

Abstract
Change in the host and/or human papillomavirus (HPV) DNA methylation profile is probably one of the main factors responsible for the malignant progression of cervical lesions to cancer. To investigate those changes we studied 173 cervical samples with different grades of cervical lesion, from normal to cervical cancer. The methylation status of nine cellular gene promoters, CCNA1, CDH1, C13ORF18, DAPK1, HIC1, RARβ2, hTERT1, hTERT2 and TWIST1, was investigated by Methylation Specific Polymerase Chain Reaction (MSP). The methylation of HPV18 L1-gene was also investigated by MSP, while the methylated cytosines within four regions, L1, 5’LCR, enhancer, and promoter of the HPV16 genome covering 19 CpG sites were evaluated by bisulfite sequencing. Statistically significant methylation biomarkers distinguishing between cervical precursor lesions from normal cervix were primarily C13ORF18 and secondly CCNA1, and those distinguishing cervical cancer from normal or cervical precursor lesions were CCNA1, C13ORF18, hTERT1, hTERT2 and TWIST1. In addition, the methylation analysis of individual CpG sites of the HPV16 genome in different sample groups, notably the 7455 and 7694 sites, proved to be more important than the overall methylation frequency. The majority of HPV18 positive samples contained both methylated and unmethylated L1 gene, and samples with L1-gene methylated forms alone had better prognosis when correlated with the host cell gene promoters’ methylation profiles. In conclusion, both cellular and viral methylation biomarkers should be used for monitoring cervical lesion progression to prevent invasive cervical cancer.
Introduction
The relationship between human papillomavirus (HPV) status and cervical cancer (CC) development is well established. There are at least 15 oncogenic or high-risk (HR)-HPV types, of which types 16 and 18 contribute up to 71% of cancer cases [1]. In contrast, DNA methylation of cellular and viral genes, resulting in silencing of gene expression, has been less studied in cervical cancerogenesis [2], even though it is one of the early events of carcinogenesis in many other cancer types [3]. DNA methylation often occurs on cytosines in CpG dinucleotides of genes for tumor suppressors, transcription factors and cell cycle regulators thus inhibiting gene expression by promoter silencing [4].
The CC occurrence is relatively rare compared with the widespread HPV infection; most women are probably infected with at least one if not several types of HPV during their sexual life, and only a small proportion will develop cervical precancerous lesions and then invasive cancer [5,6]. Factors associated with progression from precancerous cervical lesions, i.e. subclinical HPV infection to cancer are not well established. Both viral and host cells genes can be targeted by the DNA methylation machinery [4]. The fact that methylation of cellular genes can be detected even in samples taken up to seven years prior to cervical cancer diagnosis [7], was one of the reasons for this study. In addition, based on our observation the methyl groups are stable for years after DNA isolation and storage at -20°C and in line with the diagnosis taken upon sampling. Furthermore, the methylation of HPV genome could be a host defense mechanism for suppressing transcription of foreign DNA or strategy that the virus uses to maintain a long-term infection by immunosurveilance escape, or both [8,9]. We hypothesized that methylation of CpG sites in HPV16 and HPV18 genomes that represses transcription of HPV genes is one of the factors that are possibly relevant in cervical carcinogenic progression. Therefore, in this study, we focused on HPV16 and HPV18 positive cervical samples and analyzed the CpG methylation of HPV16 and HPV18 genomes as well as several key host cells genes.
There is a strong need to establish accurate diagnostic and prognostic methylation profile for a panel of genes that could discriminate between normal cervices, cervical lesions and cancer. After reviewing the literature focusing on this matter using similar sample pool and laboratory equipment [10–14], we have narrowed the choice to nine cellular genes to be investigated in this study: CCNA1 (CCNA1, cyclin A1), CDH1 (CDH1, cadherin 1, type 1, E-cadherin), C13ORF18 (KIAA0226L, KIAA0226-like), DAPK1 (DAPK1, death-associated protein kinase 1), HIC1 (HIC1, hypermethylated in cancer 1), RARβ2 (RARB, retinoic acid receptor, beta), hTERT1, hTERT2 (TERT, telomerase reverse transcriptase), and TWIST1 (TWIST1, twist family bHLH transcription factor 1). Promoter methylation of CCNA1, C13ORF18 and RARβ2 leads to disruption of cell cycle, methylation of CDH1, DAPK1, hTERT1, hTERT2 and HIC1 contribute to cancer progression by increasing proliferation, invasion, and/or metastasis while methylated TWIST1 means impaired cellular differentiation. The aim of this study was to determine the methylation profile of the host cell gene promoters and simultaneously the methylation status of HPV16 and 18 genomes to identify the best methylation diagnostic and prognostic biomarkers for cervical changes.
Results and Discussion
This is one of the few studies on a comprehensive collection number of cervical samples with well defined different cytological/histopathological diagnosis that analyzes DNA methylation of several cellular gene promoters as well as HPV16 and HPV18 genomes. Similar studies have been conducted by several authors but either on a limited number of cellular gene promoters or restricted number of analyzed HPV types [15–17].
