

Latent and active aurone synthase purified from petals of C. grandiflora (cgAUS1) were crystallized. The crystal quality of recombinantly expressed latent cgAUS1 was significantly improved by co-crystallization with the polyoxotungstate Na6[TeW6O24] within the liquid–liquid phase-separation zone.
Keywords: aurone synthase, latent proenzyme, polyphenol oxidase, liquid–liquid phase separation, polyoxometalate
Abstract
Aurone synthase (AUS), a member of a novel group of plant polyphenol oxidases (PPOs), catalyzes the oxidative conversion of chalcones to aurones. Two active cgAUS1 (41.6 kDa) forms that differed in the level of phosphorylation or sulfation as well as the latent precursor form (58.9 kDa) were purified from the petals of Coreopsis grandiflora. The differing active cgAUS1 forms and the latent cgAUS1 as well as recombinantly expressed latent cgAUS1 were crystallized, resulting in six different crystal forms. The active forms crystallized in space groups P212121 and P1211 and diffracted to ∼1.65 Å resolution. Co-crystallization of active cgAUS1 with 1,4-resorcinol led to crystals belonging to space group P3121. The crystals of latent cgAUS1 belonged to space group P1211 and diffracted to 2.50 Å resolution. Co-crystallization of recombinantly expressed pro-AUS with the hexatungstotellurate(VI) salt Na6[TeW6O24] within the liquid–liquid phase separation zone significantly improved the quality of the crystals compared with crystals obtained without hexatungstotellurate(VI).
1. Introduction
Polyphenol oxidases are type III copper enzymes and include tyrosinases (EC 1.14.18.1) and catechol oxidases (EC 1.10.3.1). Tyrosinases catalyze the ortho-hydroxylation of monophenols and the oxidation of ortho-diphenols to ortho-quinones, whereas catechol oxidases lack the monophenolase activity and exclusively catalyze the oxidation reaction. Aurone synthase (AUS) is a homologue of PPOs that exclusively possesses diphenolase activity and catalyzes the oxidative conversion of chalcones to aurones (Miosic et al., 2013 ▶; Kaintz, Molitor et al., 2014 ▶; Molitor et al., 2015 ▶). Aurones are yellow pigments found in Asteraceae species, snapdragons (Antirrhinum majus L.) and carnations (Dianthus caryophyllus) (Harborne, 1967 ▶). PPOs are expressed as latent proenzymes which are activated by proteolytic cleavage of the C-terminal domain shielding the active site (King & Flurkey, 1987 ▶; Espín et al., 1999 ▶; Marusek et al., 2006 ▶; Flurkey & Inlow, 2008 ▶; Kaintz, Maraucher et al., 2014 ▶). Two crystal structures of the active form of plant catechol oxidases, from Ipomoea batatas (PDB entry 1bt3; Klabunde et al., 1998 ▶) and Vitis vinifera (PDB entry 2p3x; Virador et al., 2010 ▶), are available. Both catechol oxidases share ∼47% sequence identity with active cgAUS1. The crystallization of the first plant PPO possessing tyrosinase activity (Escobar et al., 2008 ▶; Zekiri, Molitor et al., 2014 ▶) has recently been reported (Zekiri, Bijelic et al., 2014 ▶). However, crystal structures of latent pro-PPOs, in which the active site is shielded by the C-terminal domain, are to date limited to fungal tyrosinases from Aspergillus oryzae (PDB entry 3w6q; Fujieda et al., 2013 ▶) and Agaricus bisporus (PDB entry 4oua; Mauracher et al., 2014a ▶). In the insect tyrosinase from Manduca sexta (PDB entry 3hhs; Li et al., 2009 ▶) the active site is shielded by an N-terminal domain. Each of these protyrosinases shares less than 17% sequence identity with latent cgAUS1.
