

The crystal structures of three ternary complexes of human (h) or P. carinii (pc) dihydrofolate reductase (DHFR) with cofactor and pyrido[2,3-d]pyrimidine inhibitors are reported to define the interactions that enhance selectivity against pathogenic DHFRs. The binding conformation of the most potent inhibitor differs between the human and Pneumocystis complexes and validates the hypothesis that the N9-methyl substitution preferentially interacts with Ile123 of pcDHFR than with Val115 of hDHFR, consistent with its weaker binding affinity for hDHFR.
Keywords: dihydrofolate reductase, Pneumocystis carinii, pathogens, antifolate inhibitors, conformational analysis
Abstract
To further define the interactions that enhance the selectivity of binding and to directly compare the binding of the most potent analogue {N 6-methyl-N 6-(3,4,5-trifluorophenyl)pyrido[2,3-d]pyrimidine-2,4,6-triamine; compound 26} in the series of bicyclic pyrido[2,3-d]pyrimidine analogues of piritrexim (PTX) with native human (h), Pneumocystis carinii (pc) and Pneumocystis jirovecii (pj) dihydrofolate reductase (DHFR) enzymes, the crystal structures of hDHFR complexed with N 6-methyl-N 6-(4-isopropylphenyl)pyrido[2,3-d]pyrimidine-2,4,6-triamine (compound 22), of hDHFR complexed with compound 26 and of pcDHFR complexed with N 6-methyl-N 6-1-naphthylpyrido[2,3-d]pyrimidine-2,4,6-triamine (compound 24) are reported as ternary complexes with NADPH. This series of bicyclic pyrido[2,3-d]pyrimidines were designed in which there was a transposition of the 5-methyl group of PTX to the N9 position of the pyrido[2,3-d]pyrimidine. It was hypothesized that the N9-methyl group would preferentially interact with Ile123 of pcDHFR (and Ile123 of pjDHFR), but not with the shorter Val115 in hDHFR. Structure–activity data for this series of antifolates revealed that a trifluoro derivative (26) was the most selective against pjDHFR compared with mammalian DHFR (h/pj = 35.7). Structural data for the hDHFR–26 complex revealed that 26 binds in a different conformation from that observed in the pcDHFR–26 complex. In the hDHFR–26 complex the trifluorophenyl ring of 26 occupies a position near the cofactor-binding site, with close intermolecular contacts with Asp21, Ser59 and Ile60, whereas this ring in the pcDHFR–26 complex is positioned away from the cofactor site and near Ile65, with weaker contacts with Ile65, Phe69 and Ile123. Comparison of the intermolecular contacts between the N9-methyl group with Val115/Ile123 validates the hypothesis that the N9-methyl substituent preferentially interacts with Ile123 compared with Val115 of hDHFR, as the weaker contact with Val115 in the hDHFR structure is consistent with its weaker binding affinity compared with pcDHFR. The results for the structures of hDHFR–22 and pcDHFR–24 show that their inhibitor-binding orientation is similar to that observed in pcDHFR–26 and the pcDHFR variant (F69N) reported previously. The naphthyl moiety of 24 makes several intermolecular contacts with the active-site residues in pcDHFR that help to stabilize the binding, resulting in a more potent inhibitor.