There are some relevant literature data about methylation status of cellular genes, the following CCNA1, CDH1, C13ORF18, DAPK1, HIC1, RARβ2, hTERT1, hTERT2 and TWIST1 in cervical cancer cell lines CaSki, SiHa and HeLa [12,18–20]. Herein, methylation profiles of the nine gene promoters tested on cell lines CaSki and SiHa confirmed that all tested gene promoters were methylated in CC. In the cell line HeLa, only DAPK1 promoter was not methylated. Methylation profiles of aforementioned gene promoters were evaluated in different sample groups: samples with normal, LSIL/CIN1, HSIL/CIN2 and HSIL/CIN3 cytology, and histopathologically confirmed CC (Table 1). In the sample group with normal cytology there were 20.0% (8/40) HPV positive with one or more HR-HPVs, of which four HPV16. The majority of the samples were methylated in most tested gene promoters. CDH1 was found to be methylated in most cases (82.5%), followed by HIC1 (67.5%), RARβ2 (62.5%), and DAPK1 (55.0%). Since they have been found highly methylated in the normal cervix, these gene promoters are not ideal methylation biomarkers for cervical carcinogenesis. Contrary to our findings, Flatley et al. [21] found CDH1 promoter methylated in only 2.3% normal cervical samples. In contrast, hTERT1 was found unmethylated in all cases. The hTERT1 promoter seems to be a promising methylation biomarker being unmethylated in normal cervix. As far as this, there are contradictory findings of Iliopoulos et al. [22] who found the hTERT1 promoter methylated in a higher proportion 26.6% in normal cervix. The fact that the methylation profile of the hTERT1 promoter remains relatively low in cervical precursor changes, namely LSIL/CIN1 (12.5%), HSIL/CIN2 (5.1%) and HSIL/CIN3 (7.1%), and increases in CC samples (70.0%) favours our hypothesis (Table 1). Although, slightly less expressed similar methylation dynamics is observed with the hTERT2 and TWIST1 promoters. Accordingly, hypermethylation of all three gene promoters, hTERT1, hTERT2 and TWIST1 could be good cervical cancer biomarker, upon confirmation on larger sample pool. In the sample group with LSIL/CIN1 diagnosis there were 55.0% (22/40) HPV positive samples including six HPV16 and nine HPV18. The CDH1 and HIC1 promoters were methylated in the highest proportion of cases (75.0%, both), while TWIST1 was unmethylated in all cases. In contrast, Flatley et al. [21] have found CDH1 and HIC1 promoters methylated in much lower extent in samples with the same diagnosis, in 1.8% and 7.7% cases, respectively. In line with our finding, high HIC1 methylation (67.6%) and completely lack of methylation in TWIST1 promoter within the same diagnosis was found by Feng et al. [23]. HPV was detected in 32 of 40 samples (80.0%) with HSIL/CIN2 diagnosis, including six HPV16 and seven HPV18. Because of unsuccessful bisulfite conversion in one sample, the study group further included 39 HSIL/CIN2 samples, which are specifically classified as such by the Croatian cervical cytology classification, “Zagreb 2002” [24], in order to decipher fine differences between LSIL and HSIL. Here again, the majority of samples were methylated in most tested gene promoters, CDH1 being methylated in most cases (79.5%), while hTERT2 was unmethylated in all cases. Among 42 samples with HSIL/CIN3 diagnosis HPV was detected in 36 samples (85.7%), including 17 HPV16 and five HPV18. The highest number of methylated cases was observed in the CDH1 (76.2%), and the lowest in the hTERT2 (2.4%) promoter. Other authors reported methylation of the same genes within the same sample group but slightly different percentages of methylation per each gene promoter [21,23]. However, regarding the dynamic of the methylation profile of these two gene promoters, it seems that CDH1 promoter is constantly hypermethylated, while hTERT2 promoter is much less methylated from normal to high grade cervical lesions. In the CC group there were 81.8% (9/11) samples positive for HR-HPV, of which seven HPV16 and one HPV18. One CC sample, stage IV-A, highly necrotic, HPV-negative and unmethylated in either gene promoter was excluded from further analysis. Most of the investigated gene promoters were methylated in this sample group, ranging from 60.0% for DAPK1 to 100.0% for CDH1 promoter. Among CC diagnosis hTERT2 promoter was methylated in 60.0% of samples. In the case of hTERT higher gene expression is achieved by hypermethylation. In the study of Kumari et al. [25] treatment with 5-azacytidine and consequent demethylation of hTERT promoter led to reduction in gene expression. That was in contrast of what has been observed for conventional tumor suppressor genes. The possible explanation of this phenomenon is that methylation of hTERT promoters is heterogeneous and that only unmethylated alleles are expressed [12]. Indeed, that was observed in the study of Zinn et al. [26] where most of the cancer cell lines had hTERT promoter densely methylated, but CpGs around the transcription start site of a substantial number of hTERT alleles were unmethylated. In conclusion, herein observed hypomethylation in normal cervix means lower expression of hTERT gene and lower telomerase activity. This can explain better prognosis of patients with CC whose tumor cells lack hTERT promoter methylation [27]. The promoter methylation findings of other authors on CC samples correlate well with ours. The methylation varies over 65% for hTERT and DAPK promoters [22], 41% for DAPK, and less for CDH1, HIC and RARβ promoters [21]. For a higher number of promoters, hTERT, CDH1, DAPK, HIC1 and RARβ the methylation varies even more, ranging from 0 to 100% [27–29].
Table 1. Methylation of the host cells gene promoters in the study group (N = 171).
| Gene promoter | Number (%) of methylated samples in | ||||
|---|---|---|---|---|---|
| Normal cytology a | LSIL/CIN1 b | HSIL/CIN2 c | HSIL/CIN3 d | CC e | |
| CCNA1 | 5 (12.5) | 12 (30.0) | 9 (23.1) | 16 (38.1) | 8 (80.0) |
| CDH1 | 33 (82.5) | 30 (75.0) | 31 (79.5) | 32 (76.2) | 10 (100.0) |
| C13ORF18 | 3 (7.5) | 11 (27.5) | 10 (25.6) | 9 (21.4) | 9 (90.0) |
| DAPK1 | 22 (55.0) | 20 (50.0) | 18 (46.2) | 16 (38.1) | 6 (60.0) |
| HIC1 | 27 (67.5) | 30 (75.0) | 27 (69.2) | 24 (57.1) | 8 (80.0) |
| RARβ2 | 25 (62.5) | 14 (35.0) | 24 (61.5) | 26 (61.9) | 9 (90.0) |
| hTERT1 | 0 (0.0) | 5 (12.5) | 2 (5.1) | 3 (7.1) | 7 (70.0) |
| hTERT2 | 3 (7.5) | 5 (12.5) | 0 (0.0) | 1 (2.4) | 6 (60.0) |
| TWIST1 | 11 (27.5) | 0 (0.0) | 9 (23.1) | 9 (21.4) | 8 (80.0) |
aN = 40;
bN = 40;
cN = 39;
dN = 42;
eN = 10;
LSIL, low-grade squamous cell intraepithelial lesion; HSIL, high-grade squamous cell intraepithelial lesion; CIN, cervical intraepithelial neoplasia; CC, cervical cancer.
