

Abstract
We recently proposed a DNA replication–based mechanism of fork stalling and template switching (FoSTeS) to explain the complex genomic rearrangements associated with a dysmyelinating central nervous system disorder in humans1. The FoSTeS mechanism has been further generalized and molecular mechanistic details have been provided in the microhomology-mediated break-induced replication (MMBIR) model that may underlie many structural variations in genomes from all domains of life2. Here we provide evidence that human genomic rearrangements ranging in size from several megabases to a few hundred base pairs can be generated by FoSTeS/MMBIR. Furthermore, we show that FoSTeS/MMBIR-mediated rearrangements can occur mitotically and can result in duplication or triplication of individual genes or even rearrangements of single exons. The FoSTeS/MMBIR mechanism can explain both the gene duplication-divergence hypothesis3 and exon shuffling4, suggesting an important role in both genome and single-gene evolution.
Structural variations of the human genome, including copy number variations (CNVs) and balanced inversions generated by genomic rearrangements, represent a significant source of genetic variation5-12. Rearrangements of the human genome can be categorized into two major groups on the basis of breakpoint analysis: recurrent rearrangements, occurring in multiple unrelated individuals with clustering of breakpoints and sharing a common rearrangement interval and size, and nonrecurrent rearrangements, with variable breakpoints13. The major mechanism underlying the former is nonallelic homologous recombination (NAHR)13,14; however, the mechanism(s) for nonrecurrent rearrangements are less well established. Nonhomologous end joining (NHEJ) is a candidate recombination-based mechanism to explain some nonrecurrent rearrangements13,15.
Using high-density oligonucleotide array comparative genomic hybridization (aCGH) to study the nonrecurrent PLP1 duplication rearrangements on the human X chromosome causing Pelizaeus-Merzbacher disease (PMD; MIM312080), we recently reported a high frequency of complex rearrangements1. These complexities were not revealed by previous assays including FISH, PFGE (pulsed-field gel electrophoresis) and BAC aCGH1. The findings were inconsistent with a simple recombination-based mechanism such as NAHR or NHEJ. We proposed a new DNA replication-based mechanism termed FoSTeS to parsimoniously explain the generation of these complex rearrangements in the human genome1.
According to the FoSTeS model1, during DNA replication, the active replication fork can stall and switch templates using complementary template microhomology to anneal and prime DNA replication. The involved forks can be separated by sizeable linear distances but may be adjacent or in close proximity in three-dimensional space, perhaps within replication factories16. The mechanism enables the joining or template-driven juxtaposition of different sequences from discrete genomic positions and can result in complex rearrangements1. Because FoSTeS is a DNA replication–based model, it is predicted to occur during mitosis1. Experimental evidence for the involvement of long-distance template switching during DNA replication in stress-induced CNV formation has been found in both Escherichia coli17 and human cells18. Similar replication-based models (discussed in refs. 1,13,19) including “serial replication slippage”20 have also been proposed for smaller complex rearrangements.
The FoSTeS mechanism has been further generalized in a replicative template-switch model that may underlie structural variations in genomes from all domains of life, the MMBIR model2. MMBIR is a molecularly defined model based on experimental observations from multiple model organisms2.
Although the FoSTeS and/or MMBIR mechanism can explain the generation of complex PLP1 duplications on the human X chromosome1,2, it is uncertain to what extent this DNA replication–based mechanism may contribute to nonrecurrent rearrangements causing other genomic disorders, structural variations and genome or single-gene evolution. We hypothesized that FoSTeS/MMBIR may represent a contributing mechanism for structural variations of essentially all sizes and complexities. To test this hypothesis, we studied nonrecurrent genomic rearrangements and their sequence complexities at two disease-associated loci on the short arm of human chromosome 17: the reciprocal Potocki-Lupski microduplication syndrome (PTLS; MIM610883)/Smith-Magenis microdeletion syndrome (SMS; MIM182290) region in 17p11.2 and the reciprocal Charcot-Marie-Tooth disease type 1A (CMT1A; MIM118220) duplication/hereditary neuropathies with liability to pressure palsies (HNPP; MIM162500) deletion region in 17p12. Rearrangements were ascertained by virtue of their conveyed clinical phenotypes.
