

Supplemental Digital Content is Available in the Text.
Key Words: Qiliqiangxin, heart failure, pressure overload, cardiac remodeling
Abstract:
We previously showed that Qiliqiangxin (QL) capsules could ameliorate cardiac hypertrophy and remodeling in a mouse model of pressure overload. Here, we compared the effects of QL alone with those of QL combined with the following 3 types of antihypertensive drugs on cardiac remodeling and dysfunction induced by pressure overload for 4 weeks in mice: an angiotensin II type 1 receptor (AT1-R) blocker (ARB), an angiotensin-converting enzyme inhibitor (ACEI), and a β-adrenergic receptor (β-AR) blocker (BB). Adult male mice (C57B/L6) were subjected to either transverse aortic constriction or sham operation for 4 weeks, and the drugs (or saline) were orally administered through gastric tubes. Cardiac function and remodeling were evaluated through echocardiography, catheterization, histology, and analysis of hypertrophic gene expression. Cardiomyocyte apoptosis and autophagy, AT1-R and β1-AR expression, and cell proliferation–related molecules were also examined. Although pressure overload–induced cardiac remodeling and dysfunction, hypertrophic gene reprogramming, AT1-R and β1-AR expression, and ERK phosphorylation were significantly attenuated by QL alone, QL + ARB, QL + ACEI, and QL + BB, the attenuation was stronger in the combination treatment groups. Moreover, apoptosis was reduced to a larger extent by each combination treatment than by QL alone, whereas autophagy was more strongly attenuated by either QL + ARB or QL + ACEI. None of the treatments significantly upregulated ErbB2 or ErbB4 phosphorylation, and none significantly downregulated C/EBPβ expression. Therefore, the effects of QL on chronic pressure overload–induced cardiac remodeling may be significantly increased when QL is combined with an ARB, an ACEI, or a BB.
INTRODUCTION
Hypertension, which is one of the most common causes of heart failure, reportedly leads to the development of cardiac hypertrophy, which ultimately progresses to heart failure.1,2 Chronic pressure overload–induced adaptive cardiac hypertrophy is initially characterized by a thickened ventricular wall and by enhanced left ventricular systolic function. Excessive activation of the renin–angiotensin–aldosterone system and other neuroendocrine systems, as well as the release of angiotensin II (Ang II) and catecholamines, results in the development of irreversible chronic heart failure.3,4 Despite significant improvements in the understanding of this disease, as well as the effort expended to treat it, the prognosis of heart failure continues to be poor.5,6
Qiliqiangxin (QL) capsules contain a specific traditional Chinese medicine formulation that includes extracts from 11 types of herbs, including Radix Astragali, aconite root, Salvia miltiorrhiza, Ginseng, Semen Lepidii Apetali, Carthamus tinctorius, Cortex Periplocae Sepii Radicis, Rhizoma Alismatis, seasoned orange peel, Polygonatum Odorati, and Rumulus Ginnamomi, based on the meridian theory. Radix astragali is the principal active pharmacological component.7 QL has been demonstrated to be both a safe and efficient treatment for heart failure in both animal models and clinical trials.7–10 In 2004, QL capsules were approved by the Chinese Food and Drug Administration for the treatment of patients with heart failure. Our previous study demonstrated that QL suppressed myocardial inflammation, cardiomyocyte apoptosis, and autophagy while promoting cardiomyocyte proliferation, which resulted in the amelioration of pressure overload–induced cardiac remodeling and cardiac dysfunction.11 Other studies have revealed that QL may improve cardiac dysfunction in spontaneous hypertensive rats by inhibiting the cardiac chymase signaling pathway and that QL may have antiarrhythmic properties that enable it to regulate L-type Ca currents, Na currents, and K currents in rat ventricular myocytes.8,12
According to the 2013 AHA guidelines for the management of heart failure, diuretics, angiotensin-converting enzyme inhibitors (ACEIs), ARBs, beta-blockers, aldosterone receptor antagonists, and other agents are recommended as standard therapies for chronic heart failure.1 However, it is not clear whether combining these drugs with QL can enhance its effects on chronic heart failure. Recently, a multicenter, randomized, double-blind and placebo-controlled study revealed that QL further decreased the level of NT-proBNP in patients with chronic heart failure when used together with standard therapy. These results suggest that QL in combination with standard therapy may represent an improved means of treating chronic heart failure.7 In this study, we treated mice suffering from pressure overload with either QL alone or with QL in combination with olmesartan (ARB), captopril (ACEI) or metoprolol (BB), as each of these drugs is widely prescribed in clinical practice to treat chronic heart failure.13–15 We aimed to determine whether QL combined with these antihypertensive agents exerted superior cardioprotective effects compared with QL alone in the setting of chronic pressure overload–induced cardiac remodeling. We also attempted to determine whether the suppression of cardiomyocyte apoptosis and autophagy as well as the upregulation of cardiomyocyte proliferation as a result of QL treatment were affected by the use of the 3 aforementioned classes of drugs.
