

Abstract
One of the most fascinating discoveries in molecular oncology has been that cancer represents a disease in which genetic alterations in protein-coding, but also in non-coding genes complement each other. MicroRNAs (miRNAs) are a type of non-coding RNA (ncRNA) transcripts that can regulate gene expression primarily by disrupting messenger RNA (mRNA) translation and/or stability, or alternatively by modulating the transcription of target mRNAs. For the last decade, miRNAs have shown to be pivotal characters of every single one of the cancer hallmarks. Profiling studies have proven the significance of identifying over-expressed miRNAs (oncomiRs) causative of the activation of oncogenic pathways that lead to malignancy. Due to their crucial role in cancer, it has become a challenge to develop efficient miRNA-inhibiting strategies such as antagomiRs, locked nucleic acids or antisense oligonucleotides. However, to this date, the accessible delivery agents and their pharmacokinetic/pharmacodynamic properties are not ideal. Thus there is an urgent, unmet need to develop miRNA-based inhibitory therapeutics. Herein we present a novel therapeutic strategy that is only at the tip of the iceberg: the use of small molecule inhibitors to target specific miRNAs (SMIRs). Furthermore we describe several high-throughput techniques to screen for SMIRs both in vitro and in silico. Finally we take you through the journey that has led to discovering the handful of SMIRs that have been validated to this date.
Keywords: microRNAs, small molecules, targeted therapies, non-coding RNAs, cancer
Graphical Abstract
1. Introduction
The scientific rationalization of the human genome has to date been obscured by the (long-held) paradigm: “one gene, one protein”. The fixed mechanical orientation of having a protein-centric view of the human genome led to an increased number of subsequent assumptions such as classifying 98% of the human genome as “junk”. Nevertheless since 1950, a letter from Nobel laureate Barbara McClintock emphasized that we were letting the protein-coding gene philosophy control our reasoning; misgivings that were propagated to very recent times, until ncRNAs such as miRNAs were discovered [1]. Today, a more complex view is emerging: instead of focusing on genes, we have widened our research to non-coding genomic regions derived from what was initially relegated to the “junk pile”. Furthermore, molecular biology has evolved and redefined dogmatic terms in the field. We now consider the atomic unit for genetics “the transcript” and not the gene per se.
For over a decade, non-coding RNAs (ncRNAs) have been shown to be active participants in a broad array of regulatory events regarding cellular physiology as well as other biological processes. In 1993 the study of the gene lin-14 in C. elegans by Victor Ambros and his colleagues led to the identification of a type of small ncRNA-transcript that ultimately was able to regulate translation via an antisense RNA-RNA interaction; these were further ahead named microRNAs (miRNAs) [1]. However, it was not only until the year 2000 that the characterization of a second RNA sequence repressing protein expression elucidated the existence of a wider phenomenon concerning an unknown genomic regulatory mechanism [2].
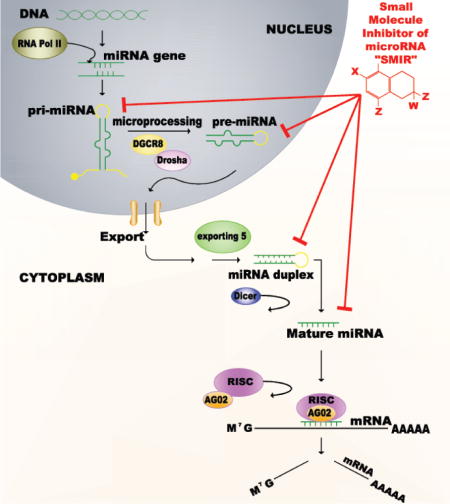
MiRNAs are a family of small ncRNAs, 19–22 nucleotides in length, which negatively regulate gene expression at a post-transcriptional level [3–5]. They are transcribed from a miRNA-coding gene by RNA polymerase II. After their transcription, miRNAs undergo a dual-processing event where they are initially a nascent transcript that folds upon itself forming a secondary hairpin structure, referred to as a long primary microRNA (pri-miRNA) [6]. This pri-miRNA is then cleaved by RNase endonuclease III Drosha, along with DGCR8 (molecular anchor part of a microprocessor complex), forming a precursor sequence (pre-miR) of about 70 nucleotides of length [7–9]. The pre-miR is translocated to the cytoplasm via Exportin 5 and RanGTP [10, 11]. Dicer, another double stranded RNA-specific cytoplasmic nuclease, defines a cleavage site that results in a 22 nucleotide long double stranded RNA transcript (dsRNA), from which the guide strand along with the RNA-induced silencing complex (RISC) targets the 3′ untranslated region (UTR) of messenger RNAs (mRNA) [12]. The end result is a decrease in the targeted protein levels (Figure 1).
Figure 1. Targeting miRNAs through a small molecule inhibitor (SMIR)-approach.
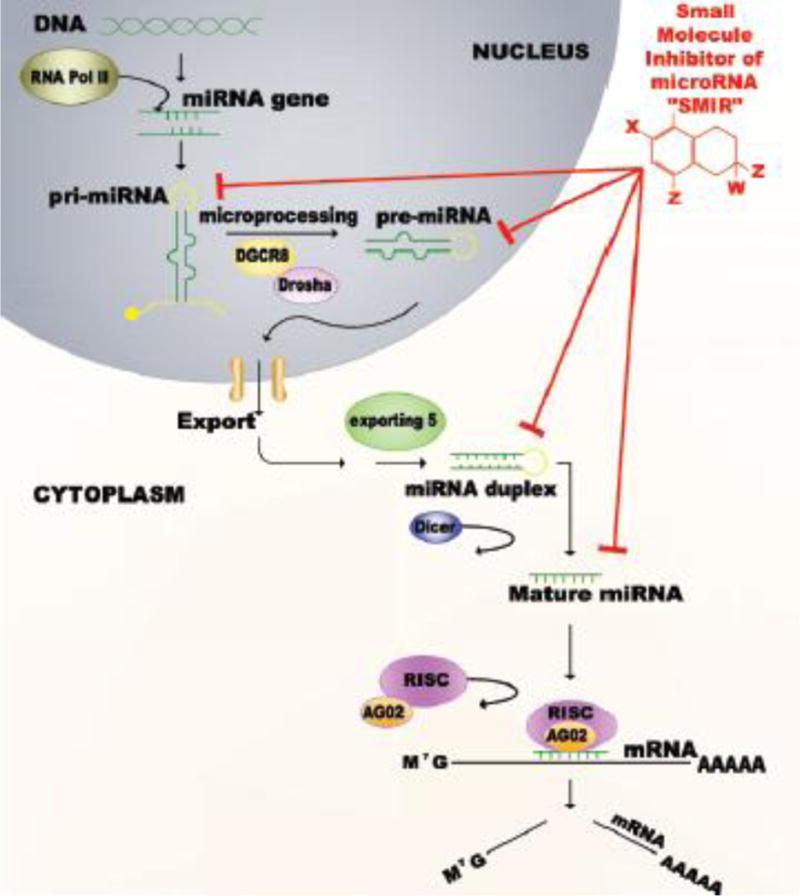
(A) MicroRNA Biogenesis. MiRNAs are transcribed by an RNA polymerase II enzyme, into a primary transcript that forms a hairpin structure referred to as pri-miRNA. It is then cleaved by RNase endonuclease III Drosha, along with DGCR8 forming a precursor sequence (pre-miR) of approximately 70 nucleotides in length. The pre-miR is then translocated to the cytoplasm via Exportin 5 where Dicer, another RNA-specific nuclease defines a cleavage site resulting in a double stranded 20 nucleotide in length transcript. From this transcript the guide strand along with the RNA-induced silencing complex (RISC) complex target the 3′ untranslated region (UTR) of messenger RNAs (mRNA). The end result is a decrease in the targeted protein levels. (B) Mechanism of the SMIR-approach. An ideal small molecules inhibitor of microRNAs (SMIRs) would be a compound that can potently bind, and therefore decrease the levels of a mature miRNA, in a specific way. Thus these elements would be targeting a mature miRNA sequence or any of its upstream precursors (primary or precursor-miRNA in the nucleus, or miRNA-duplex in the cytoplasm).
MiRNAs can regulate the expression of hundreds of genes simultaneously due to the fact that their nucleotide pairing by complementarity is imperfect [13]. In this manner, their mechanism of action implicates them in a variety of crucial processes such as tissue development, morphogenesis, apoptosis, signal transduction pathways etc [14–17]. This additionally implicates them in an array of cancer associated processes such as initiation, tumor development, invasion and metastasis. For cancer research purposes, miRNAs can be divided into two types of groups: those over-expressed, which target tumor suppressor proteins, and those with decreased expression in cells, which actually target oncogenes. The former are actually referred to as tumor suppressive miRNAs (TS-miRNAs), while the latter are called oncomiRs, which have been an attractive target for anticancer therapies during the past several years [18–20].
OncomiRs have emerged as important epigenetic regulators with causal links to the pathogenesis, maintenance and extent of cancer. The development of large-expression screens comparing miRNA levels in tumors versus normal tissues have proven useful in identifying novel miRNAs involved in cancer that could potentially become an attractive anticancer therapeutic target [21–23]. The key to small miRNA-based therapeutics lie in the antagonism of potent cellular targets such as miR-21, a miRNA that targets the mRNAs of the tumor suppressor genes (TSGs) PTEN and PDCD4 [24–27]; or miR-155, known to block the translation of CEBPβ, IL17RB, PCCD4, TCF12, ZNF652 mRNAs (of TSGs) [28, 29]. Many oncogenic miRNA-targets have been known and validated in tumor samples of different patient cohorts [21, 30, 31]. However, the truth of the matter is that we have not yet approached effective methods to treat patients this way. Shortcomings of the field lie in the fact that most of the molecules required to antagonize oncomiRs depend some kind of delivery mechanism (nanoparticles, liposomes, peptides etc.) and the majority of those already developed have proven to be ineffective or toxic [32, 33]. Thus, a nascent therapeutic concept such as antagomiRs has demonstrated to have substantial success in vitro, but in vivo, success hinges on delivery efficacy.