1. Introduction
A novel series of bicyclic pyrido[2,3-d]pyrimidines that contain an N 6-(substituted phenyl)pyrido[2,3-d]pyrimidine-2,4,6-triamine scaffold in which the substituted aniline is directly attached to the 6-position of the pyrido[2,3-d]pyrimidine ring have been shown to be selective and potent inhibitors of pathogenic sources of dihydrofolate reductase (DHFR) such as Pneumocystis jirovecii, a causative agent of pneumonia in HIV/AIDS patients (Gangjee et al., 2013 ▶). These compounds are analogues of piritrexim (PTX), a potent but nonselective DHFR inhibitor (Grivsky et al., 1980 ▶; Kovacs et al., 1988 ▶; Falloon et al., 1990 ▶). Of note are a series of analogues in which the 5-methyl group of PTX was transposed to the N9 position of the pyrido[2,3-d]pyrimidine. It was hypothesized that the N9-methyl group would make favorable contacts with Ile123 of pcDHFR (and Ile123 of pjDHFR), but not with the shorter Val115 of human DHFR. Structure–activity data for the inhibition of pyrido[2,3-d]pyrimidines against hDHFR, pcDHFR and pjDHFR enzymes revealed that a trifluro derivative (compound 26; Fig. 1 ▶) was the most selective against pjDFHR compared with the mammalian DHFR (IC50 of 0.0042 µM for pjDFHR compared with 0.15 µM for hDHFR), with a selectivity ratio of 35 (Gangjee et al., 2013 ▶).
Figure 1.

Schematics of the pyrido[2,3-d]pyrimidines under study. The numbers correspond to those used in Gangjee et al. (2013 ▶).
To further define the interactions that enhance the selectivity of binding and to directly compare the binding of the most potent analogue in the series, N 6-methyl-N 6-(3,4,5-trifluorophenyl)pyrido[2,3-d]pyrimidine-2,4,6-triamine (compound 26), with native hDHFR, pcDHFR and pjDHFR, we report crystal structure data for hDHFR in complex with N 6-methyl-N 6-(4-isopropylphenyl)pyrido[2,3-d]pyrimidine-2,4,6-triamine (compound 22), hDHFR in complex with 26 and pcDHFR in complex with N 6-methyl-N 6-1-naphthylpyrido[2,3-d]pyrimidine-2,4,6-triamine (compound 24) as ternary complexes with NADPH. Additionally, we report the IC50 binding affinity of N 6-methyl-N 6-phenylpyrido[2,3-d]pyrimidine-2,4,6-triamine (compound 16) and 26 (Fig. 1 ▶) against the double variant F69N/K37S of pcDHFR, the crystal structure of which has previously been reported, in order to verify that the activity of the variant was not significantly different from that of the native enzyme; a question that had been raised by reviewers (Gangjee et al., 2013 ▶).
2. Methods
2.1. Crystallization
Recombinant hDHFR (Gangjee et al., 2009 ▶) and pcDHFR (Gangjee et al., 2013 ▶) were expressed and purified as described previously. The hDHFR protein was washed in a Centricon-10 with 100 mM K2HPO4 buffer pH 6.9 with 30% saturated ammonium sulfate and concentrated to 7.9 mg ml−1, and the pcDHFR protein was washed with 50 mM MES buffer pH 6.0, 100 ml KCl and concentrated to 11.4–14.1 mg ml−1. All DHFR samples were incubated for 1 h on ice with a tenfold excess of NADPH and compounds 22, 24 and 26, respectively, prior to crystallization using the hanging-drop vapor-diffusion method using siliconized glass cover slips and storage at 14°C. Protein droplets of the hDHFR complexes contained K2HPO4 pH 6.9 with 30% saturated ammonium sulfate equilibrated against a reservoir solution consisting of 100 mM K2HPO4 buffer pH 6.9 with 60% saturated ammonium sulfate, 3%(v/v) ethanol, while the protein droplets for the pcDHFR complex consisted of 35% PEG 2K, 49 mM MES pH 6.0, 100 mM KCl equilibrated against a reservoir solution consisting of 0.1 M HEPES pH 7.5, 25% PEG 2000 MME. Crystals of the pcDHFR–24–NADPH ternary complex were monoclinic and belonged to space group P21. The crystals of the two hDHFR complexes were hexagonal and belonged to space group H3. Data were collected at 100 K to between 1.54 and 1.62 Å resolution for the three DHFR complexes using the remote-access robot on beamline 11-1 at the Stanford Synchrotron Radiation Laboratory (SSRL; Cohen et al., 2002 ▶; González et al., 2008 ▶; McPhillips et al., 2002 ▶). The data for these crystals were processed using the XDS program package (Kabsch, 2010 ▶). The diffraction statistics are shown in Table 1 ▶.