MATERIALS AND METHODS
Animal Models
C57BL/6 male mice (Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China) that were aged 8–10 weeks were anesthetized and underwent either a transverse aortic constriction (TAC) or a sham operation, as previously described.16,17 In brief, after anesthetization, the transverse aorta was constricted with a 7-0 nylon suture by ligating the aorta together with a blunted 27-gauge needle, which was later removed. The animal experimental protocols were carried out in compliance with the Guidelines for the Care and Use of Laboratory Animals (published by the National Academy Press: National Institutes of Health Publication No. 85-23, revised 1996) and approved by the Animal Care and Use Committee of Fudan University.
Administration of Drugs
All drugs, including Qiliqiangxin (Shijiazhuang Yiling Pharmaceutical, Shijiazhuang, China), olmesartan (Daiichi Sankyo Pharmaceutical, Shanghai, China), captopril (Bristol-Myers Squibb, Shanghai, China), and metoprolol (AstraZeneca Pharmaceutical, Shanghai, China), were purchased commercially. The mice were randomly divided into the following 6 groups: the Sham group (n = 7), the TAC group (n = 7), the QL group (n = 7), the QL + olmesartan group (n = 7), the QL + captopril group (n = 7), and the QL + metoprolol group (n = 7). Each of the drugs was dissolved in distilled water, and equal volumes of freshly prepared solution or distilled water (0.2 mL) were administered to mice daily through a gastric tube for 4 weeks. The dosages of QL, olmesartan, captopril, and metoprolol were 0.6, 5.4, 10, and 30 mg·kg·−1d·−1, respectively. The dosage chosen for each drug was based on clinically relevant concentrations and previously published data.18–20
Echocardiography and Hemodynamic Measurements
Transthoracic echocardiography was performed using a 30-MHz high-frequency scan head (VisualSonics Vevo770; VisualSonics, Toronto, Canada). The mice were anesthetized with a mixture of isoflurane (2%) and oxygen (2 L/min). All measurements were averaged over 5 consecutive cardiac cycles and were carried out by 3 technicians who were blinded to the experimental group identities. Aortic blood pressure (ABP) was evaluated as described.18 In brief, a micro-nanometer catheter (Millar 1.4F, SPR 835; Millar Instruments, Inc, Houston, TX) was inserted into the right common carotid artery and ultimately introduced into the LV, and the transducer was connected to a Power Laboratory system (AD Instruments, Castle Hill, Australia) to record ABP, LV end-systolic pressure, LV end-diastolic pressure, and dP/dT.
Morphological and Histological Analyses
The mice were killed, and the hearts were excised at 4 weeks after TAC. The excised hearts were weighed, perfused with PBS, and fixed with 4% polyformaldehyde for global morphometry before being embedded in paraffin or frozen in liquid nitrogen for further histological analysis. The paraffin-embedded hearts were sectioned at a thickness of 4 μm and stained with either hematoxylin and eosin (H&E) or Masson's trichrome. For measurements, 5 random high-power fields from each section were chosen and quantified in a blinded manner. The cross-sectional area (CSA) of the cardiomyocytes was analyzed quantitatively through morphometric analysis of the H&E-stained sections. The extent of the fibrosis was evaluated by measuring the Masson's trichrome–stained area within the entire LV wall. Five sections of each heart were examined. The images were measured using an automated image analysis system (Image-Pro Plus 5.0; Media Cybernetics, Rockville, MD).
Real-time RT-PCR
Total RNA was extracted from the heart tissues using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions, and reverse-transcription–polymerase chain reaction (RT-PCR) was performed using a TOYOBO RT-PCR kit. After purification, real-time RT-PCR analysis of the expression of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), skeletal α-actin (SAA), and sarcoplasmic reticulum Ca2+ adenosine triphosphatase (SERCA2a) was performed using a Bio-RAD IQ5 multicolor detection system (all the primers are listed in Table 1). The melting curves and quantification were analyzed using Bio-RAD software. The comparative cycle threshold method was used to determine the relative RNA expression levels. Each of the PCRs was repeated at least 3 times.
TABLE 1.
Primers for Real-time Reverse Transcription–PCR

Western Blot Analysis
Total proteins isolated from the heart tissues were size fractionated using SDS-PAGE and transferred onto Immobilon-P membranes (Millipore, Billerica, MA). The blotted membranes were incubated with antibodies against p-ERK, t-ERK, p-ErbB2, p-ErbB4, LC3b (Cell Signaling Technology, Beverly, MA), β1-AR (beta-1 adrenergic receptor; Abcam, Cambridge, MA), C/EBPβ, and AT1-R, (Santa Cruz Biotechnology Inc, Santa Cruz, CA) and with an HRP-conjugated secondary antibody (1:5000, KangChen Biotechnology, Shanghai, China). Either GAPDH or t-ERK was used as an internal control. The proteins were visualized using an ECL Western blotting detection system (GE Healthcare, catalog number RPN2106). The relative intensities of the protein bands were analyzed through densitometry with a gel documentation system using LAS-300 image analysis software. All experiments were repeated at least 3 times.
