

The A. baumannii biofilm-forming Csu fimbriae are assembled via the archaic CsuC–CsuD chaperone–usher pathway. To determine the structural basis for the biogenesis of Csu fimbriae, native and selenomethionine-substituted CsuC–CsuA/B chaperone–major subunit pre-assembly complexes were produced in Escherichia coli, purified and crystallized. Initial phases were derived from a single anomalous diffraction (SAD) experiment using the selenomethionine derivative.
Keywords: chaperone–usher pathway, archaic, fimbriae, biofilm, Acinetobacter baumannii, assembly, adhesion
Abstract
The attachment of many Gram-negative pathogens to biotic and abiotic surfaces is mediated by fimbrial adhesins, which are assembled via the classical, alternative and archaic chaperone–usher (CU) pathways. The archaic CU fimbrial adhesins have the widest phylogenetic distribution, yet very little is known about their structure and mechanism of assembly. To elucidate the biogenesis of archaic CU systems, structural analysis of the Csu fimbriae, which are used by Acinetobacter baumannii to form stable biofilms and cause nosocomial infection, was focused on. The major fimbriae subunit CsuA/B complexed with the CsuC chaperone was purified from the periplasm of Escherichia coli cells co-expressing CsuA/B and CsuC, and the complex was crystallized in PEG 3350 solution using the hanging-drop vapour-diffusion method. Selenomethionine-labelled CsuC–CsuA/B complex was purified and crystallized under the same conditions. The crystals diffracted to 2.40 Å resolution and belonged to the hexagonal space group P6422, with unit-cell parameters a = b = 94.71, c = 187.05 Å, α = β = 90, γ = 120°. Initial phases were derived from a single anomalous diffraction (SAD) experiment using the selenomethionine derivative.
1. Introduction
Most Gram-negative pathogens express fibrous adhesive virulence organelles that mediate targeting to the sites of infection. The periplasmic chaperone–usher (CU) machinery is used for the controlled assembly of the most ubiquitous and versatile class of such organelles: CU adhesive pili or fimbriae (Nuccio & Bäumler, 2007 ▶; Zavialov et al., 2007 ▶; Zav’yalov et al., 2010 ▶; Busch & Waksman, 2012 ▶). Periplasmic chaperones bind the fimbriae subunits shortly after their translocation to the periplasm and stabilize nascent subunits by forming a periplasmic binary chaperone–subunit complex. The formation of this complex represents a high-energy intermediate state (Zavialov et al., 2003 ▶, 2005 ▶) of the assembly pathway and provides the substrate for subunit polymerization (Yu, Dubnovitsky et al., 2012 ▶). The complex binds to the outer membrane assembly platform, the outer membrane usher protein (Yu, Fooks et al., 2012 ▶; Nishiyama et al., 2005 ▶), where subunits are assembled into linear fibres that are secreted through the usher pore to the cell surface (Phan et al., 2011 ▶; Yu et al., 2009 ▶).
Based on the amino-acid sequences of ushers, all fimbriae within the CU assembly class have been classified into six groups (Nuccio & Bäumler, 2007 ▶): α-fimbriae assembled via the alternate CU pathway, β-, γ-, κ- and π-fimbriae assembled via the classical CU pathway and σ-fimbriae assembled via the archaic CU pathway. σ-Fimbriae are the most widely distributed surface organelles of Gram-negative bacteria assembled using the CU pathway, comprising the largest family within the chaperone–usher assembly class (Nuccio & Bäumler, 2007 ▶). However, little is known about the structure and biogenesis of σ-fimbriae. Neither subunits nor chaperones in the archaic and classical systems have statistically significant sequence similarity, suggesting that the assembly mechanism of σ-fimbriae should differ dramatically from that of the well studied classical CU pathway. To reveal the biogenesis of σ-fimbriae, we focused on structural analysis of the Csu σ-pili system from Acinetobacter baumannii. A. baumannii belongs to the group of ESKAPE pathogens with a high rate of antibiotic resistance that are responsible for the majority of nosocomial infections (Rice, 2008 ▶). The Csu pili enable A. baumannii to form stable biofilms that are responsible for the high rate of nosocomial infections caused by the pathogen (Tomaras et al., 2003 ▶). The pilus is elaborated from four subunits, CsuA/B (16.1 kDa), CsuA (17.3 kDa), CsuB (16.9 kDa) and CsuE (33.5 kDa), using the CsuC–CsuD chaperone–usher secretion machinery (Tomaras et al., 2003 ▶). The Csu system is encoded by the csuA/B-A-B-C-D-E gene cluster, in which expression is controlled by a dedicated two-component regulatory system (Tomaras et al., 2008 ▶). Here, we report the purification and crystallization of the chaperone CsuC complexed with the major pilin subunit CsuA/B as well as the preliminary results of X-ray diffraction experiments on the crystals.