The common ~3.7-Mb PTLS/SMS-associated region in 17p11.2 is bound by two direct low copy repeats (LCRs; SMS-REPs), between which NAHR can lead to the recurrent reciprocal duplication or deletion of this region (Fig. 1). In addition to these recurrent rearrangements, we identified 14 nonrecurrent PTLS-associated duplications that varied in size from 3.5 Mb to 19.6 Mb (Fig. 1, Table 1 and Supplementary Table 1 online), in which the underlying rearrangement mechanism is unknown. We used oligonucleotide aCGH to examine these 14 nonrecurrent duplications of 17p11.2. Notably, high-resolution (averaging one interrogating oligonucleotide per 500 bp) aCGH revealed additional smaller duplications, deletions and triplications in 8 of 14 (57%) affected individuals (Fig. 2a). To determine whether these complex 17p11.2 duplications represent de novo mutations caused by multiple FoSTeS/MMBIR events or show complexity because of superimposed inherited and simple de novo rearrangement, we also tested the available parental DNAs from six affected individuals (1229, 1458, 2211, 2695 and 2711 in this study; 2337 in our previous study21). Our results showed that all of the structural changes observed in these six complex rearrangements are de novo, consistent with FoSTeS/MMBIR generating the sequence complexity at the junctions.
Figure 1.
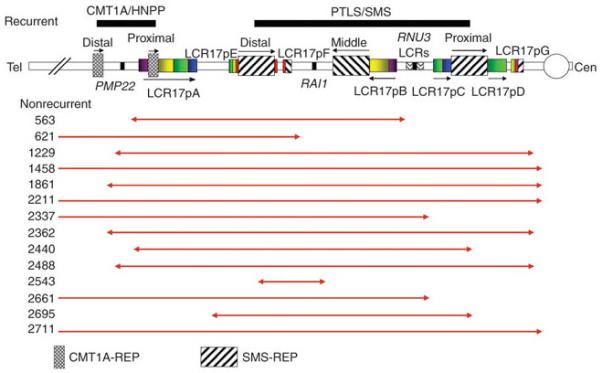
Nonrecurrent genomic duplications in 17p. Schematic representation of the short arm of human chromosome 17 with LCRs (not to scale). Recurrent reciprocal duplication/deletion between distal and proximal SMS-REPs are associated with PTLS/SMS, and recurrent reciprocal duplication/deletion between distal and proximal CMT1A-REPs are associated with CMT1A/HNPP. Fourteen nonrecurrent duplications of 17p11.2 with different sizes and breakpoints were examined in this study. The red horizontal lines depict the portion of the duplicated genomic intervals as determined by BAC aCGH, with arrowheads indicating the approximate breakpoints. Cen, centromere; Tel, telomere.
Table 1. Characteristics of the complex rearrangements in 17p.
| Scale | Subject | Rearrangement pattern | Sizea | Microhomology |
|---|---|---|---|---|
| Genomic | 2337b | del-nml-dup-nml-del-nml-dup-tri- dup-tri-dup |
19.6 Mb | NA |
| 621 | dup-tric | 15.5 Mb | CC, TTGGT | |
| 2711b | del-nml-dup | 11.3 Mb | NA | |
| 2211b | dup-nml-dup | 9.4 Mb | NA | |
| 1458b | tri-nml-dup | 9.0 Mb | NA | |
| 1229b | del-nml-dup | 6.5 Mb | ACCTTC | |
| 563 | dup-tri-dup | 5.6 Mb | CCTC, CTCCC | |
| 2695b | dup-tri-dup | 4.4 Mb | 31 bp (in AluY) | |
| 2543 | dup-nml-dup | 3.5 Mb | NA | |
| Genic | A2 | dup-tri-dup-nml-dup | 520 kb | NA |
| Exonic | A15 | del-nml-del-nml-dup | 8.7 kb | AACA, AACCT, AAG |
del, deletion; dup, duplication; nml, normal; tri, triplication; NA, not available.
The sizes of the entire genomic region involved in complex rearrangement are shown.
Entire complex rearrangement revealed by oligonucleotide aCGH is de novo based on studies of parents.
Oligonucleotide aCGH revealed a duplication-triplication rearrangement in subject 621. The sequence analysis of one of the breakpoint junction showed more complexities, some of which remain uncharacterized.
Figure 2.