Apoptotic Cell Analysis by TUNEL Labeling
TUNEL labeling was conducted in accordance with the manufacturer's protocol (In Situ Cell Death Detection kit; Merck Inc, Darmstadt, Germany). The paraffin-embedded slides were incubated with 50 μL of TUNEL reaction mixture containing TdT for 1 hour at 37°C. After washing, the DAB substrate solution was dispensed dropwise onto the slides and incubated for 5 minutes. The apoptosis-positive cells were counted in 20 randomly selected fields from each slide. The results were recorded as the number of apoptosis-positive cells per 105 cardiomyocytes.
Immunofluorescence
Autophagy and cardiomyocyte proliferation were each evaluated through immunofluorescence staining of frozen slides with anti-α-MHC (Upstate, Lake Placid, NY) and LC3b (Cell Signaling Technology) or Ki67 (Abcam). The slides were then incubated with secondary antibodies conjugated with FITC or Alexa (Invitrogen) according to the manufacturer's protocol. The LC3b and Ki67-positive aggregates in the cardiomyocytes were counted in 20 randomly selected fields from each slide and expressed as the numbers of LC3b-positive dots and Ki-67-positive cells per 105 cardiomyocytes.
Statistical Analysis
All data are expressed as the mean ± standard errors of the mean. Group mean values were compared by 1-way analysis of variance followed by an least significant difference (LSD) test. Comparisons between 2 groups were conducted using a 2-tailed Student's t test. A value of P < 0.05 was considered statistically significant.
RESULTS
Effects of QL Alone or QL in Combination With Olmesartan, Captopril, or Metoprolol on the Hemodynamic Parameters and Cardiac Function of Mice Suffering From Pressure Overload
Four weeks of TAC induced cardiac remodeling characterized by reduced cardiac contractility and a reduced ejection fraction. We investigated the improvements in cardiac remodeling induced by either QL alone or by QL in combination with olmesartan (QL + olm), captopril (QL + cap), or metoprolol (QL + met) at 4 weeks after the TAC operation. As expected, TAC induced an obvious increase in ABP, left ventricular end-systolic pressure, and left ventricular end-diastolic pressure based on the results of hemodynamic analysis. QL, QL + olm, QL + cap, and QL + met did not affect ABP or left ventricular end-systolic pressure after TAC but significantly attenuated the elevation of left ventricular end-diastolic pressure induced by TAC (Fig. 1A and see Figure 1, Supplemental Digital Content, http://links.lww.com/JCVP/A180). Both +dp/dtmax and −dp/dtmax, indices of cardiomyocyte contractility, were significantly decreased by TAC; QL induced significant increases in +dp/dtmax and −dp/dtmax after TAC. QL + olm, QL + cap, and QL + met induced higher + dp/dtmax and −dp/dtmax values than QL alone after TAC (Fig. 1B and see Figure 2, Supplemental Digital Content, http://links.lww.com/JCVP/A180). An echocardiographic analysis indicated that 4 weeks of TAC resulted in a significantly decreased left ventricular ejection fraction; QL significantly attenuated this effect, and olm, cap, and met amplified the protective effect exerted by QL (Table 2). However, there was no difference in these effects among the QL + olm, QL + cap, or QL + met groups. These data indicated that QL in combination with olmesartan, captopril, or metoprolol had superior protective effects on cardiac contractility and cardiac function compared with QL alone under similar pressure overload conditions.
FIGURE 1.

Effects of QL alone or QL in combination with olmesartan, captopril, or metoprolol on hemodynamic parameters. Mice were subjected to either a sham operation or TAC for 4 weeks and administered saline, QL (0.6 mg·kg−1·d−1), QL (0.6 mg·kg−1·d−1) plus olmesartan (5.4 mg·kg−1·d−1), QL (0.6 mg·kg−1·d−1) plus captopril (10 mg·kg−1·d−1), or QL (0.6 mg·kg−1·d−1) plus metoprolol (30 mg·kg−1·d−1). (A), Quantitative analyses of ABP, LVESP, and LVEDP are shown. (B), Quantitative analyses of +dP/dtmax and −dP/dtmax. Values are expressed as the mean ± standard errors of the mean from 7 mice. *P < 0.05 and **P < 0.01 versus the sham group; P < 0.05 versus the TAC group.
TABLE 2.
