

In order to understand the structure–function relationship of 3-deoxy-d-manno-oct-2-ulosonic acid 8-phosphate phosphatase (YrbI) from B. pseudomallei, its crystallographic study has been performed. Here, the cloning, overexpression, purification, crystallization and preliminary X-ray study of this enzyme are reported.
Keywords: 3-deoxy-d-manno-oct-2-ulosonic acid 8-phosphate phosphatase, YrbI, Burkholderia pseudomallei
Abstract
3-Deoxy-d-manno-oct-2-ulosonic acid 8-phosphate phosphatase (YrbI), the third enzyme in the pathway for the biosynthesis of 3-deoxy-d-manno-oct-2-ulosonic acid (KDO), hydrolyzes KDO 8-phosphate to KDO and inorganic phosphate. YrbI belongs to the haloacid dehalogenase (HAD) superfamily, which is a large family of magnesium-dependent phosphatase/phosphotransferase enzymes. In this study, YrbI from Burkholderia pseudomallei, the causative agent of melioidosis, has been cloned, expressed, purified and crystallized. Synchrotron X-ray data were also collected to 2.25 Å resolution. The crystal belonged to the primitive orthorhombic space group P212121, with unit-cell parameters a = 63.7, b = 97.5, c = 98.0 Å. A full structural determination is in progress to elucidate the structure–function relationship of this protein.
1. Introduction
Lipopolysaccharide (LPS), a major component of the outer membrane of Gram-negative bacteria, plays an important role in maintaining the structural integrity of the bacterial outer membrane (Kneidinger et al., 2002 ▶) and provides a barrier against the entry of toxic hydrophobic compounds into the bacterial cell (Nikaido & Vaara, 1985 ▶). LPS can be divided into three distinct structures known as lipid A, a conserved core oligosaccharide region and an O-specific polysaccharide chain. In the majority of Gram-negative bacteria, the core oligosaccharide region can be subdivided into an outer core, typically composed of hexoses and hexosamines, and an inner core made of l,d-heptose units and 3-deoxy-d-manno-oct-2-ulosonic acid (KDO; Kneidinger et al., 2002 ▶). The synthesis of LPS critically requires KDO, which links the core oligosaccharide to the lipid A moiety. It has been shown that disruption of the biosynthesis of KDO causes the accumulation of lipid A precursors and subsequent interruption of cell growth (Wu & Woodard, 2003 ▶). Thus, the enzymes involved in the biosynthetic pathway of KDO may be an alternative target for the development of new antibiotics or antibiotic adjuvants.
KDO 8-phosphate phosphatase (YrbI), the third enzyme in the pathway for the biosynthesis of KDO, hydrolyzes KDO 8-phosphate to KDO and inorganic phosphate (Wu & Woodard, 2003 ▶). YrbI belongs to the haloacid dehalogenase (HAD) superfamily, which is a large family of magnesium-dependent phosphatase/phosphotransferase enzymes. The members of the HAD superfamily share three conserved sequence motifs (Koonin & Tatusov, 1994 ▶) and a mixed α/β Rossmann fold (Morais et al., 2000 ▶). Motif I contains a DXDX(T/V) sequence, and the first aspartate of the sequence forms a phosphoaspartate intermediate with the substrate. Motif II, a conserved serine or threonine residue, is responsible for the binding of the substrate phosphate. Motif III, a (G/S)(D/S)XXX(D/N) sequence, also contains elements of the active site that bind the metal ligands and hold the substrate (Koonin & Tatusov, 1994 ▶). Although the enzymes belonging to the HAD superfamily have various biological functions, only a few HAD enzymes have been well characterized in bacteria, e.g. Escherichia coli (Kuznetsova et al., 2006 ▶), and eukaryotes, e.g. Homo sapiens (Seifried et al., 2013 ▶).
The Gram-negative soil saprophyte Burkholderia pseudomallei is notorious for its pathogenicity and causes melioidosis (Wiersinga et al., 2006 ▶). It is also classified as a potential bioterrorism agent because of its rareness in Western countries, its high accessibility, its environmental persistence, the high prevalence of severe sepsis and septic shock mortality and the fact that no vaccine is available (Gilad et al., 2007 ▶). Therefore, the development of antibiotics against melioidosis is essential to cope with a potentially disastrous situation. Since biosynthesis of KDO is essential for synthesis of LPS, YrbI of B. pseudomallei (BpYrbI) could be a good target for the development of antibiotics against melioidosis. In order to understand the structure–function relationship of BpYrbI, its crystallographic study has been performed. Here, we report the cloning, overexpression, purification, crystallization and preliminary X-ray study of this enzyme.