Table 1. Crystal data for the antifolates under study.
Values in parentheses are for the highest resolution shell.
| hDHFR22NADPH | hDHFR26NADPH | pcDHFR24NADPH | |
|---|---|---|---|
| PDB code | 4qhv | 4qjc | 4qjz |
| Data collection | |||
| Space group | H3 | H3 | P21 |
| Unit-cell parameters | |||
| a () | 84.4 | 84.5 | 36.7 |
| b () | 84.4 | 84.5 | 42.9 |
| c () | 78.3 | 77.9 | 61.0 |
| () | 90.0 | 90.0 | 90.0 |
| () | 90.0 | 90.0 | 95.3 |
| () | 120.0 | 120.0 | 90.0 |
| Beamline | 11-1, SSRL | 11-1, SSRL | 11-1, SSRL |
| Resolution () | 34.51.54 (1.621.54) | 34.41.54 (1.621.54) | 35.01.61 (1.721.61) |
| Wavelength () | 0.975 | 0.975 | 0.975 |
| R merge † (%) | 0.026 (0.79) | 0.062 (0.75) | 0.050 (0.70) |
| Completeness (%) | 92.8 (75.4) | 96.3 (83.0) | 95.8 (97.0) |
| Observed reflections | 72513 (7697) | 97495 (9939) | 61131 (9036) |
| Unique reflections | 28762 (3398) | 29910 (3757) | 23617 (3467) |
| I/(I) | 21.3 (4.5) | 10.2 (1.2) | 12.6 (1.5) |
| Multiplicity | 2.5 (2.3) | 3.3 (2.6) | 2.6 (2.6) |
| Refinement model | |||
| Resolution () | 34.51.54 | 34.41.54 | 35.01.61 |
| No. of reflections | 24506 | 24614 | 22409 |
| R factor‡ | 0.178 | 0.183 | 0.204 |
| R free § | 0.222 | 0.217 | 0.244 |
| Total protein atoms | 1757 | 1678 | 1851 |
| Total waters | 154 | 105 | 73 |
| Average B factor (2) | 19.1 | 23.2 | 22.3 |
| R.m.s.d. from ideal | |||
| Bond lengths () | 0.025 | 0.026 | 0.02 |
| Bond angles () | 2.85 | 2.52 | 2.19 |
| Luzzati plot error () | 0.168 | 0.218 | 0.205 |
| Ramachandran plot (%) | |||
| Most favored | 98.4 | 98.4 | 95.5 |
| Additional allowed | 1.1 | 1.6 | 3.0 |
| Generously allowed | 0.5 | 0.0 | 1.5 |
| Disallowed | 0.0 | 0.0 | 0.0 |
R
merge = 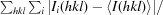
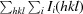 , where I(hkl) is the mean intensity of a set of equivalent reflections.
, where I(hkl) is the mean intensity of a set of equivalent reflections.
R factor = 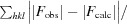
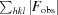 , where F
obs and F
calc are observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are observed and calculated structure-factor amplitudes, respectively.
R free was calculated as for the R factor for a random 5% subset of all reflections.
2.2. Structure solutions
The three structures were solved by molecular-replacement methods using the coordinates of pcDHFR (PDB entry 4ixe; Gangjee et al., 2013 ▶) for the pcDHFR complex and those of hDHFR (PDB entry 1u72; Cody et al., 2005 ▶) for the two hDHFR complexes in MOLREP (Vagin & Teplyakov, 2010 ▶). Inspection of the resulting difference electron-density maps was made using Coot (Emsley et al., 2010 ▶) running on an iMac workstation and revealed density for the ternary complex in all three crystals. Refinement was carried out using REFMAC5 (Murshudov et al., 2011 ▶) in the CCP4 suite of programs (Winn et al., 2011 ▶). The Ramachandran conformational parameters, based on the last refinement cycle, were generated by RAMPAGE (Lovell et al., 2002 ▶) and showed that more than 96% of the residues have the most favored conformation and that none are in disallowed regions. Coordinates for these structures have been deposited in the Protein Data Bank (hDHFR–22, PDB entry 4qhv; hDHFR–26, PDB entry 4qjc; pcDHFR–24, PDB entry 4qjz).
