δ 174.1, 169.5, 153.1, 141.2, 136.1, 133.8, 128.5, 128.4, 128.1, 127.8, 123.9, 123.2, 115.2, 75.1, 69.1, 67.0, 63.6, 56.4, 52.4, 52.1, 51.6, 44.7, 42.6, 33.4, 30.7, 30.6, 24.9, 19.5, 13.6; IR (film) υmax 3336, 2925, 1702, 1630, 1485, 1399, 1261, 748, 697 cm−1; HRMS (ESI) m/z 521.2282 [(M+H)+, C29H32N2O7 requires 521.2282].
The structure and absolute configuration of natural enantiomer (+)-8 were unambiguously established in an X-ray crystallographic assignment of this corresponding primary alcohol (+)-8b (CCDC 977767) conducted with white crystals grown from MeOH.
4.5. Compound (+)-9
A cooled (0 °C) solution of (+)-8 (247 mg, 0.389 mmol) and imidazole (24 mg) in THF (10 mL, 0.039 M) was treated with NaH (78 mg, 60% dispersion in mineral oil, 1.95 mmol, 5 equiv). The mixture was allowed to stir at 25 °C for 30 min before it was cooled to 0 °C followed by the addition of CS2 (70 µL, 1.17 mmol, 3 equiv). The reaction mixture was stirred at 25 °C for 1 h, cooled to 0 °C and then treated with MeI (73 µL, 1.17 mmol, 3 equiv). After stirring for at 25 °C for 1 h, the resulting mixture was quenched with the addition of saturated aqueous NH4Cl and diluted with H2O and EtOAc (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, 50% EtOAc–hexanes) to provide (+)-9 (263 mg, 93%) as a light yellow foam: [α]D20 +12.9 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.57 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 7.4 Hz, 2H), 7.38 (t, J = 7.4 Hz, 2H), 7.34 (t, J = 7.3 Hz, 1H), 7.23–7.26 (m, 1H), 7.17 (d, J = 7.5 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 5.34 (d, J = 12.3 Hz, 1H), 5.14 (d, J = 12.3 Hz, 1H), 4.76 (s, 1H), 4.12 (s, 1H), 3.91–3.98 (m, 1H), 3.55–3.68 (m, 3H), 3.46 (s, 3H), 2.89 (d, J = 15.7 Hz, 1H), 2.50 (s, 3H), 2.26–2.32 (m, 2H), 2.17 (dd, J = 13.1, 6.6 Hz, 1H), 2.01 (dd, J = 12.6, 9.1 Hz, 1H), 1.88 (d, J = 15.8 Hz, 1H), 1.83 (dt, J = 14.1, 4.4 Hz, 1H), 1.55–1.67 (m, 2H), 1.19–1.24 (m, 1H), 0.82 (s, 9H), −0.03 (s, 3H), −0.04 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 213.6, 171.2, 168.2, 154.0, 141.9, 135.5, 132.8, 128.9, 128.5, 128.3, 128.1, 124.3, 122.9, 117.6, 89.1, 68.32, 68.30, 63.8, 58.8, 53.8, 52.0, 44.5, 43.0, 39.5, 34.1, 33.0, 30.5, 29.8, 25.8, 19.5, 18.0, −5.5, −5.6; IR (film) υmax 2926, 1714, 1649, 1260, 1045, 732 cm−1; HRMS (ESI) m/z 725.2745 [(M+H)+, C37H48N2O7Si requires 725.2746].