2. Materials and methods
2.1. Macromolecule production
Synthetic genes for CsuC and CsuA/B were ordered from GenScript. Each of the genes was delivered on plasmid pUC57 (Table 1 ▶). The DNA fragment coding for CsuC was inserted into the pET101 expression vector (Invitrogen) using EcoRI and SacI restriction-enzyme sites to produce the plasmid pET101-CsuC. The nucleotide sequence encoding residues 29–37 of CsuA/B (residues 4–12 in the mature protein sequence) was replaced by a 6×His tag (6H)-coding fragment by performing a reverse PCR using primers ACCAGTAACAGCTGCTTGAGTATTTACC and GGCTGTACTGTAGGTGGTAGTCAAAC. The modified gene for CsuA/B (6HCsuA/B) was excised using NheI and SacI restriction enzymes and inserted into the same sites in pET101-CsuC to create the CsuC and 6HCsuA/B co-expression plasmid pET101-CsuC-6HCsuA/B. The sequence was verified by sequencing at the Finnish Microarray and Sequencing Centre.
Table 1. Macromolecule-production information.
| Sequence of wild-type CsuA/B† | MNMKNIQKSLLAALIVAGYAVNTQAAVTGQVDVKLNISTGCTVGGSQTEGNMNKFGTLNFGKTSGTWNNVLTAEVASAATGGNISVTCDGTDPVDFTVAIDGGERTDRTLKNTASADVVAYNVYRDAARTNLYVVNQPQQFTTVSGQATAVPIFGAIAPNTGTPKAQGDYKDTLLVTVNF |
| Modified sequence of CsuA/B‡ | MNMKNIQKSLLAALIVAGYAVNTQAAVTHHHHHHSTGCTVGGSQTEGNMNKFGTLNFGKTSGTWNNVLTAEVASAATGGNISVTCDGTDPVDFTVAIDGGERTDRTLKNTASADVVAYNVYRDAARTNLYVVNQPQQFTTVSGQATAVPIFGAIAPNTGTPKAQGDYKDTLLVTVNF |
| Sequence of CsuC§ | MVICMNNSAFIKNGILKSFLFASTLSLVTPVMAQATFLIWPIYPKIEANEKATAVWLQNTGKTDAMVQIRVFKWNQDGLKDNYSEQSEIIPSPPVAKIKAGEKHMLRLTKSVNLPDGKEQSYRLIVDELPIRLSDGNEQDASKVSFQMRYSIPLFAYGKGIGSGLTEESQKLNAKNALAKPVLQWSVRNNQQGQSELYLKNNGQKFARLSALKTSKTGNDISLGKAAFGYVLSNSTVKFAIDQSTAHELAKTSKIYGVDSSGIKQELIEITKMEDPS |
| Source organism | A. baumannii |
| DNA source | Oligonucleotide synthesis |
| Cloning vector | pUC57 |
| Expression vector | pET101-CsuC-6HCsuA/B |
| Expression host | E. coli strains BL21-AI and BL21(DE3) (Invitrogen) |
The secretion signal sequence is shown in italics and hypothetical hydrophobic donor-strand residues are underlined.
The 6His tag is underlined.
The secretion signal sequence is shown in italics.