3. Results
The crystal structures of three pyrido[2,3-d]pyrimidine antifolates (22, 24 and 26; Fig. 1 ▶) are reported here as ternary complexes with either native human or native P. carinii DHFR in an effort to correlate their structure–activity relationships. These complexes are compared with previously reported complexes with inhibitors 16, 22 and 26 (Fig. 1 ▶) with variant pcDHFR (Gangjee et al., 2013 ▶).
Analysis of the crystal structure of the hDHFR–26–NADPH ternary complex reported here reveals differences in the binding geometry of 26 from that in the pcDHFR–26–NADPH complex (Gangjee et al., 2013 ▶; Fig. 2 ▶, Table 2 ▶). The trifluorophenyl ring of 26 in the hDHFR complex occupies a pocket closer to the cofactor-binding site such that the 3′,4′,5′-trifluoro atoms make close intermolecular contacts with residue Ser59, whereas the trifluoro atoms make weaker contacts with the side chain of Phe69 in the pcDHFR–26–NADPH ternary complex (Fig. 2 ▶ a; Table 2 ▶; Gangjee et al., 2013 ▶). Differences in the binding geometry of compound 26 with both hDHFR and pcDHFR are also highlighted in Table 2 ▶.
Figure 2.

(a) Comparison of the contacts of the 3′,4′,5′-trifluoro atoms with the side chains of Ser59/64 and Asn64/Phe69 for hDHFR and pcDHFR, respectively. (b) Comparison of the binding of 26 in hDHFR (green; PDB entry 4qjc) and pcDHFR (yellow; PDB entry 4ixe; Gangjee et al., 2013 ▶), highlighting the contacts of the N9-methyl group with Val115 and Ile123 in hDHFR and pcDHFR, respectively.
Table 2. Comparison of F-atom contacts and bridge torsion angles for inhibitor 26 .
| hDHFR26 | pcDHFR26 | ||
|---|---|---|---|
| Trifluoro contacts () | |||
| 3FAsp21 carbonyl | 3.5 | 3FIle65CG2 | 3.9 |
| 4FSer59carbonyl | 3.4 | 4FPhe69CE | 5.8 |
| 5FIle60CG | 3.9 | 5FIle123CD1 | 6.9 |
| Bridge conformation () | |||
| C7C6N9C1 | 3.9 | C7C6N9C1 | 33.2 |
| C6N9C1C6 | 63.2 | C6N9C1C6 | 45.9 |
| C7C6N9CH3 | 167.0 | C7C6N9CH3 | 152.0 |
Comparison of the intermolecular contacts between the N9-methyl group and Val115/Ile123 validates the hypothesis that the N-methyl substituent interacts with Ile123 preferentially compared with the shorter Val115 in hDHFR (Fig. 2 ▶ b). These data show that the intermolecular contact between the N9-methyl group and Ile123 (4.8 Å) in pcDHFR is shorter than the contact in hDHFR (Val115, 5.9 Å; Table 3 ▶). The weaker contact in the hDHFR structure is consistent with its weaker binding affinity compared with pjDHFR. However, the weaker binding of pcDHFR compared with hDHFR suggests that this contact does not tell the complete story.
Table 3. Contacts () involving the inhibitor N9 CH3 with Val115 (hDHFR) or Ile123 (pcDHFR), Thr56 (hDHFR) or Thr61 (pcDHFR), and Ile60 (hDHFR) or Ile65 (pcDHFR).