The purification and characterization of latent (58.9 kDa; Fig. 1 ▶) and active (41.6 kDa; Fig. 1 ▶) cgAUS1 (A0A075DN54) from Coreopsis grandiflora has been reported by Molitor et al. (2015 ▶). Sequence analysis revealed that cgAUS1 is a member of the novel group 2 PPOs. An insertion (V237ANG240 in the cgAUS1 sequence) in a loop region near to the dinuclear copper centre is characteristic of this subgroup and might be involved in substrate docking (Molitor et al., 2015 ▶). Interestingly, phosphorylation or sulfation of a tyrosine residue with unknown function was found near to the insertion (Molitor et al., 2015 ▶). A disulfide linkage between the C-terminal domain, shielding the active site of latent PPOs, and the main core of cgAUS1 represents a novel structural feature of plant PPOs (Molitor et al., 2015 ▶). The latent cgAUS1 has to be cleaved at three different positions to result in the active form, which possesses a remaining C-terminal peptide (Fig. 1 ▶). The results of the kinetic characterization of active cgAUS1 suggest that aurone formation occurs at the chalcone aglycone stage (Molitor et al., 2015 ▶), which would constitute a differing aurone biosynthetic pathway in Asteraceae species in comparison to that described for A. majus (Plantaginaceae; Ono et al., 2006 ▶).
Figure 1.
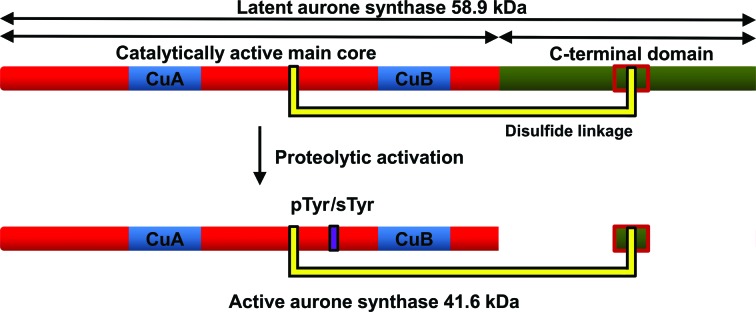
Schematic representation of the primary structure of latent and active cgAUS1. The catalytically active main core is coloured red, with the copper-binding sites coloured blue, and the C-terminal domain is shown in green. Owing to a disulfide linkage of the main core to the C-terminal domain (represented by yellow connectors), the proteolytically activated enzyme possesses a remaining C-terminal peptide (Molitor et al., 2015 ▶). The tyrosine residue Tyr230 of active cgAUS1 is partially phosphorylated or sulfated (displayed by a violet rectangle)
2. Materials and methods
2.1. Enzyme purification
The preparation of active cgAUS1 (designated cgAUS1-a1; sample 1 in Molitor et al., 2015 ▶) and latent cgAUS1 (designated cgAUS1-ln; sample 5 in Molitor et al., 2015 ▶) from petals of C. grandiflora has been described in Molitor et al. (2015 ▶). Another purification batch, starting from approximate 9 kg of frozen petal tissue, yielded 2.64 mg of highly purified active enzyme (designated cgAUS1-a2). The homogeneity of the protein samples was verified by mass determination by means of ESI-Q-TOF MS (Fig. 2 ▶; Molitor et al., 2015 ▶). The purification of recombinantly expressed cgAUS1 (designated cgAUS1-lr) has been described in Kaintz, Molitor et al. (2014 ▶). The two active forms (cgAUS1-a1 and cgAUS1-a2) were stored at a concentration of 6 mg ml−1, the latent form originating from the natural source (cgAUS1-ln) at a concentration of 10 mg ml−1 and the recombinantly expressed latent cgAUS1 (cgAUS1-lr) at a concentration of 14 mg ml−1 in 10 mM sodium acetate buffer pH 5.0 at 4°C for crystallization experiments.
Figure 2.