4.6. Compound (−)-10
A solution of (+)-9 (70.3 mg, 0.097 mmol) in anhydrous toluene (32 mL, 0.003 M) was degassed for 30 min with Ar, then placed in an oil bath at 150 °C. After stirring for 36 h at the same temperature, the resulting mixture was cooled to room temperature and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, gradient elution: 30% EtOAc–hexanes to 60% EtOAc–hexanes) to provide (−)-10 (35.8 mg, 60%) as a white foam: [α]D20 −26.5 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.75 (d, J = 8.1 Hz, 1H), 7.33–7.40 (m, 5H), 7.28 (t, J = 7.7 Hz, 1H), 7.15 (d, J = 7.3 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 5.38 (d, J = 12.2 Hz, 1H), 5.18 (d, J = 12.2 Hz, 1H), 4.10 (dd, J = 11.9, 7.6 Hz, 1H), 3.69 (s, 1H), 3.56 (s, 3H), 3.52 (dt, J = 10.5, 7.2 Hz, 1H), 3.39 (ddd, J = 10.5, 7.6, 5.3 Hz, 1H), 3.31 (td, J = 11.8, 5.9 Hz, 1H), 2.39–2.48 (m, 3H), 2.19–2.25 (m, 2H), 2.02 (td, J = 12.2, 7.8 Hz, 1H), 1.89 (dd, J = 12.5, 5.8 Hz, 1H), 1.56 (ddd, J = 13.9, 9.1, 6.3 Hz, 1H), 1.28–1.40 (m, 2H), 0.80 (s, 9H), −0.07 (s, 3H), −0.09 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 171.0, 167.1, 152.0, 149.2, 140.3, 136.2, 135.3, 128.62, 128.57, 128.5, 128.2, 124.5, 121.3, 116.1, 110.6, 68.4, 66.2, 59.1, 54.7, 51.5, 42.4, 39.3, 38.1, 37.9, 31.6, 31.3, 30.6, 25.9, 18.2, −5.59, −5.64; IR (film) υmax 2925, 1730, 1664, 1238, 751, 670 cm−1; HRMS (ESI) m/z 617.3050 [(M+H)+, C35H44N2O6Si requires 617.3041].
4.7. Compound (−)-11
A cooled (0 °C) solution of (−)-10 (68.1 mg, 0.110 mmol) in THF (6 mL, 0.018 M) was treated with Bu4NF (0.33 mL, 1.0 M in THF, 0.33 mmol, 3 equiv). After stirring for 1 h at 25 °C, the resulting mixture was quenched with the addition of saturated aqueous NH4Cl and diluted with EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, 100% EtOAc) to provide (−)-11 (54.2 mg, 98%) as a colorless oil: [α]D23 −44 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.75 (d, J = 8.1 Hz, 1H), 7.31–7.41 (m, 5H), 7.29 (t, J = 7.9 Hz, 1H), 7.16 (d, J = 7.3 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 5.39 (d, J = 12.2 Hz, 1H), 5.18 (d, J = 12.2 Hz, 1H), 4.11 (dd, J = 12.2, 7.5 Hz, 1H), 3.65 (d, J = 1.8 Hz, 1H), 3.57–3.63 (m, 1H), 3.56 (s, 3H), 3.47–3.53 (m, 1H), 3.30 (td, J = 11.8, 6.0 Hz, 1H), 2.50 (dd, J = 15.2, 2.0 Hz, 1H), 2.43 (dt, J = 16.2, 5.4 Hz, 1H), 2.38 (ddd, J = 16.1, 10.3, 5.4 Hz, 1H), 2.24 (d, J = 15.3 Hz, 1H), 2.10 (dt, J = 13.8, 5.6 Hz, 1H), 2.00–2.07 (m, 1H), 1.91 (dd, J = 12.4, 5.7 Hz, 1H), 1.60–1.64 (m, 1H), 1.43 (dt, J = 14.1, 7.0 Hz, 1H), 1.31 (dq, J = 13.5, 6.8, 6.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 170.6, 167.5, 152.0, 148.7, 136.1, 135.3, 128.6, 128.5, 128.3, 124.5, 121.2, 116.1, 110.9, 68.4, 66.4, 58.4, 54.7, 51.6, 42.4, 39.6, 38.0, 37.9, 31.3, 31.0, 30.3; IR (film) υmax 3389, 2924, 1731, 1649, 670 cm−1; HRMS (ESI) m/z 503.2156 [(M+H)+, C29H30N2O6 requires 503.2177].