In addition to the challenge of not having efficient delivery mechanisms required for known miRNA-based therapeutics, another difficulty encountered in the drug-developing processes is the high costs involved in discovering, validating and producing them. The costs of prescription drugs have represented a significant burden to patients and their families and even to the government itself. Although there are many factors that contribute to the elevated drug prices, a significant one is the cost involved in completing and obtaining approval from the Food and Drug Administration (FDA). Any drug-marketing process requires by law FDA approval, which is a rigorous, extremely expensive and time consuming process (can take over ten years to complete). Moreover, out of every 10,000 compounds discovered in research that move on to a pre-clinical stage, only 10 will complete the FDA approval process. Thus, companies are required to cover these expenses for both, approved as well as non-approved drugs (that will not be granted permission to make it to the market). In this manner, there is an urgent unmet challenge of facilitating the development of new effective drugs, in a way that can decrease the existing burdens of high costs and time constraint, both of which cancer patients simply can’t afford.
In this review, we will focus on over-expressed oncomiRs and the studies that have been published regarding the initiation and development of a new therapeutic approach for miRNA inhibition: the use of small molecule inhibitors. Small molecules have been thoroughly used with clinical applications for numerous diseases but also specifically for cancer [34, 35]. The use of chemical compounds that are already FDA approved to treat a specific disease (“X”), would accelerate the process of completing toxicological studies and clinical trials in order to apply it to another one (disease “Y”). If a compound has already been through thorough preclinical testing and FDA-related studies (ten or more years), then using the exact compound for another disease would eliminate the need of going to the process again (shortening both money expenses and time consuming processes). Through the use of small molecules, we speculate that we can achieve effective oncomiR inhibition and gain significant reconstitution of important tumor suppressor proteins that will target cancer development.
2. Targeting miRNAs for cancer therapeutics
The main function of microRNAs in normal human cells is to maintain an adequate balance of protein expression. In this sense, miRNA coding genes complement protein coding ones directly, and this event has been demonstrated in all types of cells. However, similar to proteins and an array of other types of molecules, miRNAs are dysregulated in cancer which causes the loss of cellular homeostasis and the possibility of normal cells to transform into malignant ones. Currently, a vast repertoire of oncomiRs have been predicted and validated to target tumor suppressor proteins, positively regulating processes such as cancer initiation, progression and metastasis (Table 1). Evidently this has been one of the main reasons for which miRNAs are studied the most compared to other elements of non-codifying genomic regions. Thus, the significant attention devoted to miRNA research in cancer is not just a mere coincidence; these elements are certainly very attractive agents to target!
Table 1.
Examples of oncogenic microRNAs in human cancers
| Human microRNA | Deregulation in tumors | Proven targets | References |
|---|---|---|---|
| miR-21 | Over-expressed in glioblastomas, breast, lung, prostate, colon, stomach, esophageal and cervical carcinomas; uterine leiomyosarcomas and head and neck cancers. | PTEN, RECK, E-Cadherin, PCDC4, BCL2, MAPSIN, TPM1 | [14, 24, 25, 27, 28, 36] |
| miR-155 | Over-expressed in breast, lung, colon, and pancreatic cancers, and also in Hodgkin’s disease, pediatric Burkitt lymphoma, primary mediastinal and diffuse large B-cell lymphoma. | AGTR1, AID, IKBKE, TP53INP1, VHL | [14, 29, 36] |
| miR-17/18a/19a/20a | Upregulated by MYC; overexpressed in B-cell lymphomas, medulloblastomas, colon and lung cancers. | AIB1, AML1, BIM1, CTGF, CDKN1A, E2F1, E2F2, E2F3, HIF-1A, PTEN, TGFBR2, TSP1, Rb2/P130. | [14, 36] |
| miR-10b | Over-expressed in breast, pancreatic, and esophageal cancer. | HOXD10, Tiam1, KLF4, Neurofibromin | [97, 98] |
| miR-224 | Over-expressed in cervical cancer, pancreatic ductal adenocarcinoma, breast, colorectal cancer. | SMAD4, PHLPP1, PHLPP2 | [99, 100] |
Current strategies for inhibitory-miRNA therapies are based on antisense oligonucleotides (antimiRs), locked nucleic acids (LNA), LNA-antimiR constructs, antagomirs, miRNA sponges, ribozymes/DNAzymes, small interfering RNAs (siRNAs) and short hairpin RNAs (shRNA) (for a more detailed review see [36]). Some of these have proven to be effective not only in vitro, but also in vivo. For example, systemic treatment of tumor-bearing mice with miR-10b antagomiRs markedly suppressed formation of lung metastases by increasing levels of a functionally important tumor suppressor target, the mRNA of HOXD10 [37]. In another study, medulloblastoma cells passively took up 8-mer LNA-anti-miRs and specifically inhibited targeted microRNA seed-sharing family members resulting in diminished tumor cell proliferation in vitro, and tumor growth in vivo [38]. Finally one of the most popular therapeutic agents has been Miravirsen (SPC3649), an LNA against miR-122 developed by Santaris Pharma A/S. It is the first miRNA-targeting therapy to reach clinical trials for the treatment of hepatitis C (HCV), a viral infection known to increase the odds of patients developing hepatocellular carcinoma. It recently completed phase 2a, exhibiting robust activity against viral load, including the case of a patient that reached undetectable HCV-RNA levels [39, 40]. Although all of these strategies have been truly exciting, the truth of the matter is that there are challenges involved in the delivery of these non-small-molecule agents, and even more, their pharmacodynamic and pharmacokinetic properties are not ideal for the application [32, 33]. For this reason, we are still in the need of finding new alternative therapeutic approaches to inhibit oncomiRs, and decrease their activity.
2.1 SMIRs: a fresh approach with new challenges
Drug discovery and development is currently an extremely long process that takes approximately 10–15 years. Also, drug production results in an incredible economic burden that “boosts” their final overall cost, and patients end up having to pay exaggerated prices for their treatments. Thus, time and cost are considered the main obstacles in achieving new therapeutic alternatives for cancer treatment. Both of these have led to studying different proteins, as well as also other types of molecules that could potentially be targeted in different cancer patients.
MiRNAs are main characters of cancer and they have been associated with every single one of the cancer hallmarks. Because of the continuous challenges involved in the use of nucleotide analogs to target miRNAs, the development of small-molecule drugs targeting specific miRNAs (and modulating their activities) seems to be a promising approach. The interaction of small molecules and miRNAs was termed for the first time as “SMIR” by Melo and Calin et. al. [36, 41] referring to small molecules inhibitors of specific miRNAs. The mere concept anticipated a new avenue for an even more targeted development of cancer therapies. The SMIR-approach is an appealing one, specifically because it is a way of taking the “fast-track lane” in the drug developing race, reducing the time it takes to produce/approve it and also the overall cost of the process. However, it is an approach full of risks and challenges. For example, compared to proteins, RNA transcripts have been previously neglected as drug targets because of their electronegativity and structural flexibility. Also, the paucity of X-Ray crystallography or nuclear magnetic resonance structures for miRNAs, as well as the limited availability of miRNA-Dicer or RISC complex structures [42] make their discovery that much difficult. All of these make the process daring. Nevertheless if successful, this approach would result in having an effective drug reaching the patient’s bedside incredibly faster, in addition to completely eliminating the economic burden of processing a new FDA approval.
2.2 Developing a new idea, five years ago…
The biogenesis of miRNAs has been studied in detailed, and many enzymes and proteins have been found to be required for their stepwise processing. A showcase of potential targets for small molecule-based therapeutics was actually realized for the first time while investigating the molecular details of the miRNA maturation. Furthermore, when analyzing the secondary structures of miRNAs, they appear to be “druggable” targets. The formation of stem loops found in precursors and the bulges in miRNAs facilitate targeting by small molecules [43]. Deiters, et. al. proposed that there are three basic processing stages that miRNA absolutely require in order to excel their main function, and that small molecules indeed could block miRNA maturation at all three of them: the pre-transcriptional stage, the transcription stage or the post-transcriptional stage [44].
Several ideas were envisioned as plausible. First of all, it was thought that at a pre-transcriptional level small molecules could alter miRNA expression as well as their function in cells [44]. By altering miRNA promoter regions, small molecules were thought to regulate miRNA expression indirectly. For example, miR-127, a miRNA that targets the proto-oncogene BCL6 is known to be embedded in a CpG island and therefore subject to epigenetic silencing in cancer cells [45]. Demethylating agents have proven to recover its levels of expression as part of a miRNA cluster, resulting in an overall decrease in its oncogenic target. Analogous to this, small molecules were thought to potentially inhibit deacetylation or promote hypermethylation and decrease in this way the expression of oncomiR-coding genes such as it has been shown for protein-coding genes [46, 47]. Secondly (but in a similar way), transcription factors were also thought to be good targets for small molecules, and their blockage was predicted to function as “putting the brakes” on the constitutive expression of miRNAs in the cell. For example, c-Myc was been proven to activate the expression of a cluster of six miRNAs on human chromosome 13 [48]. Two miRNAs from this cluster (oncomiRs: miR-17-5p and miR-20a) negatively regulate the expression of E2F1, thereby promoting G1-to-S phase progression in mammalian cells by activating genes involved in DNA replication and cell cycle control [48]. Small molecules were thought to be able to target oncogenic transcription factors such as E2F1, and influence a decrease in oncomiR levels and a consequent recovery of tumor suppressor proteins. Thirdly, small molecules were also hypothesized to target miRNA biogenesis post-transcriptionally by inhibiting or blocking the action of RNA-endonucleases such as Drosha or Dicer [7–9, 12], or other proteins implicated in the biogenesis of miRNA subsets or siRNAs [44]. These along with many other proteins which are crucial in managing the transcript to produce mature miRNA sequences were thought to be efficient targets to inhibit with small molecule compounds, again with the aims of decreasing downstream the expression of oncomiRs. For example, Tan et. al. developed identified a potent inhibitor (ATA, a triphenyl-methyl molecule) that selectively inhibits loading of siRNAs and/or miRNAs to Ago2 without affecting other functions of the protein [49].
Comparing miRNA regulation before, during and after transcription, the studies done five years ago led to the belief that targeting transcription was the most efficient way of inhibiting a miRNA in a specific-manner. A higher degree of specificity was supposedly obtained by targeting transcription factors presumably involved in the development and cell specific regulation of oncomiRs [44]. However, transcription factors can ubiquitously affect promoter regions of many genes simultaneously; and even more, their expression levels/mechanisms of action might be distinct depending on the tissue/cancer type. Thus, targeting this step is nowadays not considered to be specific enough.