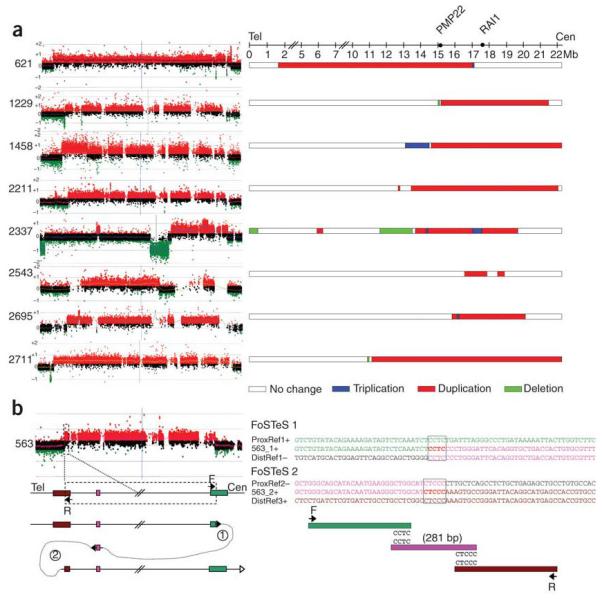
Complex 17p11.2 rearrangements revealed by oligonucleotide aCGH and sequence analysis and the underlying FoSTeS/MMBIR mechanism. (a) Oligonucleotide aCGH revealed complex rearrangements in 8 of 14 nonrecurrent 17p11.2 duplications. Array data are shown on the left. Size comparison of complex 17p11.2 rearrangements is shown on the right. Duplications are shown in red horizontal bars, deletions in green, triplications in blue, and no copy-number change in white. Figure is not drawn to scale and approximate positions are given in megabases (Mb). (b) Sequence analysis of breakpoint junctions revealed sequence complexity generated by the DNA replication FoSTeS/MMBIR mechanism in one aCGH-based apparently simple nonrecurrent 17p11.2 duplication (subject 563). This complex rearrangement was caused by two FoSTeS/MMBIR events with 4-bp and 5-bp microhomologies, respectively, at the junctions.
In subject 2543, the observed structural complexity could have been caused by two separate duplications. An alternative interpretation is that a single duplication occurred on an inversion allele that is not represented in the reference haploid human genome sequence (Supplementary Fig. 1 online). Such an inversion variation has been reported22 and can be frequent in populations, so it is more likely that the complexity in subject 2543 is generated by one simple duplication on an inversion allele rather than caused by multiple FoSTeS/MMBIR processes.
Long-range PCR assays were implemented for breakpoint sequence analysis. The proximal breakpoints of 9 out of 14 nonrecurrent 17p11.2 duplications are located in either long LCRs (>20 kb) or in the pericentromeric region (Fig. 1), making it difficult to specify a unique genomic position for their breakpoints. We successfully amplified one junction in 2 out of 6 (33%) array-based simple nonrecurrent 17p11.2 duplications and one or more junctions in 4 out of 8 (50%) complex 17p11.2 duplications.
Further complexity was detected in one of the array-based ‘simple’ duplications (Fig. 2b and Supplementary Fig. 2 online). In subject 563, two FoSTeS/MMBIR events (FoSTeS × 2) seem to have occurred, presumably reflecting the DNA template switching twice via the microhomologies (CCTC and CTCCC), yielding a complex rearrangement including a 281-bp triplication embedded within a duplication (Fig. 2b). Because the resolution of our custom CGH array is about 500 bp, such small rearrangements less than 1 kb are easily missed. By reevaluating the aCGH data, we found that only one interrogating oligonucleotide probe from our array was located in this 281-bp region and the log2(Cy5/Cy3) value was ~1, consistent with triplication. A microhomology of AT was found at the breakpoint of the simple duplication in subject 2661, consistent with either NHEJ or a single template switch event (FoSTeS × 1) (Supplementary Fig. 3 online). In the complex rearrangement of subject 621, microhomologies of CC and TTGGT were identified at the respective breakpoints or ‘join points’ for two apparent FoSTeS/MMBIR events (Supplementary Fig. 4 online). In subject 1229, a microhomology of ACCTTC was found at the junction of the 38.4-kb deletion (Supplementary Fig. 5 online). In the junction of the triplication identified in subject 2695, a 31-bp microhomology was identified between two AluY elements (Supplementary Fig. 6 online). Although this individual join point might potentially be explained by Alu-mediated recombination23,24, a simple recombination-based mechanism is inconsistent with the complexity (triplication embedded within a duplication) in subject 2695. Instead, the AluY elements could alternatively act in facilitating template switching and annealing via 31-bp microhomology between replication forks and restarting (priming) DNA replication in one of the multiple FoSTeS/MMBIR events that generated the complexity. At the deletion junction of subject 2711, a C nucleotide was found to be added at the breakpoint (Supplementary Fig. 7 online), an information scar consistent with an NHEJ event13.