2. Experimental methods
2.1. Cloning of YrbI from B. pseudomallei
The cloning primers (Genotech, Daejeon, Republic of Korea) prepared for ligation-independent cloning (LIC) were 5′-GGCGGTGGTGGCGGCATGTCCGCGCCCCCTGC-3′ for the forward strand and 5′-GTTCTTCTCCTTTGCGCCCCTAGGCTCCGCAGGCGGCC-3′ for the reverse strand. The yrbI gene was amplified by PCR using 200 ng B. pseudomallei genomic DNA template and 50 µM of primers. PrimeSTAR HS DNA polymerase with GC buffer (Takara Bio Inc., Shiga, Japan) designed for high-GC-content genomic DNA was used. The amplified LIC expression vector pB2 (Kim et al., 2005 ▶), a derivative of the pET-21a vector (Novagen, Madison, Wisconsin, USA), was incubated with T4 DNA polymerase (New England Biolabs, Beverley, Massachusetts, USA) in the presence of 1 mM dATP at 310 K for 30 min followed by incubation at 343 K for 20 min. The amplified PCR product was prepared for vector insertion using the same protocol apart from the presence of 1 mM dTTP instead of 1 mM dATP. The prepared insert was annealed into the pB2 vector, which expresses the cloned gene fused to a noncleavable N-terminal His6 tag, and was transformed into DH5α competent cells to obtain fusion clones. Clones were screened by plasmid DNA analysis and transformed into E. coli BL21 (DE3) for protein expression (Kim et al., 1998 ▶).
2.2. Growth of cultures
E. coli BL21 (DE3) cells transformed with the cloned vector harbouring the yrbI gene were grown on Luria–Bertani (LB) agar plates containing 150 µg ml−1 ampicillin. Several colonies were picked and grown in capped test tubes with 10 ml LB broth containing 150 µg ml−1 ampicillin. A cell stock of 0.85 ml culture and 0.15 ml glycerol was prepared and frozen at 193 K for use in a larger culture. The frozen cell stock was grown in 5 ml LB medium and diluted into 1000 ml fresh LB medium. The culture was incubated at 310 K with shaking until an OD600 of 0.6–0.8 was reached. At this point, expression of BpYrbI was induced using isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 1 mM. The culture was further grown at 298 K for 16 h in a shaking incubator. Cells were harvested by centrifugation at 7650g (6500 rev min−1) for 10 min in a high-speed refrigerated centrifuge at 277 K.
2.3. Protein purification
The cultured cell paste (4.09 g) was resuspended in 25 ml buffer consisting of 50 mM Tris–HCl pH 8.0, 100 mM NaCl, 10 mM imidazole, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mg ml−1 DNase I. The cell suspension was disrupted using a Digital Sonifier 450 (Branson Ultrasonics, Danbury, Connecticut, USA). Cell debris was pelleted by centrifugation at 24 900g (15 000 rev min−1) for 1 h in a high-speed refrigerated ultracentrifuge at 277 K. The supernatant was affinity-purified using a HisTrap column on an ÄKTAexplorer system (GE Healthcare, Piscataway, New Jersey, USA) at room temperature. The column was equilibrated with a buffer consisting of 50 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole. The target protein was eluted with a buffer consisting of 50 mM Tris–HCl pH 8.0, 100 mM NaCl with a gradient from 10 to 500 mM imidazole. BpYrbI was further purified by ion-exchange chromatography using a 5 ml HiTrap Q column (GE Healthcare, Piscataway, New Jersey, USA) equilibrated with buffer consisting of 20 mM Tris–HCl pH 8.0. The protein was eluted at 0.54 M NaCl using a linear NaCl gradient. SDS–PAGE showed one band around 20 kDa corresponding to the molecular weight of BpYrbI with the tag. The purified protein contained a noncleavable N-terminal His6 tag followed by five glycine residues (MHHHHHHGGGGG) and was concentrated to 5.3 mg ml−1 (using an extinction coefficient of 0.712 M −1 cm−1 at 280 nm) for crystallization in a buffer consisting of 0.54 M NaCl, 20 mM Tris–HCl pH 8.0.