Herein, we have identified five main methylation biomarker candidates, CCNA1, C13ORF18, hTERT1, hTERT2, and TWIST1, whose promoter methylation status distinguishes CC from normal and HSIL/CIN3 samples (Table 2). The progression from HSIL/CIN1 to HSIL/CIN2 seems to be related to higher methylation of RARβ2 and TWIST1 promoter, although the statistical significance is lower for RARβ2 (Fisher’s exact test p = 0.024696) than TWIST1 (Fisher’s exact test p = 0.001030). The late stage of cervical changes, CC seems to be characterized by high methylation of hTERT1, hTERT2, and TWIST1 promoter, and in a lower extent in RARβ2 promoter. We also grouped cervical precursor lesions (LSIL/CIN1, HSIL/CIN2 and HSIL/CIN3) to evaluate differences between normal, cervical lesions, and CC. In such comparison CCNA1 and C13ORF18 promoters revealed to be statistically significantly hypermethylated in cervical precursor lesions compared to normal cervices (Fisher’s exact test p = 0.023742 and 0.022603, respectively). In addition, the methylation of these two potential biomarkers compared to normal cervices seems to be an early event in cervical cancerogenesis, with C13ORF18 promoter being statistically significantly methylated in LSIL/CIN1 and HSIL/CIN2 (Fisher’s exact test p = 0.036738 in both cases), while CCNA1 promoter being statistically significantly methylated in HSIL/CIN3 (Fisher’s exact test p = 0.010994). Therefore, statistically significant methylation differences between normal cervices, lesions and CC have been observed for five gene promoters: CCNA1, C13ORF18, hTERT1, hTERT2 and TWIST. In summary, those gene promoters represent possible good methylation biomarkers for detection of cervical changes and could be useful in clinical practice. It seems that in cervical cancerogenesis there is a cascade of DNA methylation. Primarily some gene promoters are methylated, such as C13ORF18 followed by CCNA1, and then others, namely hTERT1, hTERT2 and TWIST1 (Fig 1).
Table 2. Comparison of methylation status of gene promoter in cervical samples with different diagnosis—only statistically significant (Fisher’s exact test) increased methylation changes are presented.
| Hypermethylation (N) | Gene promoter | p |
|---|---|---|
| LSIL/CIN1 (40) vs. normal (40) | C13ORF18 | 0.036738 |
| HSIL/CIN2 (39) vs. normal (40) | C13ORF18 | 0.036738 |
| HSIL/CIN3 (42) vs. normal (40) | CCNA1 | 0.010994 |
| HSIL/CIN2 (39) vs. LSIL/CIN1 (40) | RARβ2 | 0.024696 |
| TWIST1 | 0.001030 | |
| HSIL (81) vs. normal (40) | CCNA1 | 0.042593 |
| C13ORF18 | 0.043737 | |
| LSIL + HSIL (121) vs. normal (40) | CCNA1 | 0.023742 |
| C13ORF18 | 0.022603 | |
| CC (10) vs. normal (40) | CCNA1 | 0.000086 |
| C13ORF18 | 0.000001 | |
| hTERT1 | 0.000001 | |
| hTERT2 | 0.000866 | |
| TWIST1 | 0.003709 | |
| CC (10) vs. HSIL/CIN3 (42) | CCNA1 | 0.031319 |
| C13ORF18 | 0.000107 | |
| hTERT1 | 0.000090 | |
| hTERT2 | 0.000067 | |
| TWIST1 | 0.000969 | |
| CC (10) vs. LSIL + HSIL (121) | CCNA1 | 0.003007 |
| C13ORF18 | 0.000070 | |
| RARβ2 | 0.042019 | |
| hTERT1 | 0.000017 | |
| hTERT2 | 0.000026 | |
| TWIST1 | 0.000031 |
LSIL, low-grade squamous cell intraepithelial lesion; HSIL, high-grade squamous cell intraepithelial lesion; CIN, cervical intraepithelial neoplasia; CC, cervical cancer.
Fig 1. Suggested algorithm for DNA methylation potential biomarkers use in clinical practice.
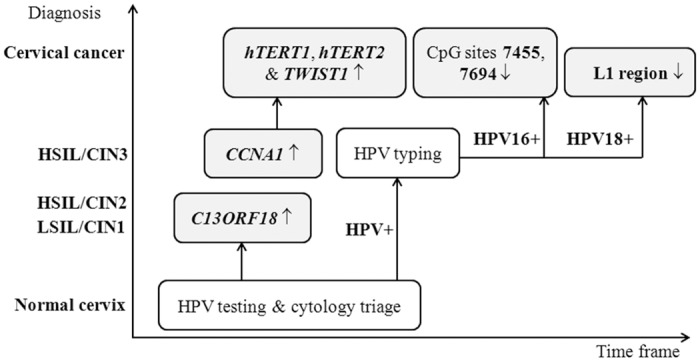
LSIL, low-grade squamous cell intraepithelial lesion; HSIL, high-grade squamous cell intraepithelial lesion; CIN, cervical intraepithelial neoplasia; ↑, hypermethylated; ↓, hypomethylated.
Other authors determined some of the selected genes as good biomarkers, but mostly as a single biomarker to distinguish a particular diagnosis. For instance, Yang et al. [13] selected CCNA1 and C13ORF18 as good methylation biomarkers to distinguish samples with CIN2+ diagnosis from normal samples. Kitkumthorn et al. [18] chose hypermethylation of CCNA1 promoter as biomarker for early diagnosis of invasive CC, De Wilde et al. [12] chose differentiated methylation of hTERT to diagnose CC, while Eijsink et al. [14] identified C13ORF18 and TERT, together with EPB41L3 and JAM3, as best methylation biomarkers for CC. Therefore, from the literature it is difficult to determine best methylation biomarkers. In a genome-wide study with Illumina Infinium HumanMethylation450 BeadChip method we confirmed the high methylation level of CCNA1, C13ORF18, hTERT1, hTERT2 and TWIST1 promoters in CC compared with the normal tissue. However, the unsupervised hierarchical clustering led to another set of genes as candidate biomarkers, RGS7, LHX8, STGALNAC5, TBX20, KCNA3, and ZSCAN18 [30], which were not evaluated herein. Furthermore, in the same study [30], using the mRNA expression data sets for external validation, we demonstrated a good coherence between DNA methylation data and the gene expression array. Therefore, we can conclude that DNA methylation is a good predictor of gene expression; consequently, DNA methylation tests are good and sufficient choice to improve diagnostics, staging and prognostics in clinical investigation because of the cost and time benefit over gene expression analysis.