Echocardiographic Parameters 4 Weeks After TAC

Inhibitory Effects of QL Alone or QL in Combination With Olmesartan, Captopril, or Metoprolol on Hypertrophic Responses Induced by Pressure Overload
Maladaptive cardiac hypertrophy results in heart failure in the setting of pressure overload.21 In this study, TAC induced cardiac hypertrophy characterized by an elevated heart weight-to-body weight ratio, increased cardiomyocyte CSA, increased LV anterior wall thickness during end-diastole, and decreased LV posterior wall thickness during end systole. QL greatly attenuated these effects; QL + olm, QL + cap, or QL + met inhibited the aforementioned hypertrophic responses to a larger extent than QL alone after TAC. Masson's trichrome staining indicated that QL reduced the fibrotic areas induced by TAC. QL + olm, QL + cap, and QL + met treatments resulted in significant decreases in fibrotic areas compared with QL alone (Figs. 2A, B). In addition, we investigated the expression of hypertrophic genes, such as atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), skeleton α-actin (SAA), and sarcoplasmic reticulum Ca2+ adenosine triphosphatase 2 (SERCA2) in heart tissue. Pressure overload resulted in the significant upregulation of ANP, BNP, and SAA gene expression and downregulation of SERCA2 gene expression; these effects were partially abolished in the QL group and in the combination therapy groups. QL + olm, QL + cap, and QL + met demonstrated superior inhibitory effects on the expression of the hypertrophic genes compared with QL alone. There was no significant difference among the QL + olm, QL + cap, or QL + met groups in terms of their effects on hypertrophic gene expression (Fig. 2C). These results indicated that QL in combination with olmesartan, captopril, or metoprolol exerted stronger inhibitory effects on cardiac hypertrophy than QL alone. This finding indicates that combination therapy may result in improvements in cardiac function that are superior to those induced by treatment with QL alone in the setting of heart failure induced by pressure overload.
FIGURE 2.

Effects of QL alone or QL in combination with olmesartan, captopril, or metoprolol on cardiac morphology, histology, echocardiography, and hypertrophic gene expression. (A), Representative images of the global heart, HE staining, Masson's trichrome staining (scale bar: 20 mm), and M-mode echocardiography. (B), The ratio of heart weight to body weight (HW/BW) and the cross-sectional and fibrotic areas of cardiomyocytes were analyzed. Values are expressed as the mean ± standard errors of the mean from 7 mice; *P < 0.05 versus the sham group; &P < 0.05 versus the TAC group; #P < 0.05 versus the QL group. (C), The expression of ANP, BNP, SAA, and SERCA2 mRNA was evaluated through real-time RT-PCR. GAPDH was used as an internal control. Values were calculated as fold changes compared with GAPDH and expressed as the mean ± standard errors of the mean from 7 mice. *P < 0.05 versus the sham group; &P < 0.05 versus the TAC group; #P < 0.05 versus the QL group.
Inhibitory Effects of QL Alone or QL in Combination With Olmesartan, Captopril, or Metoprolol on TAC-induced Cardiomyocyte Apoptosis and Autophagy
It has been demonstrated that cardiomyocyte apoptosis and autophagy are required for the transition from compensated cardiac hypertrophy to heart failure.21–24 Therefore, we examined the effects of QL and combination therapy on each of these cellular processes using TUNEL labeling and immunofluorescence staining. After 4 weeks, chronic pressure overload induced larger numbers of TUNEL-positive cells and LC3b-positive cells in the heart based on the results of the immunostaining analysis (Figs. 3A, B). The results of a Western blot were consistent with the LC3b expression levels in heart tissue. Treatment with QL significantly attenuated TAC-induced cardiac apoptosis and autophagy. QL + olm, QL + cap, and QL + met decreased the numbers of TUNEL-positive cells, and QL + olm and QL + cap decreased the numbers of LC3B-positive cells and decreased the level of LC3b expression in heart tissue to a larger extent than QL alone (Fig. 3C).
FIGURE 3.

Effects of QL alone or QL in combination with olmesartan, captopril, or metoprolol on myocardial apoptosis and autophagy. (A), Representative images of TUNEL staining (brown, scale bar: 50 mm) and immunohistological staining (scale bar: 20 mm) with antibodies against LC3b (green) and α-MHC (red); the nuclei were stained by DAPI (blue) in the LV tissues. The black arrows indicate TUNEL-positive cardiomyocytes. (B), Quantitative analysis of apoptosis and autophagy in the LV tissues. TUNEL-positive cardiomyocytes and LC3b-positive aggregates were analyzed in 20 fields that were randomly selected from each section of the LV wall. Five sections from each heart were measured, and the numbers of TUNEL-positive cardiomyocytes and LC3b-positive aggregates per 105 cardiomyocytes were expressed. *P < 0.05 versus the sham group; &P < 0.05 versus the TAC group; #P < 0.05 versus the QL group. (C), Western blot analysis of LC3b-I and LC3b-II expression; GAPDH served as a loading control. The ratio of LC3b-I LC3b-II to GAPDH was calculated. All data are expressed as the mean ± standard errors of the mean from 7 mice (n = 7). *P < 0.05 versus the sham group; &P < 0.05 versus the TAC group; #P < 0.05 versus the QL group.