ESI-Q-TOF mass spectrum of active cgAUS1-a2 and comparison with cgAUS1-a1. (a) Entire mass spectrum of cgAUS1-a2. (b) Magnified mass spectra of cgAUS1-a2 (coloured red) and cgAUS1-a1 (coloured black) in charge state z = 39.
2.2. Mass determination
Electrospray ionization mass spectrometry (ESI-MS) of purified active cgAUS1-a2 was performed on a nanoESI-QTOF mass spectrometer (maXis 4G UHR-TOF, Bruker) using 20 µl protein solution with a concentration of approximately 1 µg µl−1. Buffer exchange to 10 mM ammonium acetate pH 5.0 was performed by ultrafiltration. Just prior to the measurements, acetonitrile was added to a final concentration of 25%(v/v) and formic acid was added to a final concentration of 0.05%(v/v).
2.3. Synthesis of Na6[TeW6O24]·22H2O
The hydrated sodium salt of hexatungstotellurate(VI) was synthesized according to a modified procedure (Roy & Mishra, 1978 ▶; Schmidt et al., 1986 ▶) and is described in Mauracher et al. (2014b ▶).
2.4. Crystallization
Four different cgAUS1 samples were crystallized under several conditions: active cgAUS1-a1, active cgAUS1-a2, the latent proenzyme purified from the natural source (cgAUS1-ln) and the recombinantly expressed latent proenzyme (cgAUS1-lr).
Initial crystallization screens for active cgAUS1-a1 were performed with the Crystallization Basic Kit for Proteins from Sigma–Aldrich at 20°C by the hanging-drop vapour-diffusion technique in 15-well EasyXtal plates (Qiagen; 500 µl reservoir solution) at 293 K. Screen condition No. 9 (0.2 M ammonium acetate, 0.1 M sodium citrate pH 5.6, 30% PEG 4000) resulted in a hit. Owing to the low quality of the obtained crystals, an intensive screen for additives was necessary. The final crystallization condition (crystallization condition A in Table 1 ▶) led to high-quality single crystals of cgAUS1-a1 and cgAUS1-a2. Crystals appeared within 1 d and grew to their final dimensions (up to 500 µm in length) within 6 d (Fig. 3 ▶ a). To investigate the reaction mechanism of PPOs, active cgAUS1-a2 was co-crystallized with 1,4-resorcinol, an inhibitor of PPOs and a suicide substrate for tyrosinase (Stratford et al., 2013 ▶), which led to another crystal form (crystallization condition B in Table 1 ▶; Fig. 3 ▶ b).
Table 1. Crystallization of active and latent cgAUS1.
| Crystallization condition (resulting data set) | A (1, 2) | B (3) | C (4) | D (5) | E (6) | F † (7) |
|---|---|---|---|---|---|---|
| Protein sample | cgAUS1-a1, cgAUS1-a2 | cgAUS1-a2 | cgAUS1-ln | cgAUS1-lr | cgAUS1-lr | cgAUS1-lr |
| Protein concentration (mgml1) | 36 | 6 | 10 | 14 | 14 | 14 |
| Composition of reservoir solution | 2325% PEG 4000, 500mM NaCl, 100mM Na formate, 50mM Na citrate pH 6.4 | 22% PEG 4000, 500mM NaCl, 100mM Na formate, 50mM Na citrate pH 6.4, 100mM 1,4-resorcinol | 15% PEG 4000, 100mM MgCl2, 60mM Na citrate pH 7.4 | 15% PEG 4000, 100mM MgCl2, 60mM Na citrate pH 7.4 | 12% PEG 4000, 60mM Na citrate pH 6.4, 1mM Na6[TeW6O24] | 15% PEG 4000, 60mM Na citrate pH 5.0, 20% glycerol, 1mM Na6[TeW6O24] |
| Volume of drop (l) | 2, 3 | 2 | 2 | 2 | 2 | 2 |
| Ratio of drop (protein:reservoir) | 1:1; 1:2 | 1:1 | 1:1 | 1:1 | 1:1 | 1:1 |
| Cryoprotectant solution | 40% PEG 4000, 15% glycerol, 20mM Na citrate pH 6.4 | 40% PEG 4000, 15% glycerol, 20mM Na citrate pH 6.4, 100mM 1,4-resorcinol | 40% PEG 4000, 15% glycerol, 20mM Na citrate pH 7.4 | 40% PEG 4000, 15% glycerol, 20mM Na citrate pH 7.4 | 40% PEG 4000, 15% glycerol, 20mM Na citrate pH 6.4 | 40% PEG 4000, 15% glycerol, 20mM Na citrate pH 5.0, 5mM H2O2 |
The cryoprotectant solution contained hydrogen peroxide to generate the oxy-form of the dinuclear copper site.