4.8. Compound (−)-12
A cooled (−78 °C) solution of (−)-11 (52.3 mg, 0.104 mmol) in THF (4 mL, 0.026 M) was treated with Et3N (44 µL, 0.31 mmol) and methanesulfonyl chloride (12 µL, 0.16 mmol). After stirring for 1 h at the same temperature, sodium iodide (156 mg, 1.04 mmol) and acetone (4 mL) were added and the reaction mixture was then warmed to 50 °C. After stirring for 12 h at the same temperature, the resulting mixture was quenched with the addition of saturated aqueous NaHCO3 and diluted with H2O and hexanes (30 mL). The layers were separated, and the aqueous layer was extracted with hexanes. The combined organic layers were dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, gradient elution: 70–100% EtOAc–hexanes) to provide (−)-12 (50.9 mg, 80%) as a colorless oil: For natural (−)-12: [α]D23 −40 (c 0.51, CHCl3); for ent-(+)-12: [α]D23 +42 (c 1.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 1H), 7.28–7.43 (m, 6H), 7.09–7.16 (m, 2H), 5.39 (d, J = 12.0 Hz, 1H), 5.20 (d, J = 12.0 Hz, 1H), 4.09 (dd, J = 12.0, 7.5 Hz, 1H), 3.58 (s, 3H), 3.49 (s, 1H), 3.32 (ddd, J = 11.5, 11.5, 5.5 Hz, 1H), 3.00 (ddd, J = 13.5, 9.5, 5.0 Hz, 1H), 2.75 (ddd, J = 13.5, 9.5, 5.0 Hz, 1H), 2.50 (d, J = 15.5 Hz, 1H), 2.43 (dt, J = 16.0, 5.0 Hz, 1H), 2.33 (ddd, J = 17.0, 12.0, 5.0 Hz, 1H), 2.12 (d, J = 15.5 Hz, 1H), 1.98–2.07 (m, 2H), 1.90 (dd, J = 12.5, 6.0 Hz, 1H), 1.63–1.79 (m, 2H), 1.48 (ddd, J = 13.0, 13.0, 4.5 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 170.7, 167.0, 152.0, 149.2, 140.2, 135.8, 135.1, 128.8, 128.63, 128.55, 128.4, 124.6, 121.3, 116.2, 109.7, 68.6, 66.2, 54.7, 51.7, 42.4, 41.6, 40.5, 39.0, 30.64, 30.59, 30.1, −3.5; IR (film) υmax 2925, 1730, 1664, 1238, 751, 670 cm−1; HRMS (ESI) m/z 613.1176 [(M+H)+, C29H29IN2O5 requires 613.1194].