The plasmid was transformed into Escherichia coli strain BL21-AI (Invitrogen). Transformants were selected on agar plates with Luria–Bertani (LB) medium containing ampicillin (100 mg l−1). Selected clones were cultivated in LB medium containing ampicillin (80–100 mg l−1) at 310 K. The cells were grown at 110 rev min−1 in baffled flasks to an OD of 0.8–1 at 600 nm and were induced with 1 mM isopropyl β-d-1-thiogalactopyranoside and 0.2% arabinose for protein expression. The culture was grown for a further 2.5 h. The cells were harvested by centrifugation and suspended in 20%(w/v) sucrose in 20 mM Tris–HCl pH 8.0, 5 mM EDTA. Following a 10 min incubation at 283–293 K, the cells were harvested by centrifugation (7000g for 15 min), carefully suspended in ice-cold 5 mM MgSO4 and incubated for 10 min at 273 K. The cells were centrifuged at 7000g for 15 min and the supernatant containing periplasmic proteins was collected. The CsuC–CsuA/B complex was purified by Ni–chelate chromatography in 20 mM sodium phosphate, 0.5 M sodium chloride, 5 mM imidazole buffer pH 7.4 at 277 K. A 5–500 mM imidazole gradient was used to elute the complex. Further purification was performed by cation-exchange chromatography in 50 mM HEPES buffer pH 7.2 using a Mono S 5/50 GL column (GE Healthcare) with a 0–300 mM gradient of NaCl at 277 K. The complex eluted at approximately 120 mM NaCl. For crystallization experiments, the protein was concentrated to 23 mg ml−1 (0.54 mM) using a Vivaspin device (GE Healthcare) with a molecular-weight cutoff of 10 kDa.
To express selenomethionine (SeMet)-labelled CsuC–CsuA/B, the pET101-CsuC-CsuA/B plasmid was transformed into E. coli BL21(DE3) cells and the transformants were grown in LB medium overnight. The next morning, the cells were centrifuged at 4200g for 5 min and the cell pellet was suspended in M9 minimal medium (Doublié, 2007 ▶). The cell suspension was added to M9 minimal medium containing ampicillin (100 mg l−1) at 310 K and cultivated at 110 rev min−1 until an OD of 0.8–1 at 600 nm was reached. 30 min prior to induction, selenomethionine (50 mg l−1), lysine (100 mg l−1), phenylalanine (100 mg l−1), threonine (100 mg l−1), isoleucine (50 mg l−1), leucine (50 mg l−1) and valine (50 mg l−1) were added to the growing cells. After 30 min, 1 mM isopropyl β-d-1-thiogalactopyranoside was added and the cells were grown at 310 K for 3 h to express the protein. Expressed proteins were extracted and purified as described above.
2.2. Crystallization
The purified protein sample was concentrated to 23 mg ml−1 prior to crystallization. Crystal Screen, Crystal Screen 2 (Hampton Research) and The JCSG+ Suite (Qiagen) crystallization screening kits were used. Aliquoting was performed with a Mosquito liquid dispenser (TTP LabTech) by mixing protein solution (in 25 mM HEPES, 150 mM NaCl pH 7.2) and reservoir solution in three different ratios (0.75:1, 1:1 and 1.5:1) in a 96-well plate and equilibrating against 80 µl reservoir solution. Optimization of the crystallization conditions was carried out manually by the hanging-drop vapour-diffusion method (Table 2 ▶).
Table 2. Crystallization.
| Method | Hanging-drop vapour diffusion |
| Plate type | 24-well hanging-drop plate (Hampton Research) |
| Temperature (K) | 289 |
| Protein concentration (mgml1) | 23 |
| Buffer composition of protein solution | 25mM HEPES, 150mM NaCl pH 7.2 |
| Composition of reservoir solution | 0.1M sodium malonate pH 7.0, 15% PEG 3350 |
| Volume and ratio of drop | 4l, 1:1 |
| Volume of reservoir (l) | 1000 |
2.3. Data collection and processing
Native and SeMet-derivative crystals were soaked for 30–60 s in cryoprotection solution (well solution complemented with 18% PEG 3350 and 12% PEG 400) and then cooled by plunging them into liquid nitrogen. Diffraction data were collected under liquid-nitrogen cryoconditions at 100 K on beamlines BM14U and ID23-1 at ESRF, Grenoble, France and were processed using the Grenoble automatic dataprocessing system (GrenADeS) system at the ESRF (Monaco et al., 2013 ▶). Initial phases of SeMet-derivatized CsuC–CsuA/B were determined by the single-wavelength anomalous diffraction (SAD) phasing method and the initial model was constructed using the PHENIX software package (Adams et al., 2002 ▶).