2.3 The current aim
The principal goal of the SMIR-approach today is to find compounds that potently bind, and therefore decrease the levels of mature miRNAs in the most, specific way possible. In this sense, small molecules would be targeting a mature miRNA sequence by binding to it, or to any of its upstream precursors (primary or precursor-miRNA in the nucleus, or miRNA-duplex in the cytoplasm) (Figure 1).
If a small molecule was proven to bind to any of these sequences directly, we could achieve the goal of discovering a useful (most likely FDA-approved) drug that could be applicable for any cancer type that overexpresses that specific oncomiR. In order to achieve this, one of the first steps would be to determine potent oncogenic miRNAs that have already been proven in the literature to be crucial in the progression and advancement of cancer (most likely through previous miRNA-microarray profiling-studies in cancer). A good example of this would be miR-21. MiR-21 is over-expressed in multiple cancer types such as breast, ovaries, cervix, colon, lung, liver, brain, esophagus, prostate, pancreas and thyroid [24, 25, 27, 28]. A SMIR that specifically targets this miRNA would be useful for patients that suffer the already mentioned cancer types, which most likely have a tumor that is dependent on the expression levels of oncomiR-21.
3. Searching to discover novel SMIR’s
High Throughput Screening (HTS) is a drug-discovery process broadly used in research for many types of studies, including experimental therapeutic approaches. The term usually refers to quick methods to assess the biological or biochemical activity of numerous compounds, such as small molecules. Through these processes scientists can rapidly identify active compounds that are the basis/starting point for drug design. The technique has been used in the past years, to search for small molecule inhibitors of miRNAs [50–52]. Regarding SMIRs, HTS can be used (as an initial approach) in two ways: in vitro and in silico; and both of these have been recently explored [26, 49, 52–56]
3.1 HTS techniques in vitro
Assays detecting Fluorescence
-
-Beacons:
-
◦Fluorescent beacons are oligonucleotide hybridization probes that report the presence or the absence of specific nucleic acids in homogeneous solutions. They are usually hairpin shaped molecules that contain a quencher and a fluorophore that emits fluorescence when it binds to its nucleic acid target sequence. The technique has been proven useful in determining the levels of Dicer processed-mature miRNAs in culture cells [53, 54]. The basis of the test was that a specific sequence of a precursor miRNA would serve as the molecular fluorescent beacon, and the mRNA would be the nucleic acid-target sequence that the miRNA binds to and inhibits. To screen for SMIRs, the technique is performed by synthesizing a doubly-labeled pre-miRNA beacon containing a 5′fluorescence emitter, and a 3′ quencher. Dicer-mediated hydrolysis of the precursor results in a dissociation of the fluorescence emitter and quencher, thereby generating a Dicer-dependent increase in the fluorescence (Figure 2a).
-
◦Recent studies regarding the florescent beacon-approach have reported that the Dicer-mediated cleavage depends on the 5′ and 3′ ends of the precursor sequence. In this way, the presence of the fluorophore and the quencher might actually affect the binding and activity of the Dicer enzyme. Because of this, the technique has been revised and altered, and a new strategy has been approached and validated [52]. An alternative way to approach this pitfall was creating a DNA molecular beacon that is independent of the precursor sequence and complementary to the mature miRNA produced after the Dicer cleavage (and in this way Dicer cleavage is not expected to be altered in any way) [52]. The beacon loop is a perfect complement to the mature miRNA sequence. In this case, the precursor sequence is base pairing with the passenger strand, forming a thermodynamically stable structure, hence the beacon is not open and there is no fluorescence. When the precursor sequence is in its natural conformation, only low levels of fluorescence are observed, because of the close proximity of the quencher. However, the dicing and helicase activity of Dicer results in a mature miRNA sequence that hybridizes with the separate molecular beacon containing its perfect complement [52]. This results in opening of the beacon and increased fluorescence. In the presence of a SMIR that blocks this Dicer processing by binding to the precursor sequence, the precursor is restricted to its close conformation and a reduced fluorescence should be observed [52].
-
◦
-
-Polarization assay:
-
◦Although this assay was initially designed to perform HTS of modulators of RISC components, it can potentially have broad applications in the study of individual miRNAs, and the inhibition of their processing. The principal of the fluorescence polarization (FP) screening assay lies on the fact that labeled miRNAs are free to rotate in the absence of Ago2, resulting in a low polarization value. However, once the miRNA forms a complex with Ago2 (as well as with other proteins) it rotates more slowly, resulting in a higher polarization value [49]. This technique allows the study of a vast number of inhibitory (candidate) compounds in a short period of time.
-
◦
Figure 2. High-throughput screening techniques in vitro.
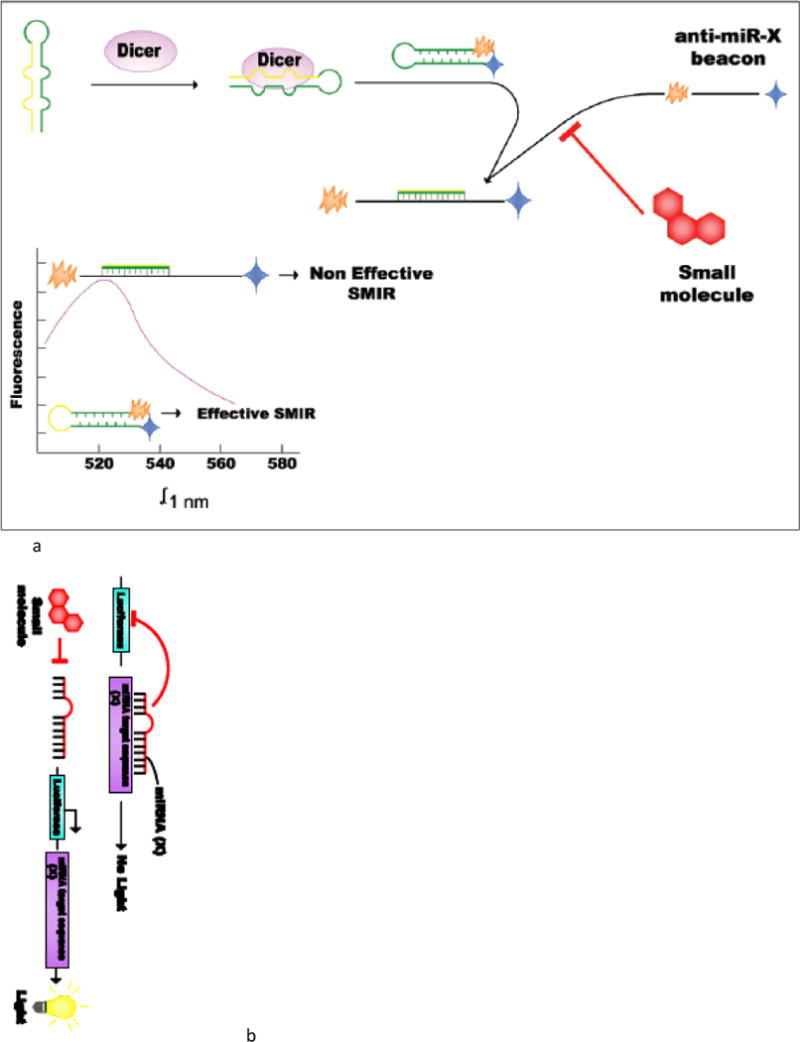
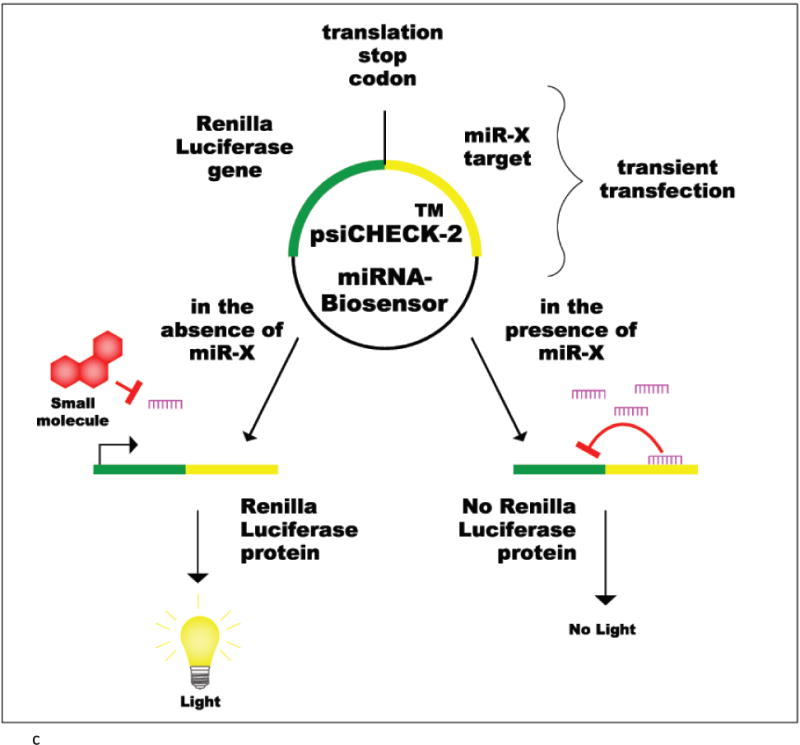
(a) Molecular based-fluorescent beacon. Fluorescent beacons are oligonucleotide hybridization probes that report the presence or the absence of specific nucleic acids in homogeneous solutions. They are usually hairpin shaped molecules that contain a quencher and a fluorophore that emits fluorescence when it binds to its nucleic acid target sequence. (b)Lenti-viral luciferase reporter vector constructs. These reporters are assembled by introducing the complementary sequence of the miRNA of interest, as well as a control miRNA (in a separate construct) that contains no detectable recognition by natural miRNAs downstream of a luciferase reporter gene. (c) psiCHECK-2 vector. It is a reporter plasmid used to clone a miRNA target sequence downstream of a Renilla luciferase (Rluc) gene. The presence or absence of a specific miRNA can be monitored by the change in expression of Rluc. The system detects a decrease in miRNA levels by an increase in Rluc expression.
Luciferase Reporter constructs
Luciferase biosensors assays provide a tool of rapid screening of miRNA activity in cells. These vectors are usually assembled by introducing the complementary sequence of a specific miRNA-(X), which is compared to a vector that has a control sequence, downstream of a luciferase reporter gene. The plasmid is then used as a sensor to detect the presence of specific mature miRNA molecules. The reporters are sequenced, validated, and introduced into culture cells by transfection. The system has been tested and proved able to detect endogenous miRNA levels when they are highly expressed and also exogenous introduction of miRNAs through precursor sequences or expression vectors. In the presence of a SMIR for miR-X, the mature sequence of the miRNA is decreased and the luciferase gene is expressed constituvely. If the molecules tested do not inhibit the miRNA, then the miRNA will bind to its target sequence and inhibit the expression of the luciferase gene.