4.9. Compound (−)-13
A cooled (0 °C) solution of a mixture of samarium(II) iodide solution (0.1 M in THF from Aldrich, 3 mL, 0.30 mmol, 19 equiv) and HMPA (0.4 mL) in a sealed vessel was treated with a solution of (−)-12 (9.7 mg, 15.8 µmol) in THF (1 mL) under an Ar atmosphere. After stirring for 1 h at the same temperature, the resulting mixture was quenched with the addition of saturated aqueous NH4Cl and diluted with H2O and EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 100% EtOAc) to provide (−)-13 (6.5 mg, 85%) as a white solid: For natural (−)-13: [α]D23 −61 (c 0.58, CHCl3); for ent-(+)-13: [α]D23 +65 (c 1.2, CHCl3); 1H NMR (500 MHz, DMSO-d6, 70 °C) δ 7.73 (br s, 1H), 7.32–7.45 (m, 5H), 7.29 (d, J = 7.3 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 5.25 (d, J = 12.1 Hz, 1H), 5.19 (d, J = 12.3 Hz, 1H), 4.05 (dd, J = 11.7, 7.5 Hz, 1H), 3.73 (s, 1H), 3.57–3.62 (m, 1H), 3.49 (s, 3H), 3.04 (td, J = 12.0, 5.1 Hz, 1H), 2.36 (dt, J = 15.5, 7.2 Hz, 1H), 2.10–2.17 (m, 1H), 1.73–1.98 (m, 5H), 1.68 (dt, J = 15.1, 7.8 Hz, 1H), 1.46–1.54 (m, 2H), 1.43 (dd, J = 13.1, 9.9 Hz, 1H), 1.23 (dd, J = 12.9, 5.1 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, 70 °C) δ 172.0, 167.5, 152.3, 140.7, 136.5, 135.7, 128.0, 127.8, 127.7, 127.5, 122.3, 121.1, 114.5, 67.7, 66.4, 63.4, 56.8, 50.9, 41.7, 41.1, 36.5, 32.3, 30.7, 30.1, 29.6, 28.4, 27.8; IR (film) υmax 2925, 1701, 1637, 1396, 1220, 731, 697 cm−1; HRMS (ESI) m/z 487.2225 [(M+H)+, C29H30N2O5 requires 487.2227].
The 1H and 13C NMR spectra of 13 in CDCl3 and CD3OD at lower temperatures (25–60 °C) led to observation of broaden peaks due to Cbz rotamers.
4.10. (−)-Kopsinine (5)
A cooled (0 °C) solution of (−)-13 (4.5 mg, 9.255 µmol) in THF (1 mL, 0.009 M) was treated with a solution of borane-tetrahydrofuran complex (111 µL, 1.0 M in THF, 111 µmol, 12 equiv). After stirring for 1 h at the same temperature, the resulting mixture was quenched with the addition of H2O and the solution was treated with 10% aqueous HCl (1 mL). After stirring for 30 min at 0 °C, 1 N aqueous NaOH was then added to the mixture until pH ~13 and diluted with EtOAc. The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by preparative thin-layer chromatography (SiO2, 30% EtOAc–hexanes) to provide (−)-14 as a white solid: For natural (−)-14: [α]D23 −93 (c 0.50, CHCl3); for ent-(+)-14: [α]D23 +95 (c 0.52, CHCl3); HRMS (ESI) m/z 473.2437 [(M+H)+, C29H32N2O4 requires 473.2435].
A solution of the above (−)-14 in EtOAc/MeOH (3:1, 1.2 mL) was treated with 10% Pd/C (2 mg) at 25 °C. After stirring for 1 h under a H2 atmosphere, the resulting mixture was filtered through a pad of Celite and washed with 2 N NaOH solution. The organic layer was dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (basic alumina, gradient elution, 100% CH2Cl2 to 10% MeOH–CH2Cl2) to provide (−)-kopsinine (5, 2.6 mg, 82% over two steps) as a white solid identical in all respects with authentic material: For natural (−)-kopsinine 5: [α]D23 −56 (c 0.15, CHCl3); for ent-(+)-kopsinine 5: [α]D23 +57 (c 0.84, CHCl3) vs lit. [α]D23 −76.9 (c 2.09, CHCl3)15a and [α]D23 −69±3 (c 0.856, CHCl3)15c for natural (−)-5; 1H NMR (600 MHz, CDCl3 (passed through a basic alumina)) δ 7.16 (d, J = 7.3 Hz, 1H), 6.98 (t, J = 7.6 Hz, 1H), 6.75 (t, J = 7.4 Hz, 1H), 6.66 (d, J = 7.7 Hz, 1H), 3.76 (s, 3H), 3.34 (q, J = 8.3 Hz, 1H), 3.12 (d, J = 13.5 Hz, 1H), 2.92–3.01 (m, 3H), 2.89 (t, J = 9.6 Hz, 1H), 2.79 (t, J = 11.7 Hz, 1H), 2.64 (t, J = 11.2 Hz, 1H), 1.87–1.96 (m, 2H), 1.52–1.62 (m, 2H), 1.35–1.45 (m, 2H), 1.19–1.34 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 174.8, 149.1, 140.7, 126.6, 121.6, 119.7, 110.8, 68.4, 66.7, 57.9, 52.0, 50.7, 47.6, 43.8, 36.5, 34.8, 33.92, 33.88, 32.1, 31.8, 17.1; IR (film) υmax 2922, 1726, 1458, 1203, 741, 670 cm−1; HRMS (ESI) m/z 339.2067 [(M+H)+, C21H26N2O2 requires 339.2067].