Figure 3.

Typical crystals of active, latent and recombinantly expressed cgAUS1. (a) Crystals of cgAUS1-a1 and cgAUS1-a2 obtained by applying crystallization condition A (data sets 1 and 2). (b) Crystals of cgAUS1-a2 co-crystallized with 1,4-resorcinol (crystallization condition B, data set 3). (c) Liquid–liquid phase separation of latent cgAUS1-lr after pre-equilibration. (d) Crystals of cgAUS1-ln (crystallization condition C, data set 4). (e) Crystals of cgAUS1-lr (crystallization condition D, data set 5). (f) Crystals of cgAUS1-lr co-crystallized with [TeW6O24]6− (crystallization condition E, data set 6).
Condition No. 9 (0.2 M ammonium acetate, 0.1 M sodium citrate pH 5.6, 30% PEG 4000) of the Crystallization Basic Kit for Proteins (Sigma–Aldrich) resulted in fully precipitated latent cgAUS1-ln. However, redissolving the precipitate in six equivalents of water and subsequent re-equilibration produced needles and bunches of needle-like crystals resembling sea urchins. Stepwise optimizations led to rod-like crystals (Table 1 ▶, crystallization condition C; Fig. 3 ▶ d). Notably, the protein started to precipitate several hours after crystallization setup and subsequently redissolved with the formation of a liquid–liquid phase-separation (LLPS) zone (Fig. 3 ▶ c). The crystallization of the recombinantly expressed enzyme (cgAUS1-lr) using crystallization condition D (Table 1 ▶) resulted in pronounced LLPS and resulted in spherulites, over nucleation and needles. After four to eight weeks, single crystals appeared (Fig. 3 ▶ e). Optimization of the crystallization conditions led to the substitution of 100 mM magnesium chloride by 1 mM hexatungstotellurate(VI) (Table 1 ▶, crystallization condition E). Crystals appeared within 2 d and the crystals grew to final dimensions of up to 500 × 50 × 50 µm within several days (Fig. 3 ▶ f). Lowering the pH of the crystallization buffer to 5.0 (Table 1 ▶, crystallization condition F) improved the stability of the crystals significantly and also enhanced crystal growth. Crystals were soaked for 30–45 min in cryoprotectant solution containing 5 mM hydrogen peroxide to generate the oxy-form of the dinuclear copper centre.
2.5. Data collection and processing
Crystals were transferred into a drop of cryoprotectant solution (Table 1 ▶) and subsequently flash-cooled in liquid nitrogen. X-ray diffraction experiments were performed using synchrotron radiation at ESRF, Grenoble, France, DESY/EMBL, Hamburg, Germany and Diamond Light Source, Oxford, England. The data sets were processed with XDS (Kabsch, 2010a ▶,b ▶). The symmetry was confirmed with POINTLESS (Evans, 2011 ▶) and Matthews parameters were calculated using MATTHEWS_COEF (Matthews, 1968 ▶), both implemented in the CCP4 suite (Winn et al., 2011 ▶).