We also investigated the CpG methylation status of HPV16 and HPV18 genomes in correlation with cytological/histopathological diagnosis and the methylation status of cellular genes. The methylation patterns of HPV16 genome has been analyzed by bisulfite sequencing for each of 19 CpG sites due to discrepant literature data. As expected, the methylation patterns of HPV16 genome in 12 samples with different diagnoses, correlated with those of the SiHa cell line (S1 Table). Most methylated CpGs in HPV16 genome of SiHa cells were detected in L1 and promoter region, while most of unmethylated CpGs in 5’LCR region and enhancer (Fig 2). Often both copies of HPV16, methylated and unmethylated were found in all diagnosis ranging from normal cervices to cervical cancer, in which the methylation profiles of CC samples were very similar to one of SiHa cell line. In almost all samples with normal cytology (N) the methylation profile was different from those in SiHa cells and CC samples. All three groups of samples with cervical precursor changes (LSIL/CIN1, HSIL/CIN2 and HSIL/CIN3) had very similar HPV16 methylation profiles. Because of this similarity of the methylation pattern, all three cervical precursor changes are shown together on Fig 3B. In addition, all CpG sites that were methylated and unmethylated in the same sample are presented as methylated (Fig 3). Our results support findings of Kalantari et al. [31] that HPV genome is normally hypermethylated and needs only one copy to be transcriptionally active. Moreover, as presented herein, for a good methylation biomarker it is more important to accurately determine the methylation percentage of specific CpG sites within HPV16 genome than the overall methylation in different sample groups. Herein, we observed that the cervical precancerous lesion group had the lowest overall methylation rate (33%), while samples with normal cytology and CC had 53% overall methylation rate, both. These results are partially in disagreement with the conclusions of other authors who claim that methylation of HPV16 is the lowest in normal and the highest in CC samples [17,32,33]. In this study, from the pool of 19 examined CpG sites of HPV16 genome, two seem to be the most significant, CpG 7455 (5’LCR) and CpG 7694 (enhancer), in which the complete lack of methylation was found in carcinoma samples and SiHa cell line, while they were highly methylated (100% of normal cervices and 83% of precursor lesions in CpG 7455) and moderately methylated (75% of normal cervices and 33% of precursor lesions in CpG 7694) in other sample groups. Further, CpGs at positions 7434 and 7461 were methylated in 50% of samples with normal cytology but no methylation was observed in precursor lesions, CC or SiHa cell line. In contrast, CpG 31 was methylated only in CC samples (50%) and SiHa cell line. CpGs at positions 37 and 52 were methylated in 75% samples with normal cytology and 100% of precursor lesions, CC and SiHa cell line. CpG 58 was methylated in 50% samples with normal cytology and 100% of precursor lesions and CC, as well as in SiHa cell line. The methylation was not observed in CpGs at positions 7535 and 7862 in neither one of the samples or SiHa cell line. In contrast, CpG 7682 was methylated in all examined samples, precursor cervical lesions, CC and SiHa cell line (Figs 2 and 3). Different methylation frequencies within HPV16 genome on the same CpG sites from our findings and different conclusions have been reported by Kalantari et al. [34]. However, similar findings on CpG methylation profiles in CC samples to ours were identified in the study of Bhattacharjee and Sengupta [35]. Thus, the complete lack or low CpG methylation in 5’LCR and enhancer, rather than in promoter region is crucial for HPV activity and consequently immortalization of host cells.
Fig 2. HPV16 methylation status of 19 CpG sites in different regions of the viral genome.
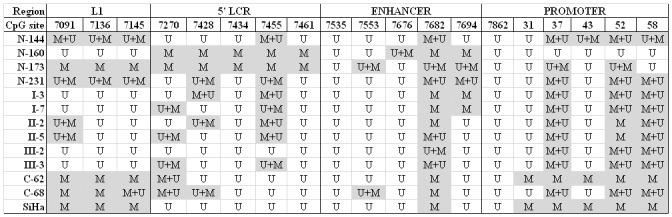
N, normal cytology; I, LSIL/CIN1 diagnosis; II, LSIL/CIN2 diagnosis; III, HSIL/CIN3 diagnosis; CC, cervical cancer; SiHa, cervical cancer cell line; M, methylated; U, unmethylated; M+U, methylated and unmethylated with predominantly methylated DNA; U+M, methylated and unmethylated with predominantly unmethylated DNA.
Fig 3. Percentage of methylation of 19 CpG sites in HPV16 genome.
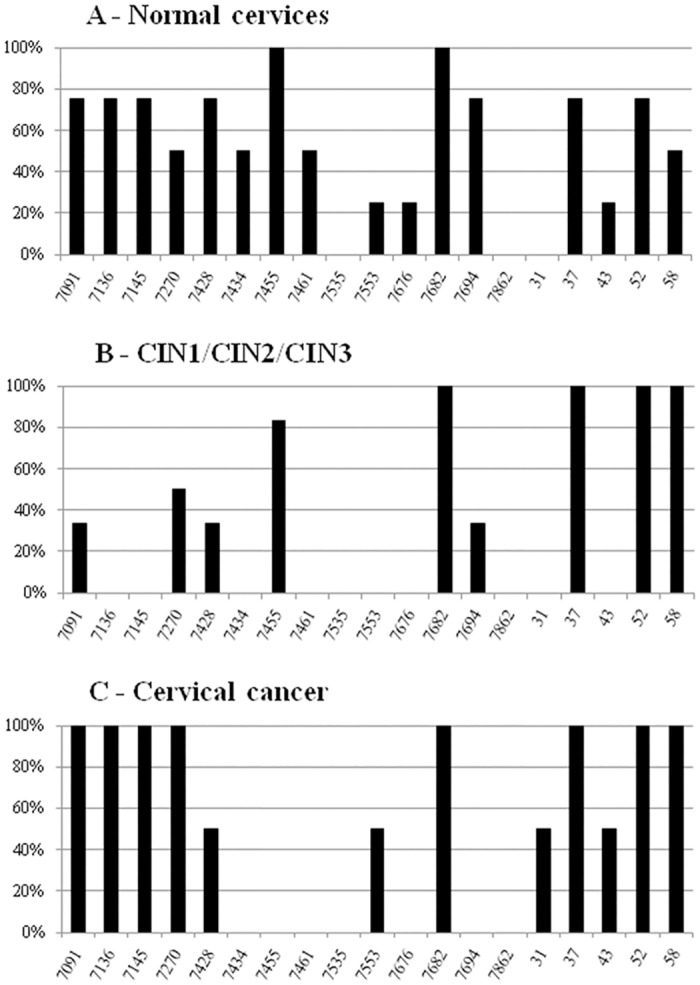
Normal cervices (N = 40); cervical precursor lesions, LSIL/CIN1, HSIL/CIN2 and HSIL/CIN3 (N = 121); cervical carcinoma samples (N = 10). CpG sites that were methylated and unmethylated in the same sample are presented herein as methylated.