Supplementary Material
Fig. 4.

X-ray crystal structure of the natural enantiomer (+)-8b defining the structure, stereochemistry, and absolute configuration (CCDC 977767).
Acknowledgements
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056). We thank Professor A. Rheingold (UCSD) for the X-ray structure determination ofthe free alcohol (+)-8b.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors maybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Copies of the 1H and 13C NMR of the synthetic intermediates 6–13 and kopsinine (5) are provided.
References and notes
- 1.Yuan ZQ, Ishikawa H, Boger DL. Org. Lett. 2005;7:741. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lajiness JP, Jiang W, Boger DL. Org. Lett. 2012;14:2078. doi: 10.1021/ol300599p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Choi Y, Ishikawa H, Velcicky J, Elliott GI, Miller MM, Boger DL. Org. Lett. 2005;7:4539. doi: 10.1021/ol051975x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Elliott GI, Velcicky J, Ishikawa H, Li Y, Boger DL. Angew. Chem. Int. Ed. 2006;45:620. doi: 10.1002/anie.200503024. [DOI] [PubMed] [Google Scholar]; (c) Ishikawa H, Elliott GI, Velcicky J, Choi Y, Boger DL. J. Am. Chem. Soc. 2006;128:10596. doi: 10.1021/ja061256t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ishikawa H, Boger DL. Hetereocycles. 2007;72:95. [Google Scholar]; (e) Kato D, Sasaki Y, Boger DL. J. Am. Chem. Soc. 2010;132:3685. doi: 10.1021/ja910695e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sasaki Y, Kato D, Boger DL. J. Am. Chem. Soc. 2010;132:13533. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Ishikawa H, Colby DA, Boger DL. J. Am. Chem. Soc. 2008;130:420. doi: 10.1021/ja078192m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gotoh H, Sears JE, Eschenmoser A, Boger DL. J. Am. Chem. Soc. 2012;134:13240. doi: 10.1021/ja306229x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishikawa H, Colby DA, Seto S, Va P, Tam A, Kakei H, Rayl TJ, Hwang I, Boger DL. J. Am. Chem. Soc. 2009;131:4904. doi: 10.1021/ja809842b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Va P, Campbell EL, Robertson WM, Boger DL. J. Am. Chem. Soc. 2010;132:8489. doi: 10.1021/ja1027748. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tam A, Gotoh H, Robertson WM, Boger DL. Bioorg. Med. Chem. Lett. 2010;20:6408. doi: 10.1016/j.bmcl.2010.09.091. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gotoh H, Duncan KK, Robertson WM, Boger DL. ACS Med. Chem. Lett. 2011;2:948. doi: 10.1021/ml200236a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Leggans EK, Barker TJ, Duncan KK, Boger DL. Org. Lett. 2012;14:1428. doi: 10.1021/ol300173v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Leggans EK, Duncan KK, Barker TJ, Schleicher KD, Boger DL. J. Med. Chem. 2013;56:628. doi: 10.1021/jm3015684. 