In aggregate, the data from oligonucleotide aCGH and breakpoint sequence analyses show that complex genomic rearrangements are frequent (at least 8 of 14 nonrecurrent PTLS-associated duplication rearrangements; Fig. 2), can be explained by the FoSTeS/MMBIR mechanism, and may require different levels of resolution (oligonucleotide aCGH and/or sequencing) to fully visualize the complexity.
Having shown that FoSTeS/MMBIR could be responsible for 65% of nonrecurrent genomic rearrangements (40–45% of total cases) associated with an X-linked genomic disorder1, and 57% of nonrecurrent genomic rearrangements (~22% of total cases including recurrent ones) of the autosomal genomic disorder PTLS, we sought to determine whether such a mechanism could generate similar rearrangements affecting single genes. Our assay for investigating potential genic structural changes depended on PMP22, the dosage-sensitive gene responsible for CMT1A/HNPP25. This second cohort consisted of anonymous samples from Athena Diagnostics. In 2007, 17.4% (743 of 4,261) of the neuropathy-associated samples assayed for the CMT1A duplication and HNPP deletion by multiplex ligation-dependent probe amplification (MLPA) were shown to have potentially pathogenic CNVs involving PMP22. Notably, 0.9% (7/743) of these CNVs had unusual MLPA patterns inconsistent with NAHR-mediated recurrent CMT1A/HNPP rearrangements.
We verified these seven nonrecurrent PMP22 rearrangements by oligonucleotide aCGH and breakpoint sequence analysis. Subject A2 had a complex rearrangement encompassing B500 kb, in which the PMP22 gene had been triplicated, and this triplication is embedded within a duplication (Fig. 3 and Table 1). In addition, four deletions within PMP22 were also confirmed by oligonucleotide aCGH: subject A11 had a deletion of exon 3, subjects A10 and A15 had deletions affecting exon 4 and subject A21 had a deletion eliminating both exons 2 and 3 (Fig. 3). The remaining two small deletions (A12 and A14) were verified by PCR amplification and sequencing and were found to involve exon 2 (deletions of 212 bp and 9 bp, respectively, Supplementary Fig. 8 online). These exonic deletions varied in size from 9 bp to 16.8 kb (Table 1 and Supplementary Table 1).
Figure 3.
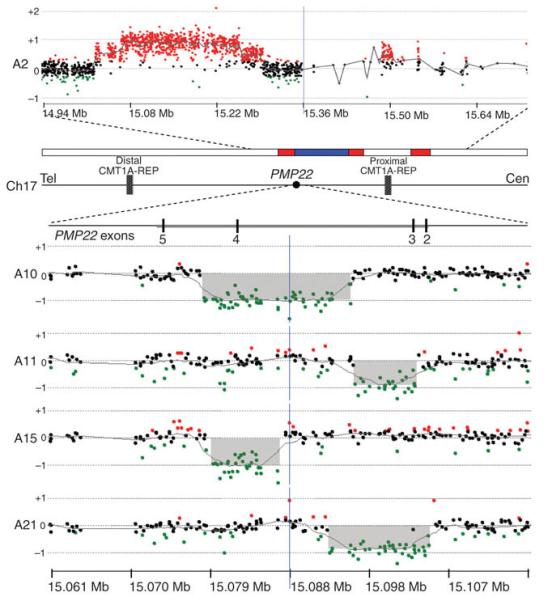
Nonrecurrent rearrangements involving the PMP22 gene were confirmed by oligonucleotide aCGH. The horizontal line in the middle of the figure depicts the 1.4-Mb CMT1A/HNPP region in 17p12, flanked by 24-kb 99% sequence identical LCRs termed CMT1A-REPs, and including the PMP22 gene with its four coding exons (numbered 2 through 5). The array data of one complex rearrangement (subject A2) are shown on the top. The PMP22 gene was triplicated in this subject and this triplication is embedded within a duplication. The array data of four small deletions (subjects A10, A11, A15, and A21) that only affect one or two exons of the PMP22 gene are shown below. Bottom, chromosome 17 genomic coordinates in megabases (NCBI Build 36).