Investigating HPV18 L1-gene methylation on 22 samples (S1 Table) by MSP method we observed that it correlates with the cytological/histopathological diagnosis, and can better predict disease prognosis. Particularly, most of the analyzed samples and HeLa cell line contained both methylated and unmethylated forms of HPV18 L1-gene (Table 3). The fact is that the L1-gene remains intact in cervical lesions and cancer samples, even when HPV genome integrates into a host cell [36]. Methylation of HPV18 L1-gene in cervical precursor lesions and CC samples was in line with the diagnosis and the methylation profile of HeLa cell line. Particularly, our samples containing only methylated form of HPV18 L1-gene had better prognosis; i.e. in those samples cellular genes were methylated at a lower extent and mostly those that have not been statistically proven as potential biomarkers. Turan et al. [37,38] also found both copies of HPV18 in HeLa cell line, with the excess of methylated form, while hypermethylation of HPV18 L1 in all carcinomas and lack of methylation in the majority of asymptomatic smears and low-grade lesions. Furthermore, Badal et al. [39] have declared that HPV18 genome hypermethylation in HeLa cell line and CC samples indicate that HPV is indeed targeted by cellular DNA methylation machinery that may affect late and early gene transcription.
Table 3. HPV18 L1-gene methylation status HPV18 in 22 samples with mild, moderate and high grade dysplasia, and cervical cancer.
| Diagnosis (N) | Number of samples (%) | ||
|---|---|---|---|
| only methylated | both methylated and unmethylated | only unmethylated | |
| LSIL/CIN1 (9) | 2 (22.2) | 7 (77.8) | 0 (0.0) |
| HSIL/CIN2 (6) | 1 (16.7) | 5 (83.3) | 0 (0.0) |
| HSIL/CIN3 (6) | 3 (50.0) | 2 (33.3) | 1 (16.7) |
| CC (1) | 0 (0.0) | 1 (100.0) | 0 (0.0) |
LSIL, low-grade squamous cell intraepithelial lesion; HSIL, high-grade squamous cell intraepithelial lesion; CIN, cervical intraepithelial neoplasia; CC, cervical cancer.
Future studies should validate the use of methylated HR-HPV as a predictive and/or diagnostic biomarker for risk of cervical cancer among HPV-positive women [32,40,41]. Particularly, MSP primers for HPV16 genome that cover CpGs 7455 and 7694 should be designed in order to avoid expensive and time consuming bisulfite sequencing, and, as such, simplify clinical applications. Our consideration of HPV genome methylation testing together with the cellular gene methylation testing as triage of women at high-risk for developing cervical cancer was also already discussed [10,27,42], but the question remains which HPV types, which HPV genome regions and which cellular genes to analyze. We believe that the results presented in this study, implied after confirmation on a larger number of samples, could resolve those concerns. Therefore, we would suggest that all HR-HPV-positive women get tested for methylation of the CCNA1, C13ORF18, hTERT1, hTERT2 and TWIST1 promoters as well as at least HPV16 CpG 7455 and 7694 sites, and HPV18 L1-gene methylation. The test results should be correlated, and in case of increased methylation of candidate biomarker genes and changed methylation profile of HPV16 and 18 genome women should be managed and subsequently monitored more thoroughly (Fig 1). In addition, based on actual knowledge on cellular and viral DNA methylation the possible use of demethylation drugs [27,43], such as DNA methyltransferases inhibitors could be considered as therapeutics but should be applied with great caution.
Material and Methods
Study group
The study group included 173 cervical specimens of women with normal cytology, precancerous lesions (squamous cell intraepithelial lesions, low-grade, LSIL and high-grade, HSIL), i.e. cervical intraepithelial neoplasia (CIN) grade 1 to 3, classified according to “Zagreb 2002”, that is in line with “NCI Bethesda System 2001” [24], and histopathologically confirmed CC [44]. From the pool of samples collected for regular diagnostics at the Rudjer Boskovic Institute, Zagreb, 40 samples with normal cytology, 40 with LSIL/CIN1, 40 with HSIL/CIN2 and 42 samples with HSIL/CIN3 diagnosis were taken randomly for methylation analyses. In addition, 11 CC (8 squamous cell carcinoma and 3 adenocarcinoma) samples were taken with cytobrush just before the surgical procedures.
Ethics Statement
Verbal patient/participant consent was obtained for each cervical specimen that was collected both for HPV diagnostic and research purposes. Written patient/participant consent was not necessary because each cervical sample is accompanied by the Laboratory service request forms, which have to be signed and approved by the practicing physician who is responsible for the verbal patient/participant consent (recorded by the clinician on the Laboratory service request forms). Thus, only samples of patients/participants who verbally agreed were included in this study. The relevant patient data (age, cytological diagnosis, HPV detection and typing result) and the extracted DNA were further encoded and processed anonymously in the laboratory. The study was achieved within the research project “Aberrant DNA methylation in HPV associated lesions” (Grant 098-0982464-2510 supported by the by the Croatian Ministry of Science, Education and Sport), which was approved as well as the sample collection procedure by the Ethical Board of the Rudjer Boskovic Institute, and the Ethical Board of Sisters of Mercy Hospital, and Clinical Hospital Center Zagreb that is in line with the Helsinki declaration (DoH/Oct2008).
DNA preparation
DNA from cervical samples was isolated on the BioRobot EZ1 (Qiagen, USA) according to the manufacturer’s instruction. After DNA extraction, the purified DNA was dissolved in 50–100 μl of tri-distillate sterile water and stored at -20°C until further analysis. Each DNA was analyzed spectrophotometrically and visually on 1% agarose gel electrophoresis, and visualized by UV irradiation on Alliance 4.7 (UVItec Cambridge, UK) [45].
HPV detection and genotyping
Detection and genotyping of HPVs was previously described by Milutin-Gasperov et al. [46] Briefly, three sets of consensus primers (PGMY09/PGMY11, L1C1/L1C2-1/L1C2-2 and GP5+/GP6+) were used for HPV detection, and type-specific primers for HPV genotyping of types 6/11, 16, 18, 31, 33, 45, 52 and 58. PCR products were analyzed by electrophoresis on 2% agarose gels stained with ethidium bromide.