Lentiviral reporter construct
These are typically assembled by introducing the complementary sequence of the miRNA of interest, as well as a control sequence (in a separate construct that contains no detectable recognition by natural miRNAs) downstream of a luciferase reporter gene (Figure 2b) [26].
psiCHECK-2 reporter plasmid
The psiCHECK2 vector (Promega) has been a useful tool to perform HTS of SMIRs. It is a reporter plasmid used to clone a miRNA target sequence downstream of a Renilla luciferase (Rluc) gene. The presence or absence of a specific miRNA can be monitored by the change in expression of Rluc. The system detects a decrease in miRNA levels by an increase in Rluc expression. On the contrary if miRNA levels increase then Rluc expression consequently decreases. With this reporter vector, there is a particular advantage; there is no need of co-transfecting a control luc-gene because it contains a Firefly luciferase (F-luc) gene which is constitutively expressed. There are several commercially available psiCHECK-2 vectors that can be acquired with a particular miRNA target sequence already cloned. An example, is the psiCHECK-2-prohibitin reporter vector which contains a very potent target sequence of miR-27a: prohibitin [52] (Figure 2c).
pEZX-MT05 plasmid
The pEZX-MT05 vector from (GeneCopoeia) also offers the opportunity of miRNA target identification. Similarly, miRNA complementary 3′ UTR sequences are inserted downstream of the secreted Gaussia luciferase (GLuc) reporter gene in its respective vector system, which is driven by SV40 promoter for expression in mammalian cells. Besides using GLuc as the miRNA 3′ UTR target reporter, a secreted Alkaline Phosphatase (SEAP) reporter driven by a CMV promoter, is also cloned and it serves as the internal control. In addition to the GLuc/SEAP dual-reporter vector system, a firefly/Renilla Duo-Luciferase reporter vector (pEZX-MT01) has also been validated and is currently available for SMIR screening [55].
Stable cell lines
Many researchers have been working in the past couple of years, on finding better high-throughput screening techniques in order to identify SMIRs. Recently, it has been proposed that using a stable cell line that constantly expresses a luciferase reporter, (instead of a transient transfection), not only will be more cost efficient and less time-consuming, but will also remove variations associated with transfection efficiency and other technical manipulations. This approach was tried by Connelly et. al.; and in their study, they recorded the steps needed to create a stable cell line with a constitutively expressed vector [51]. In it they validated the Huh 7 cell line with a constitutive expression of psiCHECK2 vector with the miR-122 target sequence [51]. This technique could be applied to any other cell line as well as any other miRNA.
3.2 HTS techniques in silico
With the advancing understanding of miRNA structure and the thermodynamics of miRNA-small-molecule interactions, in silico high-throughput screening techniques are promising because they can substantially accelerate the miRNA-targeted lead identification and optimization in a cost-efficient manner compared with the conventional drug discovery pipeline. However, targeting oncomiRs via computational approaches, is still challenging, as most computational models need recalibration to ensure the compatibility of their nucleotide target. Current in silico HTS techniques can be divided into two categories. The first category is the structure-based or receptor-based approach, which aims to identity miRNA-binding molecules that energetically fit “the pocket” in the existing or (most likely) predicted tertiary structure. The second category is the ligand-based approach, which models the quantitative structure-activity relationship (QSAR) or pharmacophore by exploiting the chemical information within a set of ligands with known experimental activities, and uses this model to identify potential actives. The predictive accuracy of the majority of in silico techniques, especially the structure-based models, have not yet been intensively benchmarked. In comparison, ligand-based approaches employing well-documented cheminformatics techniques [57] have been reviewed elsewhere [58]. Here, we will give an overview of the structure-based methods.
Due to the paucity of available miRNA 3D structures, their computational modeling is a pivotal first step for structure-based drug design. Frankly speaking, computational methods to model RNA tertiary structure are still under development due to the complicated RNA foldings (e.g., noncanonical base pairs, helices, hairpins, bulges, pseudoknots, etc) [43]. What we do know is that pri-miR and pre-miR form a hairpin fold, whereas mature miRNA is processed into a single stranded form [59]. Current methods for RNA tertiary structural modeling largely relies on the accurate prediction of RNA secondary structure. A pilot server, CompaRNA, is continuously benchmarking 28 single-sequence and 13 comparative RNA secondary structure methods for their accuracy and robustness [60, 61]. As of 2013, the best single-sequence methods are CentroidFold [62], ContextFold [63] and IPknot [64], and the best comparative methods are CentroidAlifold [65], MXScarrna [66], RNAalifold [67] and TurboFold [68]. Using the secondary structure as the input, RNA tertiary structure can now be predicted efficiently with various algorithms, including fragment assembly (MC-fold/MC-Sym pipeline [69], FARNA [70] and RNA-MoIP [71]), coarse-grained molecular dynamics (iFoldRNA [72] and NAST/C2S [73, 74]), comparative modeling (ModeRNA [75]), tertiary structure machine translation (RNAComposer [76]).
Once the RNA structure is available, molecular docking is the “production step” for in silico high-throughput virtual screening. Due to the accessibility to crystallized structures, the docking algorithms and scoring functions that predominate are those only applicable to protein targets. However, several pioneer studies have suggested that some native protein-based docking methods be useful for RNA [56, 77, 78]. Researchers in the field have acknowledged the RNA-small-molecule “scoring issue”, and implemented novel RNA-specific scoring functions such as DrugScoreRNA [79], rDock score [80, 81], iMDLScore [82] and integrative scoring [83]. Recently, Chen et al. has benchmarked five docking software with 11 scoring functions for their performances in binding mode prediction, scoring and virtual screening [82]. Based on the results, they have proposed a practical high-throughput virtual screening pipeline: GOLD::GOLDFitness-ASP-rDock_solv for rigid RNA binding site and rDock::rDock_solv-ASP-iMDLScore2 for moderately flexible RNA binding site [82]. To account for the dynamic behavior of RNA, computational (more expensive) techniques have been implemented using elastic potential grids [84], ensemble structures [78, 85] or force-field based energy minimization [86] (Figure 3).
Figure 3. High-throughput screening techniques in silico.
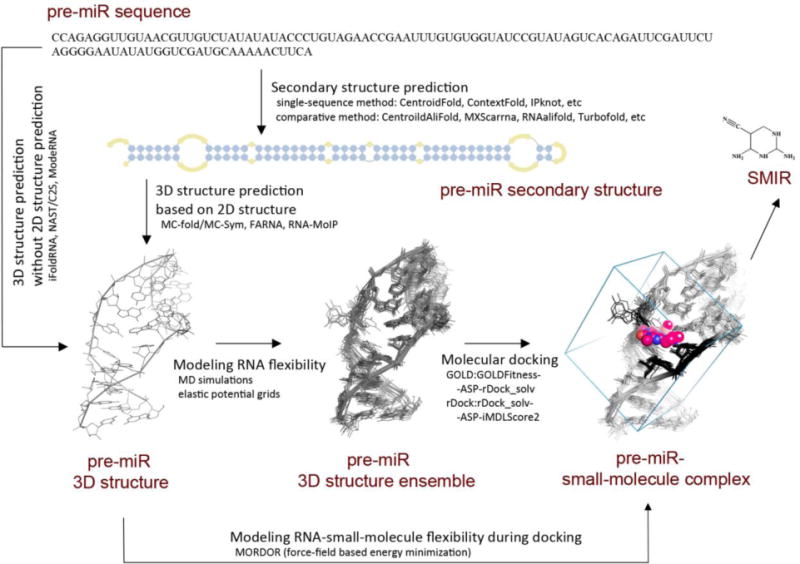
In this example, pre-miR-10b is used as reference miRNA. To model the 3D structure of this pre-miR more accurately, some tools (e.g., MC-fold/MC-Sym, FARNA and RNA-MoIP) need the secondary structure information, which can be predicted by either single-sequence method or comparative method. Other tools like iFoldRNA, NAST/C2S and ModeRNA, however, are able to predict RNA tertiary structure without 2D structure prediction. To identify the binding small-molecules with a given pre-miR, molecular docking is the pivotal technique that predicts pre-miR-small-molecule interaction and identifies potential SMIRs. The best docking combinations for RNA targets have been benchmarked. Sometimes it is necessary to model the RNA flexibility, and this can be achieved by generating structure ensemble before docking using MD simulations or elastic potential grids, or modeling the ligand-induced effect using force-field based energy minimization during docking.
As a proof of concept, Shi et al. have reported the discovery of AC1MMYR2, a potent and selective inhibitor of miR-21, based on structure-based virtual screening [50]. They used MC-fold/MC-Sym pipeline to model the 3D structure of pre-miR-21, and selected the Dicer binding site in the precursor, as the site where they wanted candidate compounds to bind to. AutoDock predicted 48 compounds with high binding affinity, and they selected five for experimental evaluation. As a result, AC1MMYR2 demonstrated the capability to block the maturation of miR-21 in a Dicer-dependent fashion and inhibit tumor proliferation, migration and invasion both in vitro and in vivo [50].
4. The trajectory
4.1 The beginning…
Ever since oligonucleotides were designed to target RNA, their therapeutic potential has been hampered by poor cellular delivery methods and their stimulation of an immunogenic response [32, 33]. Small molecule inhibitors of miRNAs have been considered important tools that would serve as therapeutic agents, and that could furthermore facilitate the discovery of novel miRNA functions in cancer (specifically). In 2006/2007 Davies and Arenz suggested for one of the first times that these non-coding RNA transcripts could be targeted with small organic molecules [54]. They hypothesized that small molecules could provide a particular advantage (compared to antisense oligonucleotides) because of their higher uptake by organisms as well as their lower manufacturing costs. Thus, they developed one of the first screening assays to detect potential inhibitors of miRNA maturation at a high-throughput format (Refer to Figure 3).