3. Results and discussion
The assembly of major subunits from classical CU systems depends on their N-terminal donor sequences. In pre-assembly chaperone–subunit complexes these sequences have no structural role and can be deleted or replaced by purification tags (Zavialov et al., 2002 ▶). Inspection of the CsuA/B N-terminal sequence (Table 1 ▶) revealed a clear pattern of alternating hydrophilic/hydrophobic residues, which is characteristic of β-strands and is typically present in the donor strands of pilins from classical systems. This characteristic motif is followed by a conserved cysteine residue that is engaged in a disulfide bond connecting the beginning of strand A to strand B in many pilins from classical systems (Berry et al., 2014 ▶; Roy et al., 2012 ▶). Assuming that this sequence plays a role in the assembly of CsuA/B subunits, we replaced nine amino acids in CsuA/B in the region between the processing site and the conserved cysteine by a 6×His tag and co-expressed the subunit with the CsuC chaperone. Both proteins accumulated to high levels in the periplasm of E. coli BL21-AI cells and were quantitatively co-purified by Ni-chelate and ion exchange chromatography (Fig. 1 ▶). Therefore, we concluded that CsuC and CsuA/B form a stable complex and that their association is unaffected by modification of the N-terminal sequence in CsuA/B.
Figure 1.
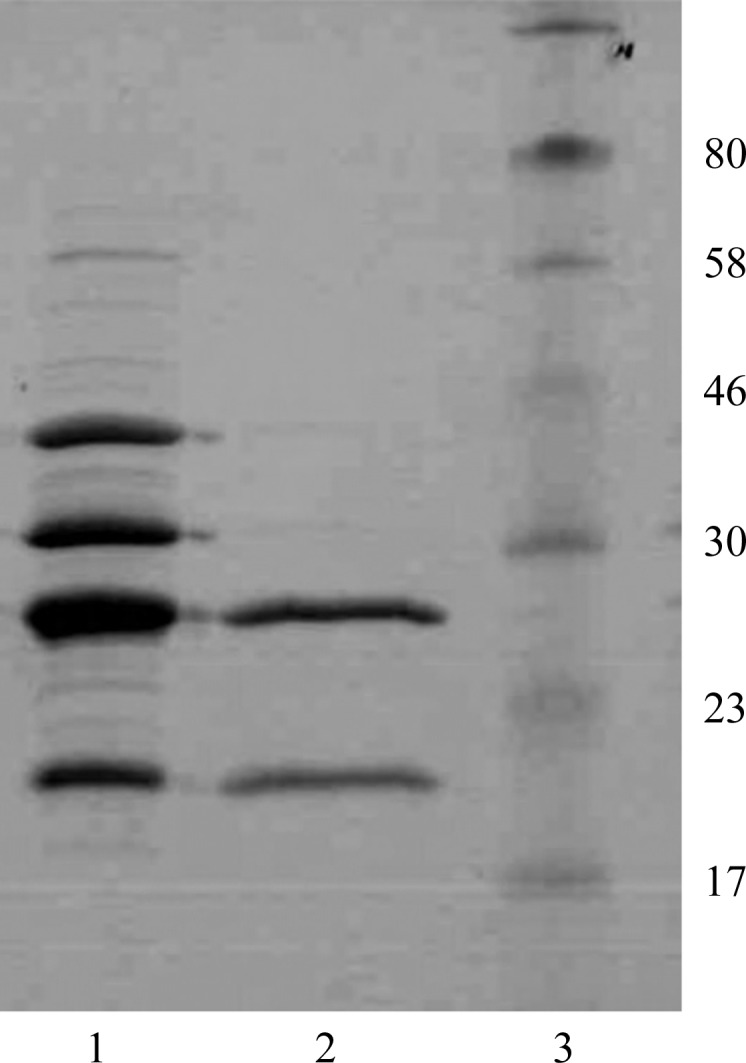
Coomassie Blue-stained SDS–polyacrylamide gel (12%) of the periplasmic extract from E. coli BL21-AI cells co-expressing CsuC and CsuA/B (lane 1), purified CsuC–CsuA/B complex (lane 2) and molecular-size marker proteins (lane 3; labelled in kDa).