2.4. Dynamic light-scattering experiment
Dynamic light scattering (DLS) was used to determine the hydrodynamic radius of native BpYrbI. It was performed using a DynaPro Titan instrument (Wyatt, Santa Barbara, California, USA). A protein concentration of 1.0 mg ml−1 was used. A micro cuvette with a volume of 12 µl and a path length of 0.15 cm was used for the measurements. The purified BpYrbI showed a single monodisperse peak corresponding to the approximate size of tetrameric BpYrbI, indicating homogeneity of the protein.
2.5. Crystallization of BpYrbI
Screening for crystallization conditions was performed at room temperature using the sparse-matrix method (Jancarik & Kim, 1991 ▶) with several screens from Hampton Research (Laguna Niguel, California, USA). A Hydra-Plus-One crystallization robot (Matrix Technologies, Hudson, New Hampshire, USA) was used to set up the screens using the sitting-drop vapour-diffusion method in a 96-well Intelli-Plate (Art Robbins Instruments, Salt Lake City, Utah, USA). Sitting drops were made by mixing 0.2 µl protein solution (5.3 mg ml−1) with 0.2 µl reservoir solution and were equilibrated against 50 µl reservoir solution. A VDX48 plate (Hampton Research, Laguna Niguel, California, USA) was used to optimize the crystallization conditions using hanging drops made by mixing 0.8 µl protein solution and 0.8 µl reservoir solution consisting of 23%(w/v) PEG 3350, 0.1 M Tris pH 8.5 and equilibrated against 200 µl reservoir solution.
2.6. Data collection and reduction
Before being flash-cooled in liquid nitrogen, crystals were soaked in the reservoir solution with an additional 15%(w/v) PEG 3350 as a cryoprotectant. X-ray diffraction data were collected at a single wavelength on beamline 7A at Pohang Light Source (PLS) using an ADSC Quantum 4 CCD detector (Area Detector Systems Corporation, Poway, California, USA) placed 300 mm from the sample. X-ray data were collected at a single wavelength (0.97933 Å) from one crystal. The oscillation range per image was 0.5°, with 1 s exposures. 240 oscillation images were collected with no overlap between contiguous images. X-ray diffraction data were processed and scaled using DENZO and SCALEPACK from the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
The yield of purified BpYrbI was 26.3 mg per litre of E. coli culture. After anion-exchange chromatography, BpYrbI appeared to be approximately 99% pure, with a prominent protein band at around 20 kDa on SDS–PAGE (Fig. 1 ▶). Diffraction-quality crystals were obtained using a reservoir solution consisting of 23%(w/v) PEG 3350, 0.1 M Tris pH 8.5. The crystals grew to dimensions of 0.12 × 0.10 × 0.04 mm within 5 d at 296 K (Fig. 2 ▶). Synchrotron data were collected to 2.25 Å resolution. The crystal belonged to the primitive orthorhombic space group P212121, with unit-cell parameters a = 63.7, b = 97.5, c = 98.0 Å. Based on the Matthews coefficient (Matthews, 1968 ▶), the asymmetric unit could contain four BpYrbI protomers with a solvent content of 35.0%. The details of the data-collection statistics are presented in Table 1 ▶. Molecular replacement (MR) was performed using PHENIX (Adams et al., 2010 ▶), with the tetrameric form of the X-ray crystal structure of the probable YrbI family phosphatase from Pseudomonas syringae pv. phaseolica 1448a (PsyYrbI; PDB entry 3mn1), which has a sequence identity of 44.2% to BpYrbI, as a search model (Daughtry et al., 2013 ▶). Unfortunately, it failed to give an MR solution. Therefore, homology models of BpYrbI were built using MODELLER 9.11 (Eswar et al., 2006 ▶). A molecular-replacement solution was found using the top model. A relatively good electron-density map was obtained after automated model building and refinement with the PHENIX AutoBuild wizard (Terwilliger et al., 2008 ▶). The electron-density map clearly showed that there are four protomers of BpYrbI in the asymmetric unit and that they form a tetramer with a similar pattern to that of PsyYrbI. The presence of a tetrameric form with an approximate size of 90 kDa was confirmed by the DLS experiment. The square-planar shape of tetrameric BpYrbI instead of a globular shape in the electron-density map explains the slightly larger size of ∼90 kDa instead of the expected ∼80 kDa in solution. The model has an R factor of 17.3% and an R free of 22.5%. Full structure determination is in progress.
Figure 1.

Coomassie-stained SDS–PAGE (12%) of BpYrbI. Lane 1, native BpYrbI (1.5 mg); lane 2, flowthrough; lane 3, BpYrbI under reducing conditions (1.5 mg); lane M, molecular-mass marker (labelled in kDa).