In their initial premise, Davies and Arenz envisioned that ligands which specifically bind pre-miRNAs could offer a therapeutic route for diseases such as cancer, where over-expression of had been linked to pathogenesis. Using pre-let-7 RNA from Drosophila melanogaster as a model, they developed a doubly-labeled pre-miRNA beacon with a fluorescent emitter and quencher to be able to detect Dicer processing (hydrolysis) through the fluorescence emitted. This way, they established the first homogeneous fluorescence assay for detecting potential inhibitors of the miRNA maturation process. However, even know their work was novel; they emphasized the need to address the “selectivity” issue with pre-miRNA binders [53, 54]. Furthermore it was proven that the location of the fluorophore molecule at the 5′ and a fluorescent quencher at the 3″ was likely to impede a proper cleavage of the pre-miRs by Dicer, since it depends on both ends of the precursor for miRNA maturation [52]. Nevertheless the concept remained very well established and consequently widened research in this field.
4.2 The first hit…
It was in the year 2008 when the first SMIR study was published in the literature, and it was mentioned for the first time that a small-molecule inhibitor of a specific miRNA was proven to be found [26]. The introduction of this concept into the miRNA therapeutic-world was huge, since until the moment specific miRNA inhibition had only been observed with antisense oligonucleotides, such as the one against BCL2, Oblimersen [87]. At the time it was only thought that miRNAs were involved in the regulation of about 30% of all genes and genetic pathways and this made them important enough to target. To this date, we know they potentially regulate a higher genome percentage (over 60%), and it has been reaffirmed that their role as potential therapeutic targets is even more important than what was initially thought [88].
Gumireddy et. al. reported a cellular screen for miRNA-pathway inhibitors and found the first small-molecule inhibitor of miRNA function. In their study they describe the development of an assay that allowed the detection of a miRNA being inhibited, after treating with small molecules that could potentially interfere and target their biogenesis pathway [26]. They selected miR-21 as the target miRNA because of its known tumorigenic properties in various cancers, such as breast, ovarian, lung, glioblastomas [16, 24, 25, 27] etc. The assay consisted on a lentiviral reporter construct that was assembled by introducing the complementary sequence to miR-21, as well as a control miRNA (−30) as a negative linker sequence downstream of a luciferase reporter gene. This plasmid was then used as a sensor to detect the presence of specific mature miRNA molecules (Refer to Figure 2b). The reporters were introduced into HeLa cells which have high miR-21 basal levels. The system proved to be able to detect miR-21 levels efficiently, as the introduction of a miR-21 expression vector yield a 90% decrease in luciferase signal [26].
Subsequently they performed a primary screen of a library containing over 1000 compounds, of which treatments were conducted at a concentration of 10uM [26]. An initial hit: diazobenzene, was found to produce over 251% increase in luciferase signal compared to solvent alone [26]. Furthermore the chemical structure of this compound enabled preliminary studies on structure-activity relationship as well as predictions on possible modifications that could increase the inhibitory strength. Structurally related compounds uncovered an alternate highly active one which actually induced a 485% increase in luciferase reporter signal [26]. The mechanism by which this compound could cause inhibition was initially unknown, since only the downstream decrease of a mature miR-21 sequence was being detected. When assessing the sequence of the pri-miR-21, an 87% decrease was detected on treated cells compared to non-treated controls [26]. These results strongly suggested that the compound was actually targeting the transcription of the miR-21 coding gene, and not the downstream processes of the common miRNA pathway [26].
4.3 Continuing discoveries…
Approximately two years after the first SMIR was described, Young, et. al. began studying miR-122 [89]. The discovery of a small molecule that could target this miRNA (specifically) seemed to be clinically relevant for pharmaceutical purposes, since its expression has been known to be necessary for hepatitis C virus (HCV) infection as well as replication. Also, HCV infection has been considered for many years a pre-malignant infection that could lead to the development of hepatocellular carcinoma, therefore, eliminating HCV replication is an important cancer-preventive approach. In support of their idea, preclinical studies had already proved that knockdown of miR-122 dramatically decreased HCV RNA in human liver cells (without any toxic effects in mice and primates), so evidently it is a very attractive non-coding RNA transcript to target [89].
For this project, a luciferase based psiCHECK-2 reporter plasmid was used to screen for SMIRs [89]. This vector contained a Firefly luciferase (F-luc) gene constitutively expressed which serves as a control, and also a Renilla luciferase (Rluc) gene which served as a reporter of miRNA levels in cells. In this way, the need of transfecting a control vector separately was eliminated. The target sequence of miR-122 was inserted downstream of the (Rluc) gene, and the change of Rluc-genetic expression would determine if there was any inhibition of the miRNA. A total of 1364 compounds were screened in a high-throughput 96 well format using Huh7 cells at a concentration of 10uM, and luciferase reporter signal was assessed after 48 hours [89]. This study led to the discovery of two potent SMIRs for miR-122, which demonstrated inhibition of HCV replication in liver cells [89]. Both of them seemed to target the transcription of the miRNA-coding gene, since the pri-miR122 sequence was proven to be reduced upon compound treatment [89]. After this study, the same group has continued to work on finding better high-throughput techniques in order to identify even more SMIRs. They have recently proposed that using a stable cell line constantly expressing the luciferase reporter, instead of a transient transfection, not only will be more cost efficient and less time-consuming, but will also remove variations associated with transfection efficiency and other technical manipulations. Their recorded steps on how they created a stable cell line (Huh 7) with a constitutively expressed psiCHECK2 vector with the miR-122 target sequence could be applied to any other cell line as well as any other miRNA [51].
Several other studies began aiming similar findings. Watashi et. al. developed a very extensive screening with the hope of discovering small molecules that could suppress miRNA function and moreover reverse tumorigenesis [90]. A total of 530 compounds, most of them with well-established pharmacokinetic (PK) and (PD) properties, were used at six different concentrations ranging from approximately 0.5–10uM [90]. Their experimental approach consisted of three screening steps. Initially human embryonic kidney 293 cells (HEK 293) were transfected with SV40 promoter driven Fluc gene, a control Rluc gene and a pGL3 promoter plasmid that expressed sh-Fluc (which provided the source of miRNA against the Fluc mRNA). Secondly, a CMV promoter driven Fluc plasmid was transfected in the same way, with the Renilla control and the shFluc. Thirdly the cells were transfected with an SV40 promoter driven Fluc and the Renilla control, but a vector coding for sh-GFP (green fluorescent protein) which codes for a miRNA that targets GFP mRNA and not Fluc mRNA. Compounds positive in the first two screenings and negative in the third one were sought. Two candidates were obtained based on these criteria: PLL, and TPF [90]. Collectively their results suggested that PLL treatment reduces pre-miRNA association with Dicer, while TPF treatment reduced the miRNA association with AGO2 [90]. Moreover they found that the two compounds could reverse the tumorigenicity of miR-93 and miR-130b over-expressing cells [90]. However, their mechanisms of action had the pitfall that they targeted the biogenesis process of miRNAs in general (not specifically). Nevertheless, in cases where over-expression is crucial for tumor maintenance, reducing the activities of these oncomiRs (albeit accompanied by commensurate reductions in other cellular miRNAs) may still be considered therapeutically efficient.
Not much time had passed when another small screening was performed, this time to find a specific inhibitor of miR-21 with better pharmacokinetic properties, and higher specificity [55]. A similar approach was used, were a luciferase-based reporter was used: pEZX-MT01 plasmid [55]. This vector has a Fluc gene, and downstream to it, the 3′ UTR of (Programmed cell death 4) PDCD4, a well-known target of miR-21 [55]. MCF7-cells (breast cancer cell line) where treated with a total of 15 small molecules (all aminoglycosides) at a concentration of 10uM. Streptomycin was found to potently inhibit miR-21 at this concentration, and even still at 5uM [55]. To furthermore validate its specificity, they tested the levels of ten other oncogenic miRNA after treating with a dose of 5uM of Streptomycin. Only one, out of the ten (miR-27a) was proven to be down-regulated in a similar manner [55]. The PDCD4 target was tested and proven to be recovered slightly (by western blot) [55]. To prove that the mechanism of inhibition was a direct one, thermal melting experiments were performed with in vitro transcribed pre-miR21, and upon addition of streptomycin, thermal stabilization was observed [55]. In summary, they showed that with these cell-based assays, streptomycin was proven to repress miR-21 levels by directly binding to its precursor sequence interfering with Dicer processing [55]. However, the treatment was not proved to be as specific as expected, since it did down-regulate one of ten other miRNAs tested [55]. Also, this study did not check for important tumor suppressor targets of miR-21 such as PTEN, RECK.
Another group of small molecules known as aza-flavanones, where the focus of a study that aimed to investigate the role of these compounds as potential miRNA inhibitors. Chandrasekhar et. al. tested the role of aza-flavanones in controlling miRNAs that specifically target proteins involved in cell death and/or proliferation [91]. In their study they focused on miR-14 from Drosophila organisms, because of its anti-apoptotic activity. A homology search of a human homologue of this miRNA led to the identification of miR-4644, a recently validated miRNA which is abundantly expressed in breast tumor tissues [91]. Since this homologue was validated, they used Drosophila flies as an in vivo model to test the inhibitory effect of different aza-flavanones using a luciferase reporter vector. The hits from the in vivo study where then tested against breast cancer cells, but with a vector for the human homologue miR-4644. Their results suggested that aza-flavanones are potentially specific towards human miR-4644 in a similar way that they are for the fly homologue miR-14. Furthermore, they demonstrated that the compounds did not have a general effect on the common miRNA biogenesis pathway nor on other miRNAs tested [91]. Their results demonstrated a novel mechanism through which miRNA inhibitors could be screened for, in order to target tumorigenic pathways such as proliferation and apoptosis (as seen with miR-4644) [91]. More so, they developed and validated a novel cross species-in vivo HTS method to identify SMIRs by using Drosophila homologues of human miRNAs. [91].
The concept of small molecule inhibitors of miRNAs began expanding in the year 2013 where several research groups worked on finding new strategies to screen for them in a high-throughput manner. A new method to screen for SMIRs in vitro was developed by Bose and colleagues in the beginning of the year 2013 [52]. With the aims of overcoming relatively time-consuming assays that do not provide information on the mechanistic details of the modulation of the SMIRs, this research group described the development of a DNA-based molecular-beacon SMIR screening method. The beacon contains a stem loop structure with a fluorophore and a dark quencher at the two ends, where the loop sequence is exactly complementary to miRNA (anti-miR). In the presence of the mature miRNA of interest the beacon “opens up” and hybridizes with it, increasing the fluorescence signal. In this method, they addressed drawbacks from the previous research groups by making use of a DNA molecular beacon (with 5′ fluorophore and 3′ quencher), independent of the pre-miRNA and complementary to the mature miRNA produced after cleavage by Dicer (in its mature form). With this approach they screened 14 commercially available aminoglycosides at a concentration of 5uM–15uM, to test if any of them inhibited miR-27a, a miRNA proven to be over-expressed in several cancer types [52]. In the screening they found that a set of them inhibited Dicer-catalyzed miR-27a maturation [52]. The positive hits were found when the compounds bound pre-miR27a and hinder Dicer processing, and the amount of mature miR-27a production was less when compared with controls [52]. The most potent SMIR they found achieved 25–30% inhibition with this assay [52]. There results were validated using a cell-based assay with the known psiCHECK-2-prohibitin reporter vector (prohibitin being a potent miR-27a target sequence) at a concentration of 20uM which yield very similar findings [52]. Their high-throughput beacon-based assay compares (favorably) to other previously described methods, as it proved to be cost-effective, sensitive and robust.