Initial screening of crystallization conditions for the complex resulted in the appearance of three-dimensional crystals in a broad range of conditions: pH 7.0–9.0 and 10–15% PEG 3350–6000 at 289 K. The largest crystals were obtained in 0.1 M sodium malonate pH 7.0, 15% PEG 3350 (Fig. 2 ▶). The crystal diffracted to 3.0 Å resolution, revealing the symmetry of a hexagonal lattice. A 96.9% complete data set was obtained with an overall R meas of 14.2% (Table 3 ▶). Analysis of the data with POINTLESS (Evans, 2006 ▶) suggested two possible space groups: P6222 and P6422. Calculation of the Matthews coefficient V M (the crystal volume per unit of protein molecular weight) assumed the presence of one molecule of the CsuC–CsuA/B complex per crystallographic asymmetric unit (V M = 2.73 Å3 Da−1).
Figure 2.

CsuC–CsuA/B crystals (grown in 0.1 M sodium malonate pH 7.0, 15% PEG 3350 at 289 K).
Table 3. Statistics of the X-ray diffraction data sets and SAD phasing.
Values in parentheses are for the outer shell.
| Native crystal | SeMet crystal | |
|---|---|---|
| Wavelength () | 0.973 | 0.979 |
| Temperature (K) | 100 | 100 |
| Detector | MAR225 | PILATUS 6M_F |
| Crystal-to-detector distance (mm) | 269.41 | 524.34 |
| Rotation range per image () | 1 | 0.1 |
| Total rotation range () | 95 | 360 |
| Exposure time per image (s) | 40 | 0.049 |
| Space group | P6422 | P6422 |
| Unit-cell parameters (, ) | a = b = 93.78, c = 167.39, = = 90, = 120 | a = b = 94.71, c = 187.05, = = 90, = 120 |
| Mosaicity () | 0.17 | 0.09 |
| Resolution range () | 50.003.00 (3.163.00) | 49.632.40 (2.532.40) |
| Total No. of reflections | 42587 (6219) | 305639 (46837) |
| No. of unique reflections | 8964 (1266) | 20138 (2869) |
| Completeness (%) | 96.9 (98.0) | 99.8 (100) |
| Multiplicity | 4.8 (4.9) | 15.2 (16.3) |
| I/(I) | 12.9 (3.2) | 37.4 (6.3) |
| R meas † | 0.142 (0.611) | 0.046 (0.571) |
| Overall B factor from Wilson plot (2) | 47.5 | 54.6 |
The anomalous signal measurability is defined as the fraction of Bijvoet-related intensity differences for which |I|/(I) > 3.0 and {I(+)/[I(+)], I()/[I()]} > 3.0 hold.
| Resolution shells () | 5.17 | 4.10 | 3.58 | 3.26 | 3.02 | 2.85 | 2.70 | 2.59 | 2.49 | 2.40 |
|---|---|---|---|---|---|---|---|---|---|---|
| Measurability | 0.56 | 0.39 | 0.26 | 0.15 | 0.08 | 0.03 | 0.02 | 0.01 | 0.002 | 0.01 |
(c).