We used PCR to amplify breakpoint junctions in all six nonrecurrent PMP22 deletions to study their underlying mechanisms. Although oligonucleotide aCGH did not detect complexity in subject A15, the breakpoint sequence analysis clearly revealed a complex rearrangement of one PMP22 exon, which can be explained by three FoSTeS/MMBIR events (FoSTeS × 3) with microhomologies of AACA, AACCT and AAG at the junctions, respectively (Fig. 4). To study the formation of this complex exonic deletion, we also examined the family members of subject A15. We identified the same heterozygous complex deletion in the affected sibling and apparent mosaicism for this complex deletion in the blood sample from the unaffected mother by PCR of the junction fragment (Fig. 4). The mosaicism was not detected by aCGH (Supplementary Fig. 8). These observations indicate that this complex deletion rearrangement is mitotic, consistent with a replicative error during mitotic DNA synthesis, as proposed for the FoSTeS/MMBIR model1,2.
Figure 4.
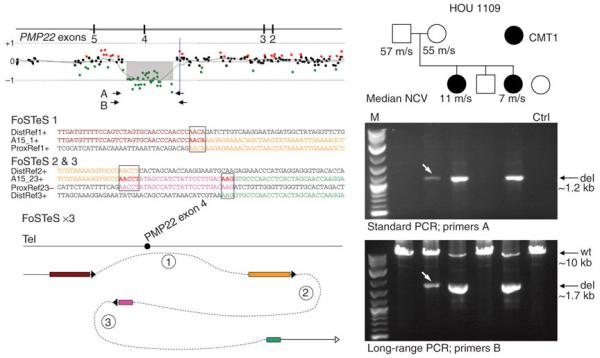
FoSTeS/MMBIR-mediated sequence complexity at the deletion breakpoint of two affected subjects (A15 and her sibling) and the mosaicism in the unaffected mother. Above left shows the PMP22 gene structure and aCGH data with the positions of the primers used to amplify the junction by standard (A) or long-range (B) PCR assays. Three FoSTeS/MMBIR events were revealed by sequence analysis of the complex deletion of PMP22 exon 4. Three microhomologies (4-bp, 5-bp and 3-bp, respectively) were found at the breakpoints. The model (not to scale) illustrating the underlying FoSTeS/MMBIR process is shown at the left bottom. In this CMT1 family (right, HOU 1109), PCR assays of the deletion breakpoint junction showed the heterozygous deletion in two subjects and the mosaicism in the unaffected mother (white arrow). In the standard PCR assay, only the deletion (del) allele can be amplified. The band for the mother is much weaker than those for two subjects. In long-range PCR assay, the smaller sized deletion allele is amplified greater than the wide-type (wt) allele in the subjects with heterozygous deletion. Only a weak band for the deletion allele is observed for the mother consistent with a low-level mosaicism for the junction specific to the complex exonic deletion rearrangement. Median nerve conduction velocity (NCV) in meters per second (m/s) is provided for the unaffected parents (normal > 38 m/s) and two subjects. M, marker; Ctrl, control.
Junction sequencing also revealed microhomologies in the five remaining simple deletions in subjects A10 (GATT), A11 (AC), A12 (GACG), A14 (GC) and A21 (C) (Supplementary Figs. 9 and 10 online). These findings of microhomology-mediated process suggest the possible involvement of single FoSTeS/MMBIR events in these simple exonic deletions, but may alternatively reflect microhomology-mediated end joining (MMEJ, also known as Ku-independent NHEJ15).
Our data show that the rearrangements generated by FoSTeS/MMBIR can be diverse in scale, from genomic duplications affecting megabases of the human genome to small deletions involving a single gene or only one exon (Table 1). These different sized rearrangements implicate FoSTeS/MMBIR in CNVs of all sizes and in the evolution of both human genomes and genes. Of note, a recent CNV study showed sequence complexities and microhomologies in 5 out of 23 (22%) CNV breakpoints12, which were consistent with two or more FoSTeS/MMBIR events (Supplementary Table 2 online). A genetic requirement for a replicative polymerase subunit Pol-32 has also been reported recently for the experimental formation of segmental duplications in yeast, whereas none of the tested recombination proteins was required26. Pol-32 has also been shown to be required for break-induced replication27 (BIR) in yeast28. These observations further support the contention that the replication-based mechanism may have an important role in CNV formation.