Cell lines
Isolated DNAs from HPV16 positive CC cell lines, CaSki (ATTC CRL-1550) and SiHa (ATCC HTB-35), and HPV18 positive, HeLa (ATTC CCL-2), were used as positive controls of aberrant methylation profiles of cellular genes and respective HPV genomes. CaSki cells contain an integrated HPV16 genome (about 600 copies per cell) as well as sequences related to HPV18. SiHa cells contain an integrated HPV16 genome (1 to 2 copies per cell), while HeLa cells contain HPV18 sequences.
Bisulfite DNA conversion
DNA of cervical samples was modified with sodium bisulfite using EpiTect Bisulfite Kit (Qiagen, USA) and then purified according to manufacturer’s instructions. Briefly, all unmethylated cytosines are converted to uracil by bisulfite treatment, while methylated cytosines remain unchanged.
Host cell gene promoters’ methylation determination
The methylation profiles of CCNA1 (Chromosome 13: 36432177–3643232), CDH1 (Chromosome 16: 68737114–6873722), C13ORF18 (Chromosome 13: 46387345–4638746), DAPK1 (Chromosome 9: 87497883–87497981), HIC1 Chromosome 7: 19117831–19118031, RARβ2 (Chromosome 3: 25428191–2542842), hTERT1 (Chromosome 5: 1295019–1295259), hTERT2 (Chromosome 5: 1294824–1295014) and TWIST1 (Chromosome 7: 19117831–19118031) gene promoters were identified. Gene promoter methylation status was obtained by Methylation Specific Polymerase Chain Reaction (MSP) with primers specific for methylated forms of gene promoters. The same method was used for detection of unmethylated forms of the same gene promoters except for the C13ORF18 promoter. The positivity of samples with unmethylated forms of gene promoters served as the internal control, since cervical smears contain mixtures of cells with diverse methylation status of the host gene promoters [17]. The MSP method and conditions for the selected genes were already described by Yang et al. [13,47], Feng et al. [23], Kitkumthorn et al. [18], and Dessain et al. [19], but the conditions were modified in most cases herein. Briefly, 5 min denaturation at 95°C was followed by 45 cycles of denaturation at 95°C for 30s, annealing at 56–61°C for 30s, elongation at 72°C for 50s, and the final 7 min extension at 72°C. The annealing temperature varied with different primers: 56°C for hTERT1 (U, unmethylated primers) and TWIST1 (M, methylated primers), 58°C for CCNA1 (M) and CCNA1 (U), 59°C for CDH1 (U), C13ORF18 (M), DAPK1 (U), HIC1 (U), RARβ2 (M) and TWIST1 (U), and 60°C for DAPK1 (M), HIC1 (M), RARβ2 (U), hTERT1 (M), hTERT2 (M) and hTERT2 (U) and 61°C for CDH1 (M). MSP was carried out for each primer-set separately in a 20 μl volume containing 0.4 mM each dNTP, 1 μM each primer, 4 mM MgCl2, 5x Green GoTaq Flexi buffer, H2O and 0.08 μl GoTaq Hot Start Polymerase (Promega, USA). Bisulfite converted specimens’ DNA or methylated/unmethylated control DNA (EpiTect Control DNA, methylated and unmethylated, Qiagen, USA) were added into reactions in the final concentrations of 25 ng/μl. Aliquots of MSP products were analyzed on a 2% agarose gel.
HPV16 genome methylation determination
Bisulfite sequencing was used to identify the number and the position of the methylated cytosines inside four regions (L1, 5’LCR, enhancer, and promoter) of the HPV16 genome covering 19 CpG sites [34]. The PCR reactions were performed in a 50 μl volume containing 1 mM each dNTP, 2.5 μM each primer, 10 mM MgCl2, 5x Green GoTaq Flexi buffer, H2O and 0.2 μl GoTaq Hot Start Polymerase. Bisulfite converted specimens’ DNA and those isolated from CaSki cell line were added into reactions in the final concentration of 25 ng/μl. The PCR conditions consisted of: 5 min denaturation at 95°C followed by 45 cycles of 30s denaturation at 95°C, 30s annealing at 50°C (16msp4F/7R primers for enhancer), 53°C (16msp3F/3R primers for L1 and 5’LCR), or 55°C (16msp5F/8R primers for promoter), and 1 min extension at 68°C with the final 7 min extension at 68°C. The amplified products were analyzed by electrophoresis on 2% agarose gels and purified with Wizard PCR and Gel Cleanup kit (Promega, USA) according to manufacturer’s instructions. Purified PCR products for bisulfite modified DNA sequencing were analyzed at the Rudjer Boskovic Institute DNA Service, Zagreb, by the ABI PRISM 3100-Avant Genetic Analyzer (Applied Biosystems, USA).
HPV18 L1-gene methylation determination
MSP with primers specific for methylated and unmethylated forms of the L1-gene (between 7017 and 7140 positions) of HPV18 was used to amplify bisulfite converted DNA [37]. Sample or HeLa bisulfite converted DNA (positive control) in the final concentrations of 25 ng/μl was used in the MSP reaction that was carried out in a 20 μl volume containing 0.4 mM each dNTP, 1 μM each primer, 4 mM MgCl2, 5x Green GoTaq Flexi buffer, H2O and 0.08 μl GoTaq Hot Start Polymerase. MSP conditions were 5 min denaturation at 95°C, followed by 45 cycles of 95°C for 30s, 58°C for 30s and 68°C for 1 min with the final 7 min extension at 68°C. The amplified products were analyzed on a 2% agarose gels.
Statistics
Since the number of positive samples in the sample groups was small, the Fisher's exact test was used to determine the correlation of methylation frequencies between different diagnoses. Statistically significant differences were considered those with p-value<0.05. The data were processed in a computer program MedCalc (version 11.4.2).