Substructure.
| Se-atom positions | ||||||
|---|---|---|---|---|---|---|
| Se atom | Assignment‡ | x | y | z | Occupancy | B (2) |
| Se1 | 114 | 7.633 | 43.423 | 130.908 | 0.67 | 13.91 |
| Se2 | 32 | 21.278 | 40.258 | 174.832 | 1.01 | 17.67 |
| Se3 | 71 | 11.945 | 45.252 | 157.659 | 1.13 | 17.35 |
| Se4 | 239 | 8.759 | 22.233 | 85.618 | 0.76 | 32.66 |
| Se5 | 32 | 21.447 | 40.333 | 177.175 | 0.43 | 23.73 |
| Se6 | 71 | 13.568 | 45.546 | 154.124 | 0.29 | 24.88 |
| Se7 | 32 | 21.723 | 40.928 | 173.064 | 0.65 | 13.52 |
| Se8 | 114 | 7.308 | 42.994 | 132.28 | 0.40 | 18.28 |
| Se9 | 114 | 8.198 | 41.493 | 130.306 | 0.78 | 16.12 |
| Se10 | 71 | 14.044 | 46.317 | 160.270 | 0.25 | 23.92 |
| Se11 | 71 | 14.127 | 45.675 | 157.400 | 0.75 | 27.37 |
| Se12 | 239 | 8.651 | 21.926 | 88.321 | 0.14 | 20.66 |
| Se13 | 239 | 9.436 | 22.271 | 83.812 | 0.27 | 24.14 |
| Se14 | 114 | 8.143 | 42.011 | 127.657 | 0.17 | 23.33 |
| Se15 | 32 | 23.676 | 41.008 | 174.627 | 0.46 | 20.70 |
| Se16 | 32 | 21.241 | 40.620 | 170.453 | 0.28 | 26.26 |
| Se17 | 32 | 20.222 | 40.251 | 172.839 | 0.43 | 14.92 |
| Se18 | 32 | 20.993 | 40.401 | 179.738 | 0.21 | 22.43 |
R
meas = 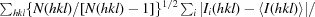
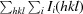 , where N(hkl) is the multiplicity.
, where N(hkl) is the multiplicity.
Several Se atoms clustered around four selenomethionine residues at positions 32, 71, 114 and 239 of CsuC.
The presence of five methionines in the complex prompted us to apply the selenomethionine (SeMet) SAD method to determine the phases. The SeMet-substituted complex was produced using the metabolic inhibition method, which requires growing cells in M9 minimum medium. Since the high concentration of glucose in the M9 minimal medium inhibited the uptake of arabinose by the E. coli BL21-AI cells, the proteins were co-expressed in E. coli BL21(DE3) cells using IPTG for induction. The SeMet-substituted complex was purified and crystallized in the same way as the native complex. Diffraction data were collected at a wavelength of 0.979 Å to a resolution of 2.4 Å (Table 3 ▶). The c axis of the unit cell of the crystal of the SeMet-substituted protein was about 20 Å longer than that of the native protein crystal, yet these crystals diffracted to higher resolution. The anomalous signal of this data set extended to 3.0 Å resolution (Table 3 ▶). The best solution found by the HySS and Phaser programs (in the PHENIX software suite) belonged to space group P6422 and contained 18 Se atoms clustered in four sites (Table 3 ▶). The sites were later assigned to four SeMet residues in the structure of CsuC (Table 3 ▶). The sum of occupancies of the atoms in each cluster exceeds 1.0, suggesting a slightly underestimated value for f′′. This could also be the reason for the low B-factor values of the atoms in the substructure. Phase improvement by density modification resulted in experimental electron density that contained recognizable features of a β-structure protein (Fig. 3 ▶). 247 residues (63%) in 14 chains and 194 side chains were placed automatically with the AutoBuild option in PHENIX. The majority of these residues were assigned to the structure of CsuC. Structure refinement is in progress.
Figure 3.

Experimental electron density computed using SAD phases. A fragment of the 2mF o − DF c (σA; Read, 1986 ▶) electron-density map contoured at 1σ is shown (produced using PyMOL; Schrödinger).