Figure 2.

Crystals of BpYrbI. These crystals appeared in the condition 23% PEG 3350, 0.1 M Tris pH 8.5. The crystals grew to approximate dimensions of 0.12 × 0.10 × 0.04 mm in 5 d.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| X-ray source | Beamline 7A, PLS |
| X-ray wavelength () | 0.97933 |
| Temperature (K) | 100 |
| Space group | P212121 |
| Unit-cell parameters () | a = 63.7, b = 97.5, c = 98.0 |
| Volume fraction of solvent (%) | 35.0 |
| V M (3Da1) | 1.89 |
| Resolution range () | 50.02.25 (2.312.25) |
| Multiplicity | 10.1 (5.9) |
| Unique reflections | 27107 (1188) |
| R merge † (%) | 6.3 (16.1) |
| Data completeness (%) | 90.1 (56.4) |
| Average I/(I) | 25.5 (7.2) |
R
merge = 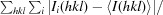
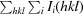 , where I
i(hkl) is the intensity of the ith observation of the unique reflection hkl and I(hkl) is the mean of the intensities of all observations of reflection hkl.
, where I
i(hkl) is the intensity of the ith observation of the unique reflection hkl and I(hkl) is the mean of the intensities of all observations of reflection hkl.
Acknowledgments
We are grateful to Dr Sarinna Tumapa at Mahidol University and Dr Sharon Peacock at the University of Cambridge for their kindness in providing us with B. pseudomallei genomic DNA and the staff at Pohang Light Source for their assistance with synchrotron data collection. This work was supported by the Basic Science Research Program (2012-0003936) through the National Research Foundation of Korea grant funded by the Korean Government (MEST). M-SK, JP and DL were supported by the Brain Korea 21 (BK21) Project.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Daughtry, K. D., Huang, H., Malashkevich, V., Patskovsky, Y., Liu, W., Ramagopal, U., Sauder, J. M., Burley, S. K., Almo, S. C., Dunaway-Mariano, D. & Allen, K. N. (2013). Biochemistry, 52, 5372–5386. [DOI] [PMC free article] [PubMed]
- Eswar, N., Webb, B., Marti-Renom, M. A., Madhusudhan, M. S., Eramian, D., Shen, M. Y., Pieper, U. & Sali, A. (2006). Curr. Protoc. Bioinformatics, Unit 5.6. 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed]
- Gilad, J., Harary, I., Dushnitsky, T., Schwartz, D. & Amsalem, Y. (2007). Isr. Med. Assoc. J. 9, 499–503. [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kim, R., Sandler, S. J., Goldman, S., Yokota, H., Clark, A. J. & Kim, S.-H. (1998). Biotechnol. Lett. 20, 207–210.
- Kim, S.-H. et al. (2005). J. Struct. Funct. Genomics, 6, 63–70. [DOI] [PubMed]
- Kneidinger, B., Marolda, C., Graninger, M., Zamyatina, A., McArthur, F., Kosma, P., Valvano, M. A. & Messner, P. (2002). J. Bacteriol. 184, 363–369. [DOI] [PMC free article] [PubMed]
- Koonin, E. V. & Tatusov, R. L. (1994). J. Mol. Biol. 244, 125–132. [DOI] [PubMed]
- Kuznetsova, E., Proudfoot, M., Gonzalez, C. F., Brown, G., Omelchenko, M. V., Borozan, I., Carmel, L., Wolf, Y. I., Mori, H., Savchenko, A. V., Arrowsmith, C. H., Koonin, E. V., Edwards, A. M. & Yakunin, A. F. (2006). J. Biol. Chem. 281, 36149–36161. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 6491–6493.
- Morais, M. C., Zhang, W., Baker, A. S., Zhang, G., Dunaway-Mariano, D. & Allen, K. N. (2000). Biochemistry, 39, 10385–10396. [DOI] [PubMed]
- Nikaido, H. & Vaara, M. (1985). Microbiol. Rev. 49, 1–32. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Seifried, A., Schultz, J. & Gohla, A. (2013). FEBS J. 280, 549–571. [DOI] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., Read, R. J. & Adams, P. D. (2008). Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed]
- Wiersinga, W. J., van der Poll, T., White, N. J., Day, N. P. & Peacock, S. J. (2006). Nature Rev. Microbiol. 4, 272–282. [DOI] [PubMed]
- Wu, J. & Woodard, R. W. (2003). J. Biol. Chem. 278, 18117–18123. [DOI] [PubMed]