The gene duplication-divergence3 and exon shuffling4 hypotheses are proposed to be responsible for the origin of new genes encoding novel functions. However, the mechanisms driving gene duplication and exon shuffling remain a mystery, although recombination- and retrotransposition-based models have been proposed for the latter29. Our observations from analyzing the PMP22 rearrangements, especially the complex rearrangement in subject A15 caused by FoSTeS × 3 that resulted in a single exon rearrangement of PMP22 (Fig. 4), suggest a DNA replication–based mechanism as a potential cause of exon shuffling.
In addition to PMP22, many exonic rearrangements that are now retrospectively consistent with a FoSTeS/MMBIR-mediated exon shuffling have been reported. By searching the complex rearrangements in the Human Gene Mutation Database (see URLs section in Online Methods) and examining the available breakpoint sequences, complex exonic rearrangements potentially generated by multiple FoSTeS/MMBIR events or other replication-based models such as serial replication slippage20 were found in at least 17 genes (Supplementary Table 3 online). Furthermore, by reevaluating other experimental evidence for exon shuffling30,31, microhomologies were also observed at the rearrangement breakpoints, a finding that can alternatively be interpreted as consistent with the FoSTeS/MMBIR mechanism (FoSTeS × 1). Given the above, FoSTeS/MMBIR provides an innovative perspective for exploring human gene and genome evolution.
Our data suggest that the FoSTeS/MMBIR mechanism can cause complex rearrangements of different (genomic, genic and exonic) scales. The sequence complexities of nonrecurrent rearrangements can be frequent, although most were likely not visualized previously owing to the limited resolution of the former experimental approaches such as FISH, PFGE and BAC aCGH. Genome-wide approaches to CNV analyses using paired-end mapping and next-generation sequencing10,11 with short sequencing reads and bioinformatic filtering may grossly underestimate potential FoSTeS/MMBIR-mediated sequence complexity. We propose that FoSTeS/MMBIR may be a key mechanism for generating structural variation, particularly nonrecurrent CNV, of the human genome1,2,32,33. The observation of mosaicism for an apparent mitotically generated, FoSTeS/MMBIR-mediated, complex PMP22 rearrangement in the unaffected mother of two children with neuropathy suggests this mechanism can have implications for genetic counseling regarding recurrence risk.
ONLINE METHODS
Subjects
We analyzed 14 PTLS-affected individuals with nonrecurrent genomic duplications in 17p11.2, some of which have been described in our previous studies21,34,35. The parental DNAs of six individuals with PTLS were also examined to confirm the de novo complex rearrangements. In addition, seven anonymous CMT1 samples and one CMT1 family with nonrecurrent genic rearrangements of the PMP22 gene in 17p12 were examined. The PTLS samples and the CMT1 family were obtained with informed consent approved by the Institutional Review Board for Human Subject Research at Baylor College of Medicine. Anonymous genomic DNAs for the PMP22 rearrangements were provided by Athena Diagnostics. The control DNAs (NA10851 and NA15510) were obtained from Coriell Cell Repositories.
Oligonucleotide aCGH
We applied aCGH by using Agilent high-density oligonucleotide-based microarrays for investigating nonrecurrent rearrangements in 17p. By Agilent eArray system (see URLs section below), we designed a 4 × 44K custom array covering 17p with resolution of 500 bp to examine nonrecurrent 17p11.2 duplications and an 8 × 15K custom array to cover the CMT1A/HNPP region and its 1-Mb flanking segments with 300-bp resolution to study PMP22 rearrangements. After digestion with AluI and RsaI, the test DNAs were labeled with Cy5-dCTP and control DNAs were labeled with Cy3-dCTP by Invitrogen BioPrime Array CGH genomic labeling kit. Purification of labeling products, array hybridization, washing, scanning and data analysis were conducted by following Agilent oligonucleotide array-based CGH protocol (version 5.0).