One of the most attractive miRNAs to find a SMIR for has been miR-21, and several groups have tried to screen for one (as previously mentioned). The fact that it is very important for the development of many different types of human cancers, such as glioblastomas, gastric tumors, breast cancers and colon cancers [16, 24, 25, 27], exalts the relevance of finding a direct specific inhibitor that could be used as therapeutic alternative in all of these. With the aims of achieving an even more specific SMIR against miR-21, Shi et. al. designed an in silico HTS technique [50]. By using the three-dimensional structure of the Dicer binding site on the pre-miR-21, they conducted a screening of 1990 compounds to find molecules that could block miR-21 maturation by MC-Fold/MC-Sym and Auto Dock programs [50]. By this method they identified a SMIR termed AC1MMYR2, which blocked the ability of Dicer to process the precursor form (in silico) [50]. In vitro, its potency was also tested, and it was found to inhibit miR-21 after 24 hours at a dose of 30uM [50]. This inhibitor also up-regulated the expression of the tumor suppressor targets of the miRNA: the mRNAs of PTEN, PDCD4 and RECK [50]. Even more, it proved to reverse epithelial-to-mesenchymal transition by reducing mesenchymal markers and reconstituting the levels of E-cadherin [50]. In vivo, the compound also demonstrated a decrease in tumor growth, invasiveness, and metastasis, increasing overall host survival with no observable cytotoxicity in orthotopic models [50]. AC1MMYR2 proved to block miR-21 maturation in a set of four different cell lines of different cancer types in a similar way [50]. Finally, the selectivity and specificity of this compound was more thoroughly assessed by measuring the levels of other miRNAs through RT-PCR [50]. Unfortunately, five out of the eleven miRNAs tested proved to also be significantly reduced with this treatment [50]. Although this was an interesting mechanistic approach, there are some issues that need to be solved regarding miR-21 specific inhibition.
On another note, two important miRNAs in gastric cancer: miR-372 and miR-373, were also the focus of investigation when searching for SMIRs, since they potently target the tumor suppressor LATS2 (large tumor suppressor kinase 2). Both of these miRNAs have been proven to be significantly over-expressed in gastric cancer cells [92]. In this screening, Vo et. al., reported the first example of multimodal RNA ligands, aiming to inhibit the biogenesis of oncomiRs [92]. The multimodal ligands are composed firstly of an artificial nucleobase designed in order to specifically recognize an RNA base pair of the double stranded region of the pre-miRNA; and secondly, an aminoglycoside known to interact in a potent manner with stem-loop RNAs with high affinity [92]. Their work was done in the AGS cell line, growth of which is dependent on high levels of miR-372 and miR-373 [92]. Some of their multimodal ligands, composed of neomycin conjugated to natural and artificial nucleobases, revealed to be able to inhibit Dicer precursor cleavage by binding to these stem-loop structured RNAs [92]. One of their compounds proved to be an efficient inhibitor in vitro, and demonstrated a dose dependent miRNA decrease [92]. Its cytostatic effect was directly correlated to the decrease of both of the oncomiRs, and this was shown by qRT-PCR and through luciferase reporter vectors [92]. Their approach consisting of the conjugation of different RNA binding motives, demonstrated to be successful for the targeting of oncomiRs, and has evidently widened the search for the rational design of precursor ligands [92]. However, they did identify significant decrease in the levels of at least 3 other oncogenic miRNAs. Nevertheless, the techniques used to perform these kinds of HTS seem very promising for new SMIR therapeutics.
Recently, a research group developed a novel strategy with the goal of identifying a small molecule that binds a precursor miRNA sequence, and inhibits its maturation. Their approach was termed Inforna, and it was validated to design lead compunds that target miRNA precursors that have been identified through sequencing and functional studies [93]. In this study researchers downloaded the sequences of all the miRNA hairpin precursors in the human transcriptome from miRbase, and their secondary structures were predicted in silico via RNA structure (considering energy minimization). The entire set of secondary structures was then parsed by Inforna, which created the output of the targetable motifs in each RNA, and the corresponding lead small molecules that could potentially bind them [93]. Inforna probed over 5 million potential interactions, requiring that the targetable motif and its closing base pairs were exact matches for motifs in the database [93]. They then redefined their lead interactions based on two criteria: the first being that the targetable motif should be between the Drosha or Dicer processing step and second that the miRNA would be causative of disease [93]. There results showed 22 lead small molecules for different precursors including some for prostate, breast, ovarian and pancreatic cancer (as well as other diseases) [93]. The most avid interaction was seen between a benzimidazole and the precursor sequence for miR-96 [93]. This compound was selective in inhibiting miR-96 biogenesis as proven by the recovery of its protein target FOXO1, inducing apoptosis in cancer cells [93]. When testing it in vitro, it showed reduced expression levels of miR-96 by 90% at a concentration of 40uM [93]. This reduction proved to be selective when testing two other miRNAs (−182 and −183, which are transcribed with −96 as a single transcript). Even more, compared to LNA, the selectivity was higher for the hit compound [93].
Regarding the binding of small molecules to precursor sequences, Murata et. al studied the synthesis and structure-activity relationships of xanthone and thioxanthone derivatives as fluorescent indicators for detecting interactions between small molecules and RNA sequences[94]. Some of the derivatives of these compounds preferentially bind to certain secondary structures of RNA such as loops or bulges, which are characteristic of precursor sequences before they undergo Dicer-mediated processing. In their study they demonstrated that the xanthone and thioxanthone derivative X2S-N,N-diMe inhibited pre-miR-29a maturation in a specific manner, by binding to the nucleotides that form part of the internal loop/bulge (and not by any interaction with Dicer itself) [94]. However, the off target effect of this compound in inhibiting other miRNAs was not investigated [94].
5. The other side of the coin: small molecule as miRNA promoting agents
Several small molecules have also been found to increase the expression of tumor suppressor miRNAs. By targeting regulatory mechanisms for the RNAi (RNA interference) pathways, small molecules have demonstrated the possibility of recovering tumor suppressor miRNAs, thereby decreasing the expression and function of oncogenic genes. Around 2008, Shan and his research group discovered a small molecule that could modulate the RNAi pathway intracellularly [95]. They used a reporter system that allowed them to monitor RNAi activity and performed a chemical screen of small molecules that enhanced RNAi. The system consisted of a cell line which stably expressed EGFP, which was infected with a lentivirus producing shRNA against EGFP. By searching for increases or decreases in fluorescence they could detect enhancers or inhibitors of the RNAi pathway. A total of 2,000 FDA-approved compounds were screened, and the results led to the identification of a small molecule named enoxacin (a fluoroquinolone) that actually enhanced the siRNA-mediated mRNA degradation with a median effect achieved at a 30uM concentration [95]. Its mechanism of action was found to be that the molecule enhances interactions between trans-activation-responsive region RNA binding protein (TRBP) and RNAs, thereby facilitating miRNA biogenesis [95]. Nevertheless no specific link to any disease was pointed out in their study. Further along the path, Melo and Calin et. al. discovered that enoxacin does enhance the production of miRNAs, but specifically so for tumor suppressor ones [41]. Treatment with enoxacin in human cell cultures and xenografted, orthotopic and metastatic mouse models revealed a TRBP-dependent and cancer-specific growth-inhibitory effect of this drug [41]. It promotes the activity of tumor suppressive miRNAs in a specific manner. These results revealed the importance not only of decreasing oncomiRs, but also of replenishing tumor suppressor miRNAs in order to recover the microRNAome that characterizes a normal cell.
In addition, other small molecule activators have also been identified via different mechanisms. With the aims of finding natural compounds which can be obtained at a low cost but at the same time were structurally diversified, Chen et. al. screened chemical blocks of compounds derived from photoreactions. By using a luciferase reporter vector, they identified a universal activator of miRNAs from the photoreaction products of naphthalene-1,4-dione with acetylenes. The small molecular activator demonstrated to significantly upregulate the expression of mature miRNAs including miR-1, miR-122 and other tumor-associated miRNAs in a non-specific manner (enhanced both: oncomiRs and tumor suppressors) [96].
6. Concluding Remarks
MiRNA expression screens that compare their levels in tumors versus normal tissues, have proven extremely useful in the past years because they have identified novel oncomiRs tightly involved in cancer. Therapeutic methods to inhibit miRNAs have demonstrated limited effectiveness regarding delivery mechanisms, as well as robust immunogenic responses [32, 33]. For years there has been a need not only of developing more effective miRNA-based therapeutics, but more so to validate them in a fast way, in order to move them from laboratory bench to the bedside. We recently envisioned that small molecule compounds could possess structural characteristics complementary to mature oncomiRs (or their upstream precursor transcripts), that could serve as an opportunity to achieve a more targeted therapeutic approach, one in which SMIRs and miRNAs would bind through a structure-specific interaction. To this date we have witnessed an extensive development of methods and techniques used to discover SMIRs. On one hand, we have validated rational computational approaches that are able to provide lead compounds that enable functional studies by predicting 3D structures of miRNAs or alternatively without even using them [57–86]. On the other, the screening techniques developed in vitro are only improving in efficiency, and represent a highly reliable approach to discover potential SMIRs as well [26, 49, 52–54]. There is absolutely no question; miRNAs are already considered a ‘magic bullet’ to target in cancer. Tumors are modernly being classified as “oncomiR-addicted” and are highly dependent on the miRNA-mediated silencing of crucial tumor suppressors proteins.