3. Results and discussion
It has been reported that crude enzyme extracts from C. grandiflora contain several active aurone synthase forms that caused several overlapping peaks during cation-exchange and anion-exchange chromatography (Molitor et al., 2015 ▶). These forms were caused by a combination of nonspecific cleavage of cgAUS1 and phosphorylation/sulfation of a tyrosine residue. Mass-spectrometric analysis of intact enzyme samples revealed that the portion of phosphorylation/sulfation was approximately identical in the different cleaved forms, as indicated by comparable intensity ratios of ∼1.4:1 for unmodified and modified cgAUS1 forms (Molitor et al., 2015 ▶). However, the newly purified enzyme (cgAUS1-a2) possessed an intensity ratio between species A (unmodified) and B (phosphorylated/sulfated) of ∼0.14:1, indicating a significantly higher level of phosphorylation or sulfation (Fig. 1 ▶ b). As the petal tissue (harvested from June to September 2011) was stored in 0.5 kg packages and no enrichment of phosphorylated/sulfated forms had been observed within the different cleaved active cgAUS1 forms (Molitor et al., 2015 ▶), it is very likely that the higher modification level is caused by unknown environmental influences.
As the initially obtained crystals of active cgAUS1-a1 were strongly intergrown, an intensive additive screen was necessary. An optimized crystallization condition at high ionic strength resulted in two differing crystal forms of comparable quality (Table 2 ▶, data sets 1 and 2). The differing modification levels of cgAUS1-a1 and cgAUS1-a2 had no effect on the crystallization of these enzyme samples. Co-crystallization of active cgAUS1-a2 with 100 mM 1,4-resorcinol resulted in smaller crystals belonging to space group P3121 (Table 2 ▶, data set 3).
Table 2. Data collection and processing.
Values in parentheses are for the outer shell.
| Data set | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Protein sample | cgAUS1-a1 | cgAUS1-a2 | cgAUS1-a2 | cgAUS1-ln | cgAUS1-lr | cgAUS1-lr | cgAUS1-lr |
| Crystallization condition | A | A | B | C | D | E, native | F, soaked in H2O2 |
| Diffraction source | P14, EMBL, DESY | I04-1, Diamond | P11, DESY | ID23, ESRF | P11, DESY | P11, DESY | I04-1, Diamond |
| Wavelength () | 1.23953 | 0.9173 | 1.0247 | 0.972499 | 1.0247 | 1.0247 | 0.9173 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Detector | PILATUS 6M-F | PILATUS 2M | PILATUS 6M | PILATUS 6M | PILATUS 6M | PILATUS 6M | PILATUS 2M |
| Crystal-to-detector distance (mm) | 223.63 | 158.8 | 301.87 | 386.862 | 301.87 | 277.77 | 236.19 |
| Rotation range per image () | 0.1 | 0.4 | 0.2 | 0.1, helical | 0.2 | 0.2 | 0.3 |
| Total rotation range () | 180 | 180 | 210 | 180 | 200 | 240 | 280 |
| Exposure time per image (s) | 0.2 | 0.4 | 0.2 | 0.043 | 0.4 | 0.2 | 0.2 |
| Space group | P212121 | P1211 | P3121 | P1211 | P1 | P1211 | P1211 |
| a, b, c () | 88.72, 90.56, 182.59 | 51.53, 183.52, 78.09 | 137.10 137.10 209.52 | 62.57, 174.11, 102.54 | 62.85, 103.79, 175.56 | 52.99, 110.41, 94.99 | 52.88, 109.75, 94.76 |
| , , () | 90.0, 90.0, 90.0 | 90.0, 94.50, 90.0 | 90.0, 90.0, 120.0 | 90.0, 105.268, 90.0 | 100.686, 91.899, 105.182 | 90, 95.761, 90 | 90, 96.297, 90 |
| Mosaicity () | 0.209 | 0.197 | 0.075 | 0.203 | 0.268 | 0.175 | 0.187 |
| Resolution range () | 45.651.62 (1.681.62) | 48.101.64 (1.701.64) | 48.921.93 (2.001.93) | 48.772.50 (2.592.50) | 47.912.93 (3.032.93) | 48.142.08 (2.152.08) | 47.411.93 (2.001.93) |
| Total No. of reflections | 1194470 (90878) | 599586 (62008) | 2019106 (201888) | 242807 (24274) | 176284 (16772) | 291243 (27660) | 305134 (30573) |
| No. of unique reflections | 186036 (18199) | 173601 (17565) | 170779 (16875) | 71373 (7074) | 88742 (8838) | 64197 (6288) | 79591 (7941) |