Acknowledgments
The authors would like to thank the staff at beamlines ID23-1 and BM14U, ESRF, Grenoble, France for their assistance in data collection. This work was supported by grants FA-136333 and FA-140959 to AZ and by an Erasmus visiting scientist stipend to NP.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Berry, A. A. et al. (2014). PLoS Pathog. 10, e1004404. [DOI] [PMC free article] [PubMed]
- Busch, A. & Waksman, G. (2012). Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1112–1122. [DOI] [PMC free article] [PubMed]
- Doublié, S. (2007). Methods Mol. Biol. 363, 91–108. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Monaco, S., Gordon, E., Bowler, M. W., Delagenière, S., Guijarro, M., Spruce, D., Svensson, O., McSweeney, S. M., McCarthy, A. A., Leonard, G. & Nanao, M. H. (2013). J. Appl. Cryst. 46, 804–810. [DOI] [PMC free article] [PubMed]
- Nishiyama, M., Horst, R., Eidam, O., Herrmann, T., Ignatov, O., Vetsch, M., Bettendorff, P., Jelesarov, I., Grütter, M. G., Wüthrich, K., Glockshuber, R. & Capitani, G. (2005). EMBO J. 24, 2075–2086. [DOI] [PMC free article] [PubMed]
- Nuccio, S. P. & Bäumler, A. J. (2007). Microbiol. Mol. Biol. Rev. 71, 551–575. [DOI] [PMC free article] [PubMed]
- Phan, G. et al. (2011). Nature (London), 474, 49–53.
- Read, R. J. (1986). Acta Cryst. A42, 140–149.
- Rice, L. B. (2008). J. Infect. Dis. 197, 1079–1081. [DOI] [PubMed]
- Roy, S. P., Rahman, M. M., Yu, X. D., Tuittila, M., Knight, S. D. & Zavialov, A. V. (2012). Mol. Microbiol. 86, 1100–1115. [DOI] [PubMed]
- Tomaras, A. P., Dorsey, C. W., Edelmann, R. E. & Actis, L. A. (2003). Microbiology, 149, 3473–3484. [DOI] [PubMed]
- Tomaras, A. P., Flagler, M. J., Dorsey, C. W., Gaddy, J. A. & Actis, L. A. (2008). Microbiology, 154, 3398–3409. [DOI] [PubMed]
- Yu, X., Dubnovitsky, A., Pudney, A. F., Macintyre, S., Knight, S. D. & Zavialov, A. V. (2012). Structure, 20, 1861–1871. [DOI] [PubMed]
- Yu, X., Fooks, L. J., Moslehi-Mohebi, E., Tischenko, V. M., Askarieh, G., Knight, S. D., Macintyre, S. & Zavialov, A. V. (2012). J. Mol. Biol. 417, 294–308. [DOI] [PubMed]
- Yu, X., Visweswaran, G. R., Duck, Z., Marupakula, S., MacIntyre, S., Knight, S. D. & Zavialov, A. V. (2009). Biochem. J. 418, 541–551. [DOI] [PubMed]
- Zavialov, A. V., Berglund, J., Pudney, A. F., Fooks, L. J., Ibrahim, T. M., MacIntyre, S. & Knight, S. D. (2003). Cell, 113, 587–596. [DOI] [PubMed]
- Zavialov, A. V., Kersley, J., Korpela, T., Zav’yalov, V. P., MacIntyre, S. & Knight, S. D. (2002). Mol. Microbiol. 45, 983–995. [DOI] [PubMed]
- Zavialov, A. V., Tischenko, V. M., Fooks, L. J., Brandsdal, B. O., Aqvist, J., Zav’yalov, V. P., Macintyre, S. & Knight, S. D. (2005). Biochem. J. 389, 685–694. [DOI] [PMC free article] [PubMed]
- Zavialov, A., Zav’yalova, G., Korpela, T. & Zav’yalov, V. (2007). FEMS Microbiol. Rev. 31, 478–514. [DOI] [PubMed]
- Zav’yalov, V., Zavialov, A., Zav’yalova, G. & Korpela, T. (2010). FEMS Microbiol. Rev. 34, 317–378. [DOI] [PubMed]