With the idea of using small molecules to inhibit oncomiRs, research has certainly aimed very high. It is very challenging to work looking for compounds that will potentially bind to a secondary/tertiary structure that we have not been able to crystallize just yet (this representing one of the biggest disadvantages). With predictions and in vitro techniques, several small molecules compounds have been found to somehow target the final levels of mature miRNA sequence. But even so, one of the hardest things to accomplish has been achieving a selective (miRNA-specific) inhibitory effect. Initially it was thought that by targeting the transcription of a specific miRNA-coding gene, the inhibitory effect would solely influence that miRNA. However, transcription factors can ubiquitously affect promoter regions of different genes; and even more, their expression levels/mechanisms of action might be distinct depending on the tissue type as well as the type of cancer. Thus, targeting transcription is simply not specific enough! A more refined strategy would be to find a compound that binds to the primary, precursor or mature sequence of a miRNA in a specific manner. In this sense, the challenge is to not affect proteins involved in the miRNA biogenesis in general, neither transcription factors nor other epigenetic regulators. The aim is to solely target the oncomiR of interest. In order to achieve this, one of the first steps would be to determine potent oncogenic miRNAs that have already been proven in the literature to be determinant molecules for the progression and advancement of cancer, for example miR-21. MiR-21 is over-expressed in multiple cancer types such as breast, ovaries, cervix, colon, lung, liver, brain, esophagus, prostate, pancreas and thyroid [16, 24, 25, 27, 28]. A SMIR that specifically targeted this miRNA would be useful and most likely applicable to patients of all of these cancer types, which most likely have a tumor development dependent on the expression levels of oncomiR-21, because of its role in negatively regulating tumor suppressive targets (PTEN, PDCD4 and RECK).
One of the disadvantages that need to be taken into consideration is that small molecule compounds can have off target effects in both: the tissue of interest or throughout the body. With this in mind, the discovery of novel SMIR’s need to undergo extensive in vitro as well as in vivo evaluation of possible side effects in order to determine if the benefits are greater than any of their undesired off target effects.
As recounted in this article, the number of validated SMIR hits is only beginning to increase, and out of the small molecules discovered, very few of them have been proven to directly bind to a miRNA or one of its precursor transcripts. Nevertheless, in particular cases where oncomiR over-expression is significantly influencing cancer development, even a non-specific could decrease cancer burden by targeting the tumor’s “oncomiR addiction”. Thus the majority of the findings in the field promise to have positive outcomes in non-coding RNA-based therapeutics.
Acknowledgments
Dr Calin is The Alan M. Gewirtz Leukemia & Lymphoma Society Scholar. Work in Dr. Calin’s laboratory is supported in part by the NIH/NCI grants 1UH2TR00943-01 and 1 R01 CA182905-01, Developmental Research Awards in Prostate Cancer, Multiple Myeloma, Leukemia (P50 CA100632) and Head and Neck (P50 CA097007) SPOREs, a SINF MDACC_DKFZ grant in CLL, a SINF grant in colon cancer, a Kidney Cancer Pilot Project, the Duncan Family Institutional Seed Funds, The Blanton-Davis Ovarian Cancer – 2013 Sprint for Life Research Award, the Laura and John Arnold Foundation, the RGK Foundation and the Estate of C. G. Johnson, Jr and by the CLL Global Research Foundation and a Department of Defense Breast Cancer Idea Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 2.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 3.Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- 4.Lai EC. microRNAs: runts of the genome assert themselves. Curr Biol. 2003;13:R925–936. doi: 10.1016/j.cub.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. Rna. 2003;9:112–123. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 8.Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 9.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 12.Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 13.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 14.Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer, Nature reviews. Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 15.Spizzo R, Nicoloso MS, Croce CM, Calin GA. SnapShot: MicroRNAs in Cancer. Cell. 2009;137:586–586. e581. doi: 10.1016/j.cell.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Lee CG. MicroRNA and cancer–focus on apoptosis. J Cell Mol Med. 2009;13:12–23. doi: 10.1111/j.1582-4934.2008.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer, Nature reviews. Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 18.Blenkiron C, Miska EA. miRNAs in cancer: approaches, aetiology, diagnostics and therapy. Hum Mol Genet. 2007;16(Spec No 1):R106–113. doi: 10.1093/hmg/ddm056. [DOI] [PubMed] [Google Scholar]
- 19.Bader AG, Brown D, Stoudemire J, Lammers P. Developing therapeutic microRNAs for cancer. Gene Ther. 2011;18:1121–1126. doi: 10.1038/gt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monroig PD, Calin GA. MicroRNA and Epigenetics: Diagnostic and Therapeutic Opportunities. Curr Pathobiol Rep. 2013;1:43–52. doi: 10.1007/s40139-013-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferdin J, Kunej T, Calin GA. Non-coding RNAs: identification of cancer-associated microRNAs by gene profiling. Technol Cancer Res Treat. 2010;9:123–138. doi: 10.1177/153303461000900202. [DOI] [PubMed] [Google Scholar]
- 22.Munker R, Calin GA. MicroRNA profiling in cancer. Clin Sci (Lond) 2011;121:141–158. doi: 10.1042/CS20110005. [DOI] [PubMed] [Google Scholar]
- 23.Di Leva G, Croce CM. The Role of microRNAs in the Tumorigenesis of Ovarian Cancer. Front Oncol. 2013;3:153. doi: 10.3389/fonc.2013.00153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Lou Y, Yang X, Wang F, Cui Z, Huang Y. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. International journal of molecular medicine. 2010;26:819–827. doi: 10.3892/ijmm_00000530. [DOI] [PubMed] [Google Scholar]
- 25.Yan LX, Huang XF, Shao Q, Huang MY, Deng L, Wu QL, Zeng YX, Shao JY. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. Rna. 2008;14:2348–2360. doi: 10.1261/rna.1034808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang Q, Deiters A. Small-molecule inhibitors of microrna miR-21 function. Angew Chem Int Ed Engl. 2008;47:7482–7484. doi: 10.1002/anie.200801555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sicard F, Gayral M, Lulka H, Buscail L, Cordelier P. Targeting miR-21 for the therapy of pancreatic cancer. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:986–994. doi: 10.1038/mt.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czyzyk-Krzeska MF, Zhang X. MiR-155 at the heart of oncogenic pathways. Oncogene. 2014;33:677–678. doi: 10.1038/onc.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zadran S, Remacle F, Levine RD. miRNA and mRNA cancer signatures determined by analysis of expression levels in large cohorts of patients. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:19160–19165. doi: 10.1073/pnas.1316991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parrella P, Barbano R, Pasculli B, Fontana A, Copetti M, Valori VM, Poeta ML, Perrone G, Righi D, Castelvetere M, Coco M, Balsamo T, Morritti M, Pellegrini F, Onetti-Muda A, Maiello E, Murgo R, Fazio VM. Evaluation of microRNA-10b prognostic significance in a prospective cohort of breast cancer patients. Molecular cancer. 2014;13:142. doi: 10.1186/1476-4598-13-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nature reviews Drug discovery. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nature reviews Cancer. 2011;11:59–67. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nature reviews Drug discovery. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nature reviews Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 36.Zhang S, Chen L, Jung EJ, Calin GA. Targeting microRNAs with small molecules: from dream to reality. Clin Pharmacol Ther. 2010;87:754–758. doi: 10.1038/clpt.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–347. doi: 10.1038/nbt.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy BL, Obad S, Bihannic L, Ayrault O, Zindy F, Kauppinen S, Roussel MF. Silencing of the miR-17~92 cluster family inhibits medulloblastoma progression. Cancer Res. 2013;73:7068–7078. doi: 10.1158/0008-5472.CAN-13-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindow M, Kauppinen S. Discovering the first microRNA-targeted drug. J Cell Biol. 2012;199:407–412. doi: 10.1083/jcb.201208082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 41.Melo S, Villanueva A, Moutinho C, Davalos V, Spizzo R, Ivan C, Rossi S, Setien F, Casanovas O, Simo-Riudalbas L, Carmona J, Carrere J, Vidal A, Aytes A, Puertas S, Ropero S, Kalluri R, Croce CM, Calin GA, Esteller M. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4394–4399. doi: 10.1073/pnas.1014720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang HW, Noland C, Siridechadilok B, Taylor DW, Ma E, Felderer K, Doudna JA, Nogales E. Structural insights into RNA processing by the human RISC-loading complex. Nat Struct Mol Biol. 2009;16:1148–1153. doi: 10.1038/nsmb.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 44.Deiters A. Small molecule modifiers of the microRNA and RNA interference pathway. AAPS J. 2010;12:51–60. doi: 10.1208/s12248-009-9159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–443. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 46.Pop MS, Stransky N, Garvie CW, Theurillat JP, Hartman EC, Lewis TA, Zhong C, Culyba EK, Lin F, Daniels DS, Pagliarini R, Ronco L, Koehler AN, Garraway LA. A small molecule that binds and inhibits the ETV1 transcription factor oncoprotein. Molecular cancer therapeutics. 2014;13:1492–1502. doi: 10.1158/1535-7163.MCT-13-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 49.Tan GS, Chiu CH, Garchow BG, Metzler D, Diamond SL, Kiriakidou M. Small molecule inhibition of RISC loading. ACS chemical biology. 2012;7:403–410. doi: 10.1021/cb200253h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shi Z, Zhang J, Qian X, Han L, Zhang K, Chen L, Liu J, Ren Y, Yang M, Zhang A, Pu P, Kang C. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013;73:5519–5531. doi: 10.1158/0008-5472.CAN-13-0280. [DOI] [PubMed] [Google Scholar]
- 51.Connelly CM, Thomas M, Deiters A. High-throughput luciferase reporter assay for small-molecule inhibitors of microRNA function. J Biomol Screen. 2012;17:822–828. doi: 10.1177/1087057112439606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bose D, Jayaraj GG, Kumar S, Maiti S. A molecular-beacon-based screen for small molecule inhibitors of miRNA maturation. ACS chemical biology. 2013;8:930–938. doi: 10.1021/cb300650y. [DOI] [PubMed] [Google Scholar]