Supporting Information
(DOC)
Acknowledgments
We are especially grateful to Jasminka Golubić Talić and Mario Glavač for their valuable technical assistance and to Pau Marc Muñoz Torres for his expertise in bioinformatics.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the Croatian Ministry of Science, Education and Sport (grant 098-0982464-2510). MG was also supported by the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement No 316289. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. De Sanjose S, Quint WG, Alemany L, Geraets DT, Klaustermeier JE, Lloveras B, et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 2010;11: 1048–1056. 10.1016/S1470-2045(10)70230-8 [DOI] [PubMed] [Google Scholar]
- 2. Virmani AK, Muller C, Rathi A, Zoechbauer-Mueller S, Mathis M, Gazdar AF. Aberrant methylation during cervical carcinogenesis. Clin Cancer Res. 2001;7: 584–589. [PubMed] [Google Scholar]
- 3. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21: 5400–5413. 10.1038/sj.onc.1205651 [DOI] [PubMed] [Google Scholar]
- 4. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16: 6–21. 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 5. Burchell AN, Rodrigues A, Moravan V, Tellier P-P, Hanley J, Coutlée F, et al. Determinants of prevalent human papillomavirus in recently-formed heterosexual partnerships: A dyadic-level analysis. J Infect Dis. 2014; 10.1093/infdis/jiu200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370: 890–907. 10.1016/S0140-6736(07)61416-0 [DOI] [PubMed] [Google Scholar]
- 7. Steenbergen RDM, Kramer D, Braakhuis BJM, Stern PL, Verheijen RHM, Meijer CJLM, et al. TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J Natl Cancer Inst. 2004;96: 294–305. [DOI] [PubMed] [Google Scholar]
- 8. Remus R, Kämmer C, Heller H, Schmitz B, Schell G, Doerfler W. Insertion of foreign DNA into an established mammalian genome can alter the methylation of cellular DNA sequences. J Virol. 1999;73: 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Badal V, Chuang LSH, Tan EH-H, Badal S, Villa LL, Wheeler CM, et al. CpG Methylation of Human Papillomavirus Type 16 DNA in Cervical Cancer Cell Lines and in Clinical Specimens: Genomic Hypomethylation Correlates with Carcinogenic Progression. J Virol. 2003;77: 6227–6234. 10.1128/JVI.77.11.6227-6234.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woodman CBJ, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7: 11–22. 10.1038/nrc2050 [DOI] [PubMed] [Google Scholar]
- 11. Wentzensen N, Sherman ME, Schiffman M, Wang SS. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol Oncol. 2009;112: 293–299. 10.1016/j.ygyno.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Wilde J, Kooter J, Overmeer R, Claassen-Kramer D, Meijer C, Snijders P, et al. hTERT promoter activity and CpG methylation in HPV-induced carcinogenesis. BMC Cancer. 2010;10: 271 10.1186/1471-2407-10-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang N, Eijsink JJH, Lendvai A, Volders HH, Klip H, Buikema HJ, et al. Methylation Markers for CCNA1 and C13ORF18 Are Strongly Associated with High-Grade Cervical Intraepithelial Neoplasia and Cervical Cancer in Cervical Scrapings. Cancer Epidemiol Biomarkers Prev. 2009;18: 3000–3007. 10.1158/1055-9965.EPI-09-0405 [DOI] [PubMed] [Google Scholar]
- 14. Eijsink JJH, Yang N, Lendvai A, Klip HG, Volders HH, Buikema HJ, et al. Detection of cervical neoplasia by DNA methylation analysis in cervico-vaginal lavages, a feasibility study. Gynecol Oncol. 2011;120: 280–283. 10.1016/j.ygyno.2010.10.029 [DOI] [PubMed] [Google Scholar]
- 15. Sun C, Reimers LL, Burk RD. Methylation of HPV16 genome CpG sites is associated with cervix precancer and cancer. Gynecol Oncol. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel DA, Rozek LS, Colacino JA, Van Zomeren-Dohm A, Ruffin MT, Unger ER, et al. Patterns of cellular and HPV 16 methylation as biomarkers for cervical neoplasia. J Virol Methods. 2012;184: 84–92. 10.1016/j.jviromet.2012.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalantari M, Osann K, Calleja-Macias IE, Kim S, Yan B, Jordan S, et al. Methylation of human papillomavirus 16, 18, 31, and 45 L2 and L1 genes and the cellular DAPK gene: Considerations for use as biomarkers of the progression of cervical neoplasia. Virology. 2014;448: 314–321. 10.1016/j.virol.2013.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kitkumthorn N, Yanatatsanajit P, Kiatpongsan S, Phokaew C, Triratanachat S, Trivijitsilp P, et al. Cyclin A1 promoter hypermethylation in human papillomavirus-associated cervical cancer. BMC Cancer. 2006;6: 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dessain SK, Yu H, Reddel RR, Beijersbergen RL, Weinberg RA. Methylation of the human telomerase gene CpG island. Cancer Res. 2000;60: 537 [PubMed] [Google Scholar]
- 20. Ivanova T, Petrenko A, Gritsko T, Vinokourova S, Eshilev E, Kobzeva V, et al. Methylation and silencing of the retinoic acid receptor-beta 2 gene in cervical cancer. BMC Cancer. 2002;2: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Flatley JE, McNeir K, Balasubramani L, Tidy J, Stuart EL, Young TA, et al. Folate Status and Aberrant DNA Methylation Are Associated With HPV Infection and Cervical Pathogenesis. Cancer Epidemiol Biomarkers Prev. 2009;18: 2782–2789. 10.1158/1055-9965.EPI-09-0493 [DOI] [PubMed] [Google Scholar]
- 22. Iliopoulos D, Oikonomou P, Messinis I, Tsezou A. Correlation of promoter hypermethylation in hTERT, DAPK and MGMT genes with cervical oncogenesis progression. Oncol Rep. 2009;22: 199–204. [DOI] [PubMed] [Google Scholar]
- 23. Feng Q, Balasubramanian A, Hawes SE, Toure P, Sow PS, Dem A, et al. Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst. 2005;97: 273–282. [DOI] [PubMed] [Google Scholar]
- 24. Ovanin-Rakić A, Pajtler M, Stanković T, Audy-Jurković S, Ljubojević N, Grubišić G, et al. The classification of cytologic findings of cervix uteri “Zagreb 2002”: The Modification of the “Zagreb 1990” and “NCI Bethesda System 2001” Classifications. Gynaecol Perinatol. 2003;12: 148–153. [Google Scholar]