PCR amplification and DNA sequencing analysis
To amplify breakpoint junction, we designed outward-facing primers for presumed tandem duplications or triplications and used inward-facing primers for deletions1. Different orientations and combinations of primers were also tested for complex rearrangements. Long-range PCR was conducted using TaKaRa LA Taq polymerase, whereas standard PCR used Qiagen HotStar Taq polymerase. PCR products that potentially contained breakpoint junctions (Supplementary Table 4 online) were submitted to SeqWright DNA Technology Services (Houston, Texas) for sequencing by the Sanger dideoxy method. DNA sequences were analyzed by comparing to reference sequences using the UCSC Genome Browser (see URLs section below).
URLs
Human Gene Mutation Database, http://www.hgmd.cf.ac.uk/ac/index.php; Agilent eArray, https://earray.chem.agilent.com/earray/; UCSC Genome Browser, http://genome.ucsc.edu/.
Supplementary Material
ACKNOWLEDGMENTS
We thank all participating subjects and families for their kind cooperation in the study. We also thank W. Bi, W. Gu, J.A. Lee and P. Stankiewicz for their critical reviews and C.M.B. Carvalho and M.A. Withers for their assistance. This work was supported in part by the Charcot Marie Tooth Association and the National Institute of Neurological Disorders and Stroke (NINDS, NIH).
Footnotes
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS
F.Z., M.K. and J.R.L. designed and interpreted the experiments; F.Z. and M.K. performed the experiments. A.M.C. provided clinical data; A.M.C., C.F.T. and S.D.B. provided subject samples; C.F.T. and S.D.B. provided MLPA data; F.Z. and J.R.L. wrote the manuscript.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
References
- 1.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 2.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohno S. Evolution by Gene Duplication. Springer-Verlag; Berlin: 1970. [Google Scholar]
- 4.Gilbert W. Why genes in pieces? Nature. 1978;271:501. doi: 10.1038/271501a0. [DOI] [PubMed] [Google Scholar]
- 5.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 6.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 7.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 9.Flores M, et al. Recurrent DNA inversion rearrangements in the human genome. Proc. Natl. Acad. Sci. USA. 2007;104:6099–6106. doi: 10.1073/pnas.0701631104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perry GH, et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. PathoGenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 15.Lieber MR. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 16.Kitamura E, Blow JJ, Tanaka TU. Live-cell imaging reveals replication of individual replicons in eukaryotic replication factories. Cell. 2006;125:1297–1308. doi: 10.1016/j.cell.2006.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006;2:e48. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arlt MF, et al. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009;84:339–350. doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F, Carvalho CMB, Lupski JR. Complex human chromosomal and genomic rearrangements. Trends Genet. doi: 10.1016/j.tig.2009.05.005. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JM, Chuzhanova N, Stenson PD, Ferec C, Cooper DN. Complex gene rearrangements caused by serial replication slippage. Hum. Mutat. 2005;26:125–134. doi: 10.1002/humu.20202. [DOI] [PubMed] [Google Scholar]
- 21.Vissers LE, et al. Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum. Genet. 2007;121:697–709. doi: 10.1007/s00439-007-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 23.Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003;73:823–834. doi: 10.1086/378594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen SK, et al. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lupski JR, Chance PF. Hereditary motor and sensory neuropathies involving altered dosage or mutation of PMP22: the CMT1A duplication and HNPP deletion. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy. Elsevier Science; Philadelphia: 2005. pp. 1659–1680. [Google Scholar]
- 26.Payen C, Koszul R, Dujon B, Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 28.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 29.Long M. Evolution of novel genes. Curr. Opin. Genet. Dev. 2001;11:673–680. doi: 10.1016/s0959-437x(00)00252-5. [DOI] [PubMed] [Google Scholar]
- 30.van Rijk AA, de Jong WW, Bloemendal H. Exon shuffling mimicked in cell culture. Proc. Natl. Acad. Sci. USA. 1999;96:8074–8079. doi: 10.1073/pnas.96.14.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones JM, et al. The mouse neurological mutant flailer expresses a novel hybrid gene derived by exon shuffling between Gnb5 and Myo5a. Hum. Mol. Genet. 2000;9:821–828. doi: 10.1093/hmg/9.5.821. [DOI] [PubMed] [Google Scholar]
- 32.Bi W, et al. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carvalho CM, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Potocki L, et al. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doco-Fenzy M, et al. The clinical spectrum associated with a chromosome 17 short arm proximal duplication (dup 17p11.2) in three patients. Am. J. Med. Genet. A. 2008;146:917–924. doi: 10.1002/ajmg.a.32195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.