The shortest distances for each complex are given for comparison.
| DHFR | N9 CH3Val115/Ile123 | N9 CH3Thr56/Thr61 | N9 CH3Ile60/Ile65 |
|---|---|---|---|
| hDHFR22 | Carbonyl O, 5.3; CG1, 6.9 | CG2, 4.6 | CG1, 4.5 |
| pcDHFR22 (F69N) | Carbonyl O, 4.7; CG1, 4.8 | CG2, 3.8 | CG1, 4.7 |
| hDHFR26 | Carbonyl O, 5.2; CG2, 5.8 | CG2, 3.9 | CG1, 4.5 |
| pcDHFR26 | Carbonyl O, 4.5; CG1, 5.4 | CG2, 3.6 | CG1, 5.2 |
| pcDHFR24 | Carbonyl O, 4.5; CD1, 4.3 | CG2, 3.6 | CG1, 4.2 |
Interestingly, analysis of the 2F o − F c electron-density map of the hDHFR complex reveals a trace of positive density (3.5σ) that is occupied by the trifluorophenyl ring in the pcDHFR complex (Fig. 3 ▶). During refinement, a partial occupancy model of the trifluorophenyl ring in two alternate conformations was refined to incorporate this density; however, there was insufficient density to build up the rest of the phenyl ring to warrant accepting this as a final model. Additionally, the final refinement has the cofactor NADPH present at half-occupancy, with the phosphate positions in the backbone of the cofactor also occupied by sulfate from the crystallization medium.
Figure 3.

Stereo comparison of the 2F o − F c difference electron-density map (1σ) for the hDHFR–26–NADPH ternary complex (yellow) with the fit of the pcDHFR–26–NADPH complex (green). Also shown is the 3.5σ F o − F c density (green), which has a partial fit for an alternate conformation of the trifluorophenyl ring as observed in the pcDHFR complex (green).
The data for hDHFR–22–NADPH shows that the binding of compound 22 is similar to that observed in pcDHFR-F69N–22–NADPH (Gangjee et al., 2013 ▶). The small variation in the conformation about the N9-methyl bridge results in a much weaker contact between the N9-methyl group and Val115 (7.1 Å) in the hDHFR complex than the same contact to Ile123 (4.8 Å) in the pcDHFR-F69N complex (Fig. 4 ▶). These structural results are consistent with the activity of 22 against hDHFR and pcDHFR. Unlike compounds 24 and 26, there are few interactions of the 4-propyl group with the active-site residues.
Figure 4.

Comparison of hDHFR–22–NADPH (green; PDB entry 4qhv) with pcDHFR-F69N–22–NADPH (yellow; PDB entry 4ixf; Gangjee et al., 2013 ▶), highlighting the contact of the N9-methyl group with Val115 (green) and Ile123 (yellow) of the pcDHFR complex.
Analysis of the structural data for the pcDHFR–24–NADPH ternary complex indicates that the binding orientation of 24 is similar to that in pcDHFR–26–NADPH (Fig. 5 ▶) and that the contact of the N9-methyl group is the shortest (4.4 Å) of the pcDHFR complexes in this series. These data show that the naphthyl moiety makes several intermolecular contacts with 4.5 Å of the side chains of residues Phe69, Pro66, Ile65 and Ile33. These hydrophobic interactions help stabilize the binding.
Figure 5.

Comparison of pcDHFR–24–NADPH (green; PDB entry 4qjz) with pcDHFR–26–NAPDH (yellow; PDB entry 4ixe; Gangjee et al., 2013 ▶).
Activity data for the pcDHFR F69N/K37S double variant reveal that there is a slight decrease in binding affinity of 16 for the variant (IC50 of 0.10 and 0.076 µM for mutant and wild-type pcDHFR, respectively), whereas there is an increase in the binding affinity of 26 for the pcDHFR variant (IC50 of 0.094 and 0.23 µM for mutant and wild-type pcDHFR, respectively). However, these differences in IC50 are not significant. These data suggest that the F69N/K37S variants do not have a significant influence on binding kinetics and that the crystal structures of these variants (Gangjee et al., 2013 ▶) is representative of the binding to the native DHFR enzyme.
4. Discussion
The crystal structures of three pyrido[2,3-d]pyrimidine antifolates are reported in complex with either human or P. carinii DHFR in an effort to understand their structure–activity relationships. The activity data reported for these analogues (Gangjee et al., 2013 ▶), as well as for a variant form of pcDHFR (F69N/K37S), revealed that compound 26 was 35-fold more selective for binding P. jirovecii DHFR (pjDHFR) than the human enzyme. These structural results validate the hypothesis that the N9-methyl group interacts more favorably with Ile123 (present in both pcDHFR and pjDHFR) than with Val115 in hDHFR. These contacts range from 4.4 to 4.8 Å for pcDHFR and from 5.9 to 7.1 Å for hDHFR (Table 3 ▶).
Structure–activity correlations are more difficult to evaluate based on other active-site interactions. These data reveal that enzyme inhibition is at least tenfold greater for pjDHFR than for pcDHFR and that inhibition is somewhat weaker for hDHFR. The one anomaly is the potency of 24 with hDHFR.
Supplementary Material
PDB reference: human dihydrofolate reductase, complex with pyrido[2,3-d]pyrimidine inhibitor, 4qhv
PDB reference: 4qjc
PDB reference: Pneumocystis carinii dihydrofolate reductase, complex with pyrido[2,3-d]pyrimidine inhibitor, 4qjz
Acknowledgments
This work was supported in part by National Institutes of Health Institute of Allergy and Infectious Diseases grant AI069966 (AG) and by the Greater Buffalo Community Foundation (VC).
References
- Cody, V., Luft, J. R. & Pangborn, W. (2005). Acta Cryst. D61, 147–155. [DOI] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst. 35, 720–726. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Falloon, J. et al. (1990). Clin. Res. 38, 361A.
- Gangjee, A., Li, W., Lin, L., Zeng, Y., Ihnat, M., Warnke, L. A., Green, D. W., Cody, V., Pace, J. & Queener, S. F. (2009). Bioorg. Med. Chem. 17, 7324–7336. [DOI] [PMC free article] [PubMed]
- Gangjee, A., Namjoshi, O. A., Raghavan, S., Queener, S. F., Kisliuk, R. L. & Cody, V. (2013). J. Med. Chem. 56, 4422–4441. [DOI] [PMC free article] [PubMed]
- González, A., Moorhead, P., McPhillips, S. E., Song, J., Sharp, K., Taylor, J. R., Adams, P. D., Sauter, N. K. & Soltis, S. M. (2008). J. Appl. Cryst. 41, 176–184.
- Grivsky, E. M., Lee, S., Sigel, C. W., Duch, D. S. & Nichol, C. A. (1980). J. Med. Chem. 23, 327–329. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kovacs, J. A., Allegra, C. J., Swan, J. C., Drake, J. C., Parrillo, J. E., Chabner, B. A. & Masur, H. (1988). Antimicrob. Agents Chemother. 32, 430–433. [DOI] [PMC free article] [PubMed]
- Lovell, S. C., Davis, I. W., Arendell, W. B. III, de Baker, P. L. W., Word, J. M., Prisant, M. G., Richardson, J. S. & Richardson, D. C. (2002). Proteins, 50, 437–450. [DOI] [PubMed]
- McPhillips, T. M., McPhillips, S. E., Chiu, H.-J., Cohen, A. E., Deacon, A. M., Ellis, P. J., Garman, E., Gonzalez, A., Sauter, N. K., Phizackerley, R. P., Soltis, S. M. & Kuhn, P. (2002). J. Synchrotron Rad. 9, 401–406. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human dihydrofolate reductase, complex with pyrido[2,3-d]pyrimidine inhibitor, 4qhv
PDB reference: 4qjc
PDB reference: Pneumocystis carinii dihydrofolate reductase, complex with pyrido[2,3-d]pyrimidine inhibitor, 4qjz