| Completeness (%) | 99.64 (98.42) | 98.64 (99.65) | 99.99 (99.98) | 97.84 (97.68) | 98.49 (97.9) | 98.44 (97.56) | 98.67 (99.11) |
| Multiplicity | 6.42 (4.99) | 3.45 (3.53) | 11.82 (11.96) | 3.4 (3.43) | 1.99 (1.90) | 4.54 (4.40) | 3.83 (3.85) |
| I/(I) | 11.6 (2.0) | 8.6 (2.0) | 12.2 (2.0) | 7.6 (2.0) | 4.9 (2.0) | 11.4 (2.0) | 10.7 (2.0) |
| R p.i.m. † | 0.045 (0.397) | 0.065 (0.426) | 0.043 (0.463) | 0.093 (0.545) | 0.091 (0.417) | 0.051 (0.454) | 0.056 (0.389) |
| CC1/2 ‡ | 0.998 (0.693) | 0.993 (0.604) | 0.999 (0.676) | 0.986 (0.563) | 0.979 (0.691) | 0.996 (0.765) | 0.996 (0.754) |
| Overall B factor from Wilson plot (2) | 14.88 | 16.04 | 28.18 | 47.54 | 48.28 | 36.49 | 26.11 |
| No. of molecules per asymmetric unit | 4 | 4 | 6 | 4 | 8 | 2 | 2 |
| Matthews coefficient (3Da1) | 2.20 | 2.21 | 2.28 | 2.29 | 2.30 | 2.35 | 2.32 |
| Solvent content (%) | 44.23 | 44.43 | 46.02 | 46.25 | 46.47 | 47.62 | 47.02 |
R
p.i.m. = 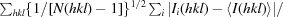
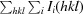 , where Ii(hkl) is the ith observation of reflection hkl and I(hkl) is the weighted average intensity for all observations of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and I(hkl) is the weighted average intensity for all observations of reflection hkl.
CC1/2 is defined as the correlation coefficient between two random half data sets, as described by Karplus Diederichs (2012 ▶).
Only two crystals of latent cgAUS1-ln were obtained, which diffracted to 2.50 Å resolution (helical data-collection mode; Table 2 ▶, data set 4). The crystallization was not reproducible as nucleation and crystal growth were difficult to control within the LLPS. The phase separation was even more pronounced in crystallization setups of recombinantly expressed cgAUS1-lr. Crystals diffracting to 2.93 Å resolution and belonging to space group P1 were only obtained after several weeks (Table 2 ▶, data set 5). It has recently been shown that the use of the polyoxometalate hexatungstotellurate(VI) as a crystallization additive (Bijelic & Rompel, 2015 ▶) improved the crystallization of tyrosinase from A. bisporus (Mauracher et al., 2014b ▶) and that this compound was involved in crystal packing (Mauracher et al., 2014a ▶). Similar observations have been reported for the co-crystallization of this compound with HEWL (Bijelic et al., 2015 ▶). Substitution of 100 mM magnesium chloride by 1 mM hexatungstotellurate(VI) (Na6[TeW6O24]) drastically improved the nucleation and crystal growth of cgAUS1-lr within the LLPS. Crystals diffracting to 2.08 Å resolution were obtained within several days (Table 2 ▶, data set 6). However, the crystals were very unstable in the cryoprotectant solution and on addition of reservoir solution to the crystallization drop, as they rapidly generated cracks and dissolved. However, one data set (Table 2 ▶, data set 6) was obtained from an unscathed crystal by reducing the contact time with the cryoprotectant solution to a minimum (∼1 s) before flash-cooling. Lowering the pH of the crystallization condition improved the stability of the crystals without any significant change in the unit-cell parameters. These crystals were soaked with hydrogen peroxide to obtain the oxy-form of the dinuclear copper centre (Table 2 ▶, data set 7). The successful crystallization of recombinantly expressed cgAUS1 enables crystallization of the mutants of cgAUS1 reported recently (Kaintz et al., 2015 ▶). Crystallographic data and X-ray data-collection statistics are summarized in Table 2 ▶. The crystal structure of active cgAUS1-a1 was solved by molecular replacement using the crystal structure of catechol oxidase from I. batatas as a model (∼47% sequence identity; PDB entry 1bt3). The crystal structure of latent cgAUS1-ln was solved by molecular replacement followed by automated model building. Refinement of the obtained models is in progress.