- 25. Kumari A, Srinivasan R, Wig JD. Effect of c-MYC and E2F1 Gene Silencing and of 5-Azacytidine Treatment on Telomerase Activity in Pancreatic Cancer-Derived Cell Lines. Pancreatology. 2009;9: 360–368. 10.1159/000212094 [DOI] [PubMed] [Google Scholar]
- 26. Zinn RL, Pruitt K, Eguchi S, Baylin SB, Herman JG. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007;67: 194–201. 10.1158/0008-5472.CAN-06-3396 [DOI] [PubMed] [Google Scholar]
- 27. Dueñas-González A, Lizano M, Candelaria M, Cetina L, Arce C, Cervera E. Epigenetics of cervical cancer. An overview and therapeutic perspectives. Mol Cancer. 2005;4: 38 10.1186/1476-4598-4-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Narayan G, Arias-Pulido H, Koul S, Vargas H, Zhang FF, Villella J, et al. Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: its relationship to clinical outcome. Mol Cancer. 2003;2: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Widschwendter A, Gattringer C, Ivarsson L, Fiegl H, Schneitter A, Ramoni A, et al. Analysis of aberrant DNA methylation and human papillomavirus DNA in cervicovaginal specimens to detect invasive cervical cancer and its precursors. Clin Cancer Res. 2004;10: 3396–3400. [DOI] [PubMed] [Google Scholar]
- 30. Farkas SA, Milutin-Gašperov N, Grce M, Nilsson TK. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics. 2013;8: 1213–1225. 10.4161/epi.26346 [DOI] [PubMed] [Google Scholar]
- 31. Kalantari M, Lee D, Calleja-Macias IE, Lambert PF, Bernard HU. Effects of cellular differentiation, chromosomal integration and 5-aza-2′-deoxycytidine treatment on human papillomavirus-16 DNA methylation in cultured cell lines. Virology. 2008;374: 292–303. 10.1016/j.virol.2007.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mirabello L, Schiffman M, Ghosh A, Rodriguez AC, Vasiljevic N, Wentzensen N, et al. Elevated methylation of HPV16 DNA is associated with the development of high grade cervical intraepithelial neoplasia. Int J Cancer. 2013;132: 1412–1422. 10.1002/ijc.27750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lorincz AT, Brentnall AR, Vasiljević N, Scibior-Bentkowska D, Castanon A, Fiander A, et al. HPV16 L1 and L2 DNA methylation predicts high-grade cervical intraepithelial neoplasia in women with mildly abnormal cervical cytology: HPV16 DNA methylation in cervical lesions. Int J Cancer. 2013;133: 637–644. 10.1002/ijc.28050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kalantari M, Calleja-Macias IE, Tewari D, Hagmar B, Lie K, Barrera-Saldana HA, et al. Conserved methylation patterns of human papillomavirus type 16 DNA in asymptomatic infection and cervical neoplasia. J Virol. 2004;78: 12762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bhattacharjee B, Sengupta S. CpG methylation of HPV 16 LCR at E2 binding site proximal to P97 is associated with cervical cancer in presence of intact E2. Virology. 2006;354: 280–285. [DOI] [PubMed] [Google Scholar]
- 36. Matovina M, Sabol I, Grubisic G, Gasperov N, Grce M. Identification of human papillomavirus type 16 integration sites in high-grade precancerous cervical lesions. Gynecol Oncol. 2009;113: 120–127. 10.1016/j.ygyno.2008.12.004 [DOI] [PubMed] [Google Scholar]
- 37. Turan T, Kalantari M, Cuschieri K, Cubie HA, Skomedal H, Bernard HU. High-throughput detection of human papillomavirus-18 L1 gene methylation, a candidate biomarker for the progression of cervical neoplasia. Virology. 2007;361: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turan T, Kalantari M, Calleja-Macias IE, Cubie HA, Cuschieri K, Villa LL, et al. Methylation of the human papillomavirus-18 L1 gene: A biomarker of neoplastic progression? Virology. 2006;349: 175–183. [DOI] [PubMed] [Google Scholar]
- 39. Badal S, Badal V, Calleja-Macias IE, Kalantari M, Chuang LSH, Li BFL, et al. The human papillomavirus-18 genome is efficiently targeted by cellular DNA methylation. Virology. 2004;324: 483–492. [DOI] [PubMed] [Google Scholar]
- 40. Clarke MA, Wentzensen N, Mirabello L, Ghosh A, Wacholder S, Harari A, et al. Human Papillomavirus DNA Methylation as a Potential Biomarker for Cervical Cancer. Cancer Epidemiol Biomarkers Prev. 2012;21: 2125–2137. 10.1158/1055-9965.EPI-12-0905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wentzensen N, Sun C, Ghosh A, Kinney W, Mirabello L, Wacholder S, et al. Methylation of HPV18, HPV31, and HPV45 Genomes and Cervical Intraepithelial Neoplasia Grade 3. JNCI J Natl Cancer Inst. 2012;104: 1738–1749. 10.1093/jnci/djs425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cuzick J, Bergeron C, von Knebel Doeberitz M, Gravitt P, Jeronimo J, Lorincz AT, et al. New Technologies and Procedures for Cervical Cancer Screening. Vaccine. 2012;30: F107–F116. 10.1016/j.vaccine.2012.05.088 [DOI] [PubMed] [Google Scholar]
- 43. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429: 457–463. 10.1038/nature02625 [DOI] [PubMed] [Google Scholar]
- 44. Vrdoljak E, Haller H, Corusić A, Jelavić TB, Matković V, Strinić T, et al. [Clinical recommendations for diagnosing, treatment and monitoring of patients with uterine cervical cancer--Croatian Oncology Society and Croatian Society for Gynecology and Obstetrics as Croatian Medical Association units and Croatian Society of Gynecological Oncology]. Liječnički Vjesn. 2013;135: 225–229. [PubMed] [Google Scholar]
- 45. Sambrook J, Frisch EF, Maniatis T. Molecular cloning—A laboratory manual. 2nd edn Cold Sprind Harbor Laboratory, Cold Spring Harbor, New York; 1982. [Google Scholar]
- 46. Milutin-Gasperov N, Sabol I, Halec G, Matovina M, Grce M. Retrospective study of the prevalence of high-risk human papillomaviruses among Croatian women. Coll Antropol. 2007;31 Suppl 2: 89–96. [PubMed] [Google Scholar]
- 47. Yang HJ, Liu V, Wang Y, Tsang P, Ngan H. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer. 2006;6: 212 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.