2013, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Schleicher KD, Sasaki Y, Tam A, Kato D, Duncan KK, Boger DL. J. Med. Chem. 2013;56:483. doi: 10.1021/jm3014376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Campbell EL, Skepper CK, Sankar K, Duncan KK, Boger DL. Org. Lett. 2013;15:5306. doi: 10.1021/ol402549n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Barker TJ, Duncan KK, Otrubova K, Boger DL. ACS Med. Chem. Lett. 2013;4:985. doi: 10.1021/ml400281w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Turner TC, Shibayama K, Boger DL. Org. Lett. 2013;15:1100. doi: 10.1021/ol400135n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wilkie GD, Elliott GI, Blagg BSJ, Wolkenberg SE, Soenen DR, Miller MM, Pollack S, Boger DL. J. Am. Chem. Soc. 2002;124:11292. doi: 10.1021/ja027533n. [DOI] [PubMed] [Google Scholar]; (b) Elliott GI, Fuchs JR, Blagg BSJ, Ishikawa H, Tao H, Yuan ZQ, Boger DL. J. Am. Chem. Soc. 2006;128:10589. doi: 10.1021/ja0612549. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wolkenberg SE, Boger DL. J. Org. Chem. 2002;67:7361. doi: 10.1021/jo020437k. [DOI] [PubMed] [Google Scholar]
- 8.Reviews: Margetic D, Troselj P, Johnston MR. Mini-Rev. Org. Chem. 2011;8:49. Boger DL. Tetrahedron. 1983;39:2869. Boger DL. Chem. Rev. 1986;86:781.
- 9.(a) Boger DL, Brotherton CE. J. Org. Chem. 1984;49:4050. [Google Scholar]; (b) Mullican MD, Boger DL. J. Org. Chem. 1984;49:4033. [Google Scholar]; (c) Mullican MD, Boger DL. J. Org. Chem. 1984;49:4045. [Google Scholar]
- 10.Campbell EL, Zuhl AM, Liu CM, Boger DL. J. Am. Chem. Soc. 2010;132:3009. doi: 10.1021/ja908819q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee K, Boger DL. J. Am. Chem. Soc. 2014;136:3312. doi: 10.1021/ja500548e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie J, Wolfe AL, Boger DL. Org. Lett. 2013;15:868. doi: 10.1021/ol303573f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corey EJ, Howe WJ, Orf HW, Pensak DA, Petersson G. J. Am. Chem. Soc. 1975;97:6116. [Google Scholar]
- 14.(a) Kuehne ME, Seaton PJ. J. Org. Chem. 1985;50:4790. [Google Scholar]; (b) Ogawa M, Kitagawa Y, Natsume M. Tetrahedron Lett. 1987;28:3985. [Google Scholar]; (c) Wenkert E, Pestchanker MJ. J. Org. Chem. 1988;53:4875. [Google Scholar]; (d) Magnus P, Brown P. J. Chem. Soc. Chem. Commun. 1985:184. [Google Scholar]; (e) Jones SB, Simmons B, Mastracchio A, MacMillan DWC. Nature. 2011;475:183. doi: 10.1038/nature10232. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Harada S, Sakai T, Takasu K, Yamada K, Yamamoto Y, Tomioka K. Chem. Asian J. 2012;7:2196. doi: 10.1002/asia.201200575. [DOI] [PubMed] [Google Scholar]
- 15.(a) Crow WD, Michael M. Aust. J. Chem. 1955;8:129. [Google Scholar]; (b) Crow WD, Michael M. Aust. J. Chem. 1962;15:130. [Google Scholar]; (c) Kump WG, Schmid H. Helv. Chim. Acta. 1961;44:1503. [Google Scholar]; (d) Kump WG, Count DJL, Battersby AR, Schmid H. Helv. Chim. Acta. 1962;45:854. [Google Scholar]; (e) Kump WG, Patel MB, Rowson JM, Schmid H. Helv. Chim. Acta. 1964;47:1497. [Google Scholar]
- 16.DePuy CH, King RW. Chem. Rev. 1960;60:431. [Google Scholar]
- 17.Molander GA, Harris CR. J. Org. Chem. 1997;62:7418. doi: 10.1021/jo971047e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.