Acknowledgments
The authors are grateful for financial support by the Austrian Science Fund (FWF) P25217 and P27534, and the Deutsche Forschungsgemeinschaft (Ro 1084/8-1). The EU Cost action CM1203 PoCheMoN is acknowledged. We acknowledge Professor Andreas Rizzi and Claudia Michael for their support during ESI-Q-TOF MS experiments. We thank Professor Kristina Djinovic-Carugo and Georg Mlynek (MFPL, Vienna Biocenter) for their kind support during crystallization experiments and for the opportunity to perform X-ray diffraction experiments to assess the quality of the obtained crystals. We thank the beamline scientists Elspeth Gordon (ESRF ID23-1, mx1450), Anja Burkhardt (DESY P11, I-20120633 EC) and Alice Douangamath (Diamond Light Source I04-1, MX8476) for their generous support during the allocated beam times. Special thanks to Gleb Bourenkov and Victor S. Lamzin (DESY/EMBL, Hamburg, Germany) for the opportunity of data collection at beamline P14 during the ‘European School for Macromolecular Crystallography (ESMAX) 2012’. The authors are truly grateful to Dr Rami Al-Oweini and Professor Dr Ulrich Kortz from Jacobs University, Bremen, Germany, who initially supplied Na6[TeW6O24]·22H2O. The authors’ home page is at http://www.bpc.univie.ac.at.
References
- Bijelic, A., Molitor, C., Mauracher, S. G., Al-Oweini, R., Kortz, U. & Rompel, A. (2015). Chembiochem, 16, 233–241. [DOI] [PMC free article] [PubMed]
- Bijelic, A. & Rompel, A. (2015). Coord. Chem. Rev. 299, 22–38. [DOI] [PMC free article] [PubMed]
- Escobar, M. A., Shilling, A., Higgins, P., Uratsu, S. L. & Dandekar, A. M. (2008). J. Am. Soc. Hortic. Sci. 133, 852–858.
- Espín, J. C., van Leeuwen, J. & Wichers, H. J. (1999). J. Agric. Food Chem. 47, 3509–3517. [DOI] [PubMed]
- Evans, P. R. (2011). Acta Cryst. D67, 282–292. [DOI] [PMC free article] [PubMed]
- Flurkey, W. H. & Inlow, J. K. (2008). J. Inorg. Biochem. 102, 2160–2170. [DOI] [PubMed]
- Fujieda, N., Yabuta, S., Ikeda, T., Oyama, T., Muraki, N., Kurisu, G. & Itoh, S. (2013). J. Biol. Chem. 288, 22128–22140. [DOI] [PMC free article] [PubMed]
- Harborne, J. B. (1967). Comparative Biochemistry of the Flavonoids. London, New York: Academic Press.