- 53.Davies BP, Arenz C. A fluorescence probe for assaying micro RNA maturation. Bioorg Med Chem. 2008;16:49–55. doi: 10.1016/j.bmc.2007.04.055. [DOI] [PubMed] [Google Scholar]
- 54.Davies BP, Arenz C. A homogenous assay for micro RNA maturation. Angew Chem Int Ed Engl. 2006;45:5550–5552. doi: 10.1002/anie.200601332. [DOI] [PubMed] [Google Scholar]
- 55.Bose D, Jayaraj G, Suryawanshi H, Agarwala P, Pore SK, Banerjee R, Maiti S. The tuberculosis drug streptomycin as a potential cancer therapeutic: inhibition of miR-21 function by directly targeting its precursor. Angew Chem Int Ed Engl. 2012;51:1019–1023. doi: 10.1002/anie.201106455. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Shen J, Sun X, Li W, Liu G, Tang Y. Accuracy assessment of protein-based docking programs against RNA targets. Journal of chemical information and modeling. 2010;50:1134–1146. doi: 10.1021/ci9004157. [DOI] [PubMed] [Google Scholar]
- 57.Jamal S, Periwal V, Consortium O, Scaria V. Computational analysis and predictive modeling of small molecule modulators of microRNA. Journal of cheminformatics. 2012;4:16. doi: 10.1186/1758-2946-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin M, II, Cronin J, Dearden P, Gramatica YC, Martin R, Todeschini V, Consonni VE, Kuz’min R, Cramer R, Benigni C, Yang J, Rathman L, Terfloth J, Gasteiger A, Richard A. Tropsha, QSAR modeling: where have you been? Where are you going to? Journal of medicinal chemistry. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nature reviews Molecular cell biology. 2013;14:475–488. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 60.Puton T, Kozlowski LP, Rother KM, Bujnicki JM. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic acids research. 2014;42:5403–5406. doi: 10.1093/nar/gku208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puton T, Kozlowski LP, Rother KM, Bujnicki JM. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic acids research. 2013;41:4307–4323. doi: 10.1093/nar/gkt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato K, Hamada M, Asai K, Mituyama T. CENTROIDFOLD: a web server for RNA secondary structure prediction. Nucleic acids research. 2009;37:W277–280. doi: 10.1093/nar/gkp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zakov S, Goldberg Y, Elhadad M, Ziv-Ukelson M. Rich parameterization improves RNA structure prediction. Journal of computational biology : a journal of computational molecular cell biology. 2011;18:1525–1542. doi: 10.1089/cmb.2011.0184. [DOI] [PubMed] [Google Scholar]
- 64.Sato K, Kato Y, Hamada M, Akutsu T, Asai K. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics. 2011;27:i85–93. doi: 10.1093/bioinformatics/btr215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamada M, Sato K, Asai K. Improving the accuracy of predicting secondary structure for aligned RNA sequences. Nucleic acids research. 2011;39:393–402. doi: 10.1093/nar/gkq792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tabei Y, Asai K. A local multiple alignment method for detection of non-coding RNA sequences. Bioinformatics. 2009;25:1498–1505. doi: 10.1093/bioinformatics/btp261. [DOI] [PubMed] [Google Scholar]
- 67.Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harmanci AO, Sharma G, Mathews DH. TurboFold: iterative probabilistic estimation of secondary structures for multiple RNA sequences. BMC bioinformatics. 2011;12:108. doi: 10.1186/1471-2105-12-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 70.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reinharz V, Major F, Waldispuhl J. Towards 3D structure prediction of large RNA molecules: an integer programming framework to insert local 3D motifs in RNA secondary structure. Bioinformatics. 2012;28:i207–214. doi: 10.1093/bioinformatics/bts226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma S, Ding F, Dokholyan NV. iFoldRNA: three-dimensional RNA structure prediction and folding. Bioinformatics. 2008;24:1951–1952. doi: 10.1093/bioinformatics/btn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonikas MA, Radmer RJ, Altman RB. Knowledge-based instantiation of full atomic detail into coarse-grain RNA 3D structural models. Bioinformatics. 2009;25:3259–3266. doi: 10.1093/bioinformatics/btp576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jonikas MA, Radmer RJ, Laederach A, Das R, Pearlman S, Herschlag D, Altman RB. Coarse-grained modeling of large RNA molecules with knowledge-based potentials and structural filters. Rna. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rother M, Rother K, Puton T, Bujnicki JM. ModeRNA: a tool for comparative modeling of RNA 3D structure. Nucleic acids research. 2011;39:4007–4022. doi: 10.1093/nar/gkq1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, Blazewicz J, Adamiak RW. Automated 3D structure composition for large RNAs. Nucleic acids research. 2012;40:e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Detering C, Varani G. Validation of automated docking programs for docking and database screening against RNA drug targets. Journal of medicinal chemistry. 2004;47:4188–4201. doi: 10.1021/jm030650o. [DOI] [PubMed] [Google Scholar]
- 78.Moitessier N, Westhof E, Hanessian S. Docking of aminoglycosides to hydrated and flexible RNA. Journal of medicinal chemistry. 2006;49:1023–1033. doi: 10.1021/jm0508437. [DOI] [PubMed] [Google Scholar]
- 79.Pfeffer P, Gohlke H. DrugScoreRNA–knowledge-based scoring function to predict RNA-ligand interactions. Journal of chemical information and modeling. 2007;47:1868–1876. doi: 10.1021/ci700134p. [DOI] [PubMed] [Google Scholar]
- 80.Morley SD, Afshar M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using Ribodock. Journal of computer-aided molecular design. 2004;18:189–208. doi: 10.1023/b:jcam.0000035199.48747.1e. [DOI] [PubMed] [Google Scholar]
- 81.Ruiz-Carmona S, Alvarez-Garcia D, Foloppe N, Garmendia-Doval AB, Juhos S, Schmidtke P, Barril X, Hubbard RE, Morley SD. rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS computational biology. 2014;10:e1003571. doi: 10.1371/journal.pcbi.1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen L, Calin GA, Zhang S. Novel insights of structure-based modeling for RNA-targeted drug discovery. Journal of chemical information and modeling. 2012;52:2741–2753. doi: 10.1021/ci300320t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: combining techniques to model RNA-small molecule complexes. Rna. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kruger DM, Bergs J, Kazemi S, Gohlke H. Target Flexibility in RNA-Ligand Docking Modeled by Elastic Potential Grids. ACS medicinal chemistry letters. 2011;2:489–493. doi: 10.1021/ml100217h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nature chemical biology. 2011;7:553–559. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guilbert C, James TL. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. Journal of chemical information and modeling. 2008;48:1257–1268. doi: 10.1021/ci8000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moreira JN, Santos A, Simoes S. Bcl-2-targeted antisense therapy (Oblimersen sodium): towards clinical reality. Reviews on recent clinical trials. 2006;1:217–235. doi: 10.2174/157488706778250050. [DOI] [PubMed] [Google Scholar]
- 88.Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiological reviews. 2011;91:827–887. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 89.Young DD, Connelly CM, Grohmann C, Deiters A. Small molecule modifiers of microRNA miR-122 function for the treatment of hepatitis C virus infection and hepatocellular carcinoma. J Am Chem Soc. 2010;132:7976–7981. doi: 10.1021/ja910275u. [DOI] [PubMed] [Google Scholar]
- 90.Watashi K, Yeung ML, Starost MF, Hosmane RS, Jeang KT. Identification of small molecules that suppress microRNA function and reverse tumorigenesis. J Biol Chem. 2010;285:24707–24716. doi: 10.1074/jbc.M109.062976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chandrasekhar S, Pushpavalli SN, Chatla S, Mukhopadhyay D, Ganganna B, Vijeender K, Srihari P, Reddy CR, Janaki Ramaiah M, Bhadra U. aza-Flavanones as potent cross-species microRNA inhibitors that arrest cell cycle. Bioorganic & medicinal chemistry letters. 2012;22:645–648. doi: 10.1016/j.bmcl.2011.10.061. [DOI] [PubMed] [Google Scholar]
- 92.Vo DD, Staedel C, Zehnacker L, Benhida R, Darfeuille F, Duca M. Targeting the Production of Oncogenic MicroRNAs with Multimodal Synthetic Small Molecules. ACS chemical biology. 2014;9:711–721. doi: 10.1021/cb400668h. [DOI] [PubMed] [Google Scholar]
- 93.Velagapudi SP, Gallo SM, Disney MD. Sequence-based design of bioactive small molecules that target precursor microRNAs. Nature chemical biology. 2014;10:291–297. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Murata A, Fukuzumi T, Umemoto S, Nakatani K. Xanthone derivatives as potential inhibitors of miRNA processing by human Dicer: targeting secondary structures of pre-miRNA by small molecules. Bioorganic & medicinal chemistry letters. 2013;23:252–255. doi: 10.1016/j.bmcl.2012.10.108. [DOI] [PubMed] [Google Scholar]
- 95.Shan G, Li Y, Zhang J, Li W, Szulwach KE, Duan R, Faghihi MA, Khalil AM, Lu L, Paroo Z, Chan AW, Shi Z, Liu Q, Wahlestedt C, He C, Jin P. A small molecule enhances RNA interference and promotes microRNA processing. Nat Biotechnol. 2008;26:933–940. doi: 10.1038/nbt.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen X, Huang C, Zhang W, Wu Y, Chen X, Zhang CY, Zhang Y. A universal activator of microRNAs identified from photoreaction products. Chemical communications. 2012 doi: 10.1039/c2cc32157b. [DOI] [PubMed] [Google Scholar]
- 97.Setoyama T, Zhang X, Natsugoe S, Calin GA. microRNA-10b: a new marker or the marker of pancreatic ductal adenocarcinoma? Clin Cancer Res. 2011;17:5527–5529. doi: 10.1158/1078-0432.CCR-11-1477. [DOI] [PubMed] [Google Scholar]
- 98.Moriarty CH, Pursell B, Mercurio AM. miR-10b targets Tiam1: implications for Rac activation and carcinoma migration. J Biol Chem. 2010;285:20541–20546. doi: 10.1074/jbc.M110.121012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liao WT, Li TT, Wang ZG, Wang SY, He MR, Ye YP, Qi L, Cui YM, Wu P, Jiao HL, Zhang C, Xie YJ, Wang JX, Ding YQ. microRNA-224 promotes cell proliferation and tumor growth in human colorectal cancer by repressing PHLPP1 and PHLPP2. Clin Cancer Res. 2013;19:4662–4672. doi: 10.1158/1078-0432.CCR-13-0244. [DOI] [PubMed] [Google Scholar]
- 100.Zhang GJ, Zhou H, Xiao HX, Li Y, Zhou T. Up-regulation of miR-224 promotes cancer cell proliferation and invasion and predicts relapse of colorectal cancer. Cancer Cell Int. 2013;13:104. doi: 10.1186/1475-2867-13-104. [DOI] [PMC free article] [PubMed] [Google Scholar]