These results suggested that QL in combination with olmesartan, captopril, or metoprolol exerted greater inhibitory effects on cardiac apoptosis and autophagy compared with QL alone.
Underlying Molecular Mechanism Involved in the Improvement Induced by QL Alone or QL in Combination With Olmesartan, Captopril, or Metoprolol in the Setting of TAC-induced Cardiac Hypertrophy, Apoptosis, and Autophagy
The activation and upregulation of both AT1-R and β1-AR reportedly contributes to the development of cardiac hypertrophy, apoptosis, and autophagy in the setting of pressure overload. In this study, we examined the protein expression of AT1-R and β1-AR in heart tissue. The administration of QL suppressed the upregulation of the 2 proteins induced by TAC. QL + olm and QL + cap induced responses similar to QL treatment alone, and these treatments exerted similar effects on the expression of β1-AR; however, QL + olm and QL + cap reduced the expression level of AT1-R in the myocardium to a greater extent than QL alone. In addition, QL + met reduced the expression of β1-AR to a greater extent than QL alone; however, QL + met and QL inhibited the expression of AT1-R to a similar extent. The activation of either AT1-R or β1-AR induced the phosphorylation of ERK (p-ERK), which contributes to the hypertrophic response commonly observed in the setting of pressure overload.25,26 QL significantly suppressed the upregulation of p-ERK that was induced by TAC; QL + olm, QL + cap, and QL + met enhanced the inhibitory effect exerted by QL on the TAC-induced upregulation of p-ERK. There was no significant difference among the QL + olm, QL + cap, or QL + met groups in terms of their effects on the above-mentioned processes (Fig. 4).
FIGURE 4.
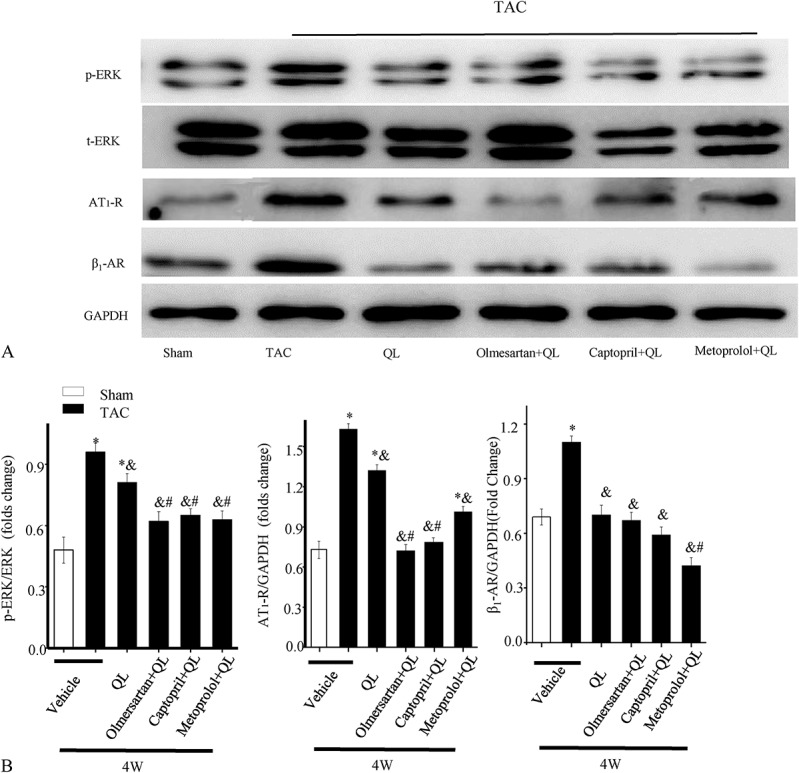
Changes in cardiac remodeling-associated protein expression levels induced by QL alone or QL in combination with olmesartan, captopril, or metoprolol. (A), Western blot analysis of phosphorylated ERK (p-ERK), ERK, AT1-R, and β1-AR; GAPDH served as a loading control. (B), Quantitative analysis of the ratio of p-ERK to ERK and the expression of AT1-R and β1-AR (expressed as fold changes compared with GAPDH). The data are expressed as the mean ± standard errors of the mean from 7 mice (n = 7). *P < 0.05 versus the sham group; &P < 0.05 versus the TAC group; #P < 0.05 versus the QL group.
The addition of olmesartan, captopril, or metoprolol to QL enhances the effects exerted by QL alone on pressure overload–induced cardiac dysfunction.
Effects of QL Alone or QL in Combination With Olmesartan, Captopril, or Metoprolol on Cardiomyocyte Proliferation After TAC
Our previously published data indicated that ErbB family receptors and C/EBPβ may be involved in the effects of QL on cardiac remodeling and subsequent cardiac dysfunction.11 Four weeks of TAC exerted only limited effects on the expression of p-ErbB2, p-ErbB4, and C/EBPβ compared with the sham group (Fig. 5A). QL alone or in combination with olmesartan, captopril, or metoprolol upregulated the expression of both p-ErbB2 and p-ErbB4 and downregulated the expression of C/EBPβ compared with TAC. QL + olm and QL + cap treatment resulted in a higher p-ErbB2 expression level, and QL + cap and QL + met induced an increase in p-ErbB4 expression that was greater than the increase facilitated by QL alone, although this difference was not statistically significant. We then determined the numbers of Ki67-positive cardiomyocytes in the myocardium through double immunostaining methods (Ki67 is an index of proliferation). There were only limited numbers of Ki67-positive cardiomyocytes in both the sham and TAC groups. Both QL alone and QL in combination with olmesartan, captopril, or metoprolol increased the number of Ki67-positive cardiomyocytes in the myocardium after TAC. However, there was no significant difference among the QL, QL + olm, QL + cap, or QL + met groups (Fig. 5B).
FIGURE 5.

Effects of QL alone or in combination with olmesartan, captopril, or metoprolol on cardiomyocyte proliferation during 4 weeks of TAC. (A), Quantitative analysis and representative images of Western blots of C/EBPβ, pErbB2, and pErbB4. GAPDH served as a loading control. Values were calculated for the ratio of C/EBPβ, ErbB2, or ErbB4 to GAPDH. (B), All data are expressed as the mean ± standard errors of the mean from 7 mice (n = 7). *P < 0.05 versus the sham group; &P < 0.05 versus the TAC group. (B), Representative images of immunofluorescence staining for Ki67 (green) and α-MHC (red) in LV sections from heart tissue of the QL treatment group (the white arrow indicates 1 Ki67-positive cardiomyocyte; scale bar: 10 mm).
These results suggested that QL alone and QL in combination with olmesartan, captopril, or metoprolol induced cardiomyocyte proliferation after TAC. However, the increases in cardiomyocyte proliferation noted among these groups were not significantly different.
DISCUSSION
This study has demonstrated both the safety and the efficacy of QL combination therapy with an ARB (olmesartan), an ACEI (captopril), and a BB (metoprolol) in the treatment of chronic pressure overload–induced cardiac hypertrophy in mice. Combination therapy exhibited superior protective effects on cardiac remodeling and dysfunction compared with QL treatment alone. Mechanistically, cardiomyocyte apoptosis was reduced to a larger extent by each of the combination treatments, whereas autophagy was attenuated more significantly by the combination of QL with either an ARB or an ACEI, but not by the combination of QL with a BB. In addition, the expression levels of AT1-R and β1-AR were downregulated more significantly by the combination of QL with either an ARB or an ACEI as well as by the combination of QL with a BB. However, the increase in cardiomyocyte proliferation was not significantly different among the QL, QL + ARB, QL + ACEI, and QL + BB groups.
The AngII/AT1-R system plays a pivotal role in the progression of cardiac hypertrophy and the development of heart failure.27–29 Blocking the generation of AngII or the activation of AT1-R with a renin inhibitor, an ACEI, or an ARB ameliorates cardiac remodeling and dysfunction.30–32 However, therapeutic approaches that involve the combination of an ACEI with an ARB remain controversial. The combination of an ACEI and an ARB did not significantly improve the morbidity and mortality of cerebrovascular disease or congestive heart failure in the CHARM-Added trial,33 However, in the ONTARGET trial, the combination of the 2 therapies worsened renal function compared with the use of an ACEI or an ARB alone.34 This finding suggests that significant risk may be associated with this form of combination therapy. Interestingly, in this study, QL in combination with either olmesartan or captopril improved the cardiac dysfunction induced by pressure overload. Recently, QL reportedly facilitated decreases in the level of NT-proBNP in patients with chronic heart failure as a result of treatment with an ARB or an ACEI. These results suggest that QL in combination with either olmesartan or captopril may exert superior cardioprotective effects compared with an ARB or an ACEI. Metoprolol, one of the most commonly prescribed beta-blockers, exerts its pharmacological effects through the inhibition of adrenergic receptors.35 Many large-scale clinical trials, including the CIBISII, the MERIT-HF, and the COPERNICUS trial, have demonstrated that the long-term use of a BB in patients with heart failure reduces overall mortality, cardiovascular mortality, and the risk of sudden cardiac death.36
During the early phase of pressure overload, adaptive cardiac hypertrophy is beneficial because it enables the heart to retain its normal level of function. However, excessive cardiac hypertrophy and fibrosis lead to irreversible heart failure during late-phase pressure overload.21 In this study, QL in combination with olmesartan, captopril, or metoprolol had a significant inhibitory effect on cardiac hypertrophy and fibrosis in mice in the setting of pressure overload. This effect was greater than that exerted by QL alone and was characterized by decreases in the heart weight-to-body weight ratio, cardiomyocyte CSA, expression of hypertrophic genes, and fibrosis area. These findings may partially explain why QL combined with metoprolol may be more effective for treating patients with heart failure.
Autophagy and apoptosis are 2 self-destructive processes that play an important role in the maintenance of cardiac function in the pathogenesis of heart failure.21,37,38 The crosstalk between these 2 process was only partially uncovered. A number of studies have confirmed that a variety of common upstream stimuli (including pressure overload) can trigger both autophagy and apoptosis.17 Notably, autophagy is a lysosomal degradation procedure, which can be beneficial or detrimental. Mostly, autophagy makes cells to adapt to stress, but massive autophagy can also induce cell death.39 Comparably, apoptosis is a process of programmed cell death by which the targeted cells can be disposed by multicellular organisms.37 Emerging data confirmed that autophagy and apoptosis could interact with each other regarding cell survival and death.40 For example, ingredients of the apoptotic pathways can regulate autophagy process through crosstalk with autophagy-related proteins.41 Similarly, activation of autophagy pathways can reduce apoptotic cell death during certain cellular stages.42 In this study, we observed that pressure overload–induced autophagy and apoptosis of cardiomyocytes were significantly reduced at 4 weeks in the QL group, and cardiomyocyte apoptosis was reduced to a larger extent by each of the combination treatments than by QL alone, whereas autophagy was more strongly attenuated only in combination treatment with olmesartan or captopril. In our previous study, we revealed that autophagy induced by the pressure overload at 4 weeks in mice can be regulated by the AT1-R-mediated p38-MAPK pathway,22 and the expression change of autophagy mark protein LC3b was similar with that of AT1-R in the combination groups. Thus, we inferred that the reasonable explanation for this result may be that treatment with metoprolol has no further influence on the expression of AT1-R, which can regulate the autophagy process induced by the pressure overload in mice. However, we still cannot exclude that the activation of AT1-R or β1-AR could affect the crosstalk between autophagy and apoptosis induced by pressure overload. Indeed, the relationship between these 2 processes seems extremely complex, and insights of the interconnections between the autophagy and apoptosis in the pathogenesis of heart failure induced by pressure overload are required for the further clarification of their common roles in heart failure and cardiac remodeling.
It has been demonstrated that pressure overload may trigger cardiac hypertrophy, fibrosis, apoptosis, or autophagy through the AT1-R-mediated ERK, JNK, or p38-MAPK pathways.22 Beta-adrenergic receptors mediate these signaling pathways primarily through the cAMP/PKA or the ERK pathway.43 In this study, the upregulation of AT1-R and β1-AR as a result of pressure overload was significantly inhibited by both QL and QL combination therapy. QL in combination with either olmesartan or captopril decreased the expression of AT1-R in the myocardium compared with QL alone; however, the effects of these combination treatments on β1-AR expression were similar to those of QL alone. QL in combination with metoprolol inhibited the upregulation of AT1-R, as did QL; however, the combination of the 2 agents decreased the expression of β1-AR to a larger extent than QL alone. The activation of either AT1-R or β1-AR induced the phosphorylation of ERK (p-ERK), which contributes to both the hypertrophic response and to the fibrosis observed in the setting of pressure overload.25,26 In this study, QL significantly suppressed the upregulation of p-ERK induced by TAC, and QL in combination with olmesartan, captopril, or metoprolol decreased the level of p-ERK to a greater extent than QL alone. QL in combination with either olmesartan or captopril exerted an inhibitory effect on cardiac hypertrophy and fibrosis through the downregulation of both AT1-R expression and p-ERK levels, whereas QL in combination with metoprolol achieved similar results by inhibiting β1-AR expression and decreasing p-ERK levels.
Recently, some studies have demonstrated that cardiomyocytes have the potential to proliferate in response to specific stimuli,44,45 and ErbB receptors belong to the epidermal growth factor receptor family.46 The binding of its agonist, Neuregulin1, to ErbB4 increases its kinase activity, induces heterodimerization with either ErbB2 or ErbB4, and stimulates intracellular signal transduction pathways46 that contribute to myocardial regeneration. C/EBPβ, a member of the bHLH family of DNA-binding transcription factors, plays a pivotal role in cell proliferation and differentiation in many tissues and cells, including cardiomyocytes.47 The downregulation of cardiac C/EBPβ levels curtailed the development of pressure overload–induced heart failure in mice.45 Our results indicated that QL treatment increased the phosphorylation of both ErbB2 and ErbB4 and reduced the expression of C/EBPβ compared with vehicle treatment in mice in the setting of pressure overload. However, combination therapy with olmesartan, captopril, and metoprolol did not cause any significant changes in the expression of these proteins compared with QL treatment, suggesting that the signaling pathway mediated by the AT1-R and the beta-adrenergic receptors exerted only a minimal effect on the cardiac regeneration signaling pathway mediated by the ErbB receptor and by CEBP/β at 4 weeks after TAC.
Our present study compared the effects of QL alone and QL combined with an ARB, an ACEI, or a BB on cardiac hypertrophy, remodeling, and dysfunction, each of which may be induced by chronic pressure overload. The results of our study indicated that combination therapy facilitated greater improvements in cardiac hypertrophy, fibrosis, and dysfunction, as well as cardiomyocyte apoptosis and autophagy, in the setting of pressure overload compared with QL alone, which may be indicative of the stronger cardioprotective role played by combination therapy. The mechanism underlying the effects of combination therapy may be related to the downregulation of AT1-R or β1-AR. In addition, both QL alone and combination therapy induced cardiomyocyte proliferation by regulating ErbB family receptors and CEBP/β in the setting of pressure overload; however, there was no difference in the effect exerted by QL alone and the effect exerted by QL in combination with olmesartan, captopril, or metoprolol. Although this study design does not fully reflect the complexity of QL's ability to treat heart failure in clinical practice, it has demonstrated that QL is both a safe and effective therapy for pressure overload–induced cardiac hypertrophy and remodeling in mice. However, the exact molecular mechanisms underlying the cardioprotective effects of QL remain unknown.
In conclusion, these results suggest that compared with QL alone, QL in combination with standard therapies may exert more beneficial effects in the setting of chronic pressure overload–induced cardiac hypertrophy, remodeling, and dysfunction.
ACKNOWLEDGMENTS
The authors thank both Guoping Zhang and Jianguo Jia for their excellent technical support and for their assistance with the experiments.
Footnotes
Supported by grants from the National Natural Science Foundation of China (81220108003, 81370258, and 81200185), the National Basic Research Program of China (973 Program, 2012CB518605), the Science and Technology Commission of Shanghai Municipality (11JC1402400 and 13JC1401703), and the China Doctoral Foundation (20110071110051).
The authors report no conflicts of interest.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jcvp.org).
Y. Ye, H. Gong, and X. Wang have contributed equally.
REFERENCES
- 1.Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–e239. [DOI] [PubMed] [Google Scholar]
- 2.McMurray JJ, Adamopoulos S, Anker SD, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14:803–869. [DOI] [PubMed] [Google Scholar]
- 3.Chinese Society of Cardiology of Chinese Medical A, Editorial Board of Chinese Journal of C. Guidelines for the diagnosis and management of chronic heart failure [in Chinese]. Zhonghua Xin Xue Guan Bing Za Zhi. 2007;35:1076–1095. [PubMed] [Google Scholar]
- 4.Al-Mohammad A, Mant J. The diagnosis and management of chronic heart failure: review following the publication of the NICE guidelines. Heart. 2011;97:411–416. [DOI] [PubMed] [Google Scholar]
- 5.de Virginy DR. Novel and potential future biomarkers for assessment of the severity and prognosis of chronic heart failure: a clinical review. Heart Fail Rev. 2006;11:333–334. [DOI] [PubMed] [Google Scholar]
- 6.Eichhorn EJ, Bristow MR. Medical therapy can improve the biological properties of the chronically failing heart. A new era in the treatment of heart failure. Circulation. 1996;94:2285–2296. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Zhang J, Huang J, et al. A multicenter, randomized, double-blind, parallel-group, placebo-controlled study of the effects of qili qiangxin capsules in patients with chronic heart failure. J Am Coll Cardiol. 2013;62:1065–1072. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, Chen J, Xu T, et al. Qiliqiangxin improves cardiac function in spontaneously hypertensive rats through the inhibition of cardiac chymase. Am J Hypertens. 2012;25:250–260. [DOI] [PubMed] [Google Scholar]
- 9.Xiao H, Song Y, Li Y, et al. Qiliqiangxin regulates the balance between tumor necrosis factor-alpha and interleukin-10 and improves cardiac function in rats with myocardial infarction. Cell Immunol. 2009;260:51–55. [DOI] [PubMed] [Google Scholar]
- 10.Chen F, Wu JL, Fu GS, et al. Chronic treatment with qiliqiangxin ameliorates aortic endothelial cell dysfunction in diabetic rats. J Cardiovasc Pharmacol Ther. 2014;20:230– 240. [DOI] [PubMed] [Google Scholar]
- 11.Zou YZ, Lin L, Ye Y, et al. Qiliqiangxin inhibits the development of cardiac hypertrophy, remodeling, and dysfunction during 4 weeks of pressure overload in mice. J Cardiovasc Pharmacol. 2012;59:268–280. [DOI] [PubMed] [Google Scholar]
- 12.Wei YD, Liu XY, Wei