- Kabsch, W. (2010a). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kaintz, C., Mauracher, S. G. & Rompel, A. (2014). Adv. Protein Chem. Struct. Biol. 97, 1–35. [DOI] [PubMed]
- Kaintz, C., Mayer, R. L., Jirsa, F., Halbwirth, H. & Rompel, A. (2015). FEBS Lett. 589, 789–797. [DOI] [PMC free article] [PubMed]
- Kaintz, C., Molitor, C., Thill, J., Kampatsikas, I., Michael, C., Halbwirth, H. & Rompel, A. (2014). FEBS Lett. 588, 3417–3426. [DOI] [PMC free article] [PubMed]
- Karplus, P. A. & Diederichs, K. (2012). Science, 336, 1030–1033. [DOI] [PMC free article] [PubMed]
- King, R. S. & Flurkey, W. H. (1987). J. Sci. Food Agric. 41, 231–240.
- Klabunde, T., Eicken, C., Sacchettini, J. C. & Krebs, B. (1998). Nature Struct. Mol. Biol. 5, 1084–1090. [DOI] [PubMed]
- Li, Y., Wang, Y., Jiang, H. & Deng, J. (2009). Proc. Natl Acad. Sci. USA, 106, 17002–17006. [DOI] [PMC free article] [PubMed]
- Marusek, C. M., Trobaugh, N. M., Flurkey, W. H. & Inlow, J. K. (2006). J. Inorg. Biochem. 100, 108–123. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mauracher, S. G., Molitor, C., Al-Oweini, R., Kortz, U. & Rompel, A. (2014a). Acta Cryst. D70, 2301–2315. [DOI] [PMC free article] [PubMed]
- Mauracher, S. G., Molitor, C., Al-Oweini, R., Kortz, U. & Rompel, A. (2014b). Acta Cryst. F70, 263–266. [DOI] [PMC free article] [PubMed]
- Miosic, S., Knop, K., Hölscher, D., Greiner, J., Gosch, C., Thill, J., Kai, M., Shrestha, B. K., Schneider, B., Crecelius, A. C., Schubert, U. S., Svatoš, A., Stich, K. & Halbwirth, H. (2013). PLoS One, 8, e61766. [DOI] [PMC free article] [PubMed]
- Molitor, C., Mauracher, S. G., Pargan, S., Mayer, R. L., Halbwirth, H. & Rompel, A. (2015). Planta, 10.1007/s00425-015-2261-0. [DOI] [PMC free article] [PubMed]
- Ono, E., Fukuchi-Mizutani, M., Nakamura, N., Fukui, Y., Yonekura-Sakakibara, K., Yamaguchi, M., Nakayama, T., Tanaka, T., Kusumi, T. & Tanaka, Y. (2006). Proc. Natl Acad. Sci. USA, 103, 11075–11080. [DOI] [PMC free article] [PubMed]
- Roy, S. K. & Mishra, H. C. (1978). J. Indian Chem. Soc. 55, 188–195.
- Schmidt, K. J., Schrobilgen, G. J. & Sawyer, J. F. (1986). Acta Cryst. C42, 1115–1118.
- Stratford, M. R., Ramsden, C. A. & Riley, P. A. (2013). Bioorg. Med. Chem. 21, 1166–1173. [DOI] [PubMed]
- Virador, V. M., Reyes Grajeda, J. P., Blanco-Labra, A., Mendiola-Olaya, E., Smith, G. M., Moreno, A. & Whitaker, J. R. (2010). J. Agric. Food Chem. 58, 1189–1201. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zekiri, F., Bijelic, A., Molitor, C. & Rompel, A. (2014). Acta Cryst. F70, 832–834. [DOI] [PMC free article] [PubMed]
- Zekiri, F., Molitor, C., Mauracher, S. G., Michael, C., Mayer, R. L., Gerner, C. & Rompel, A. (2014). Phytochemistry, 101, 5–15. [DOI] [PMC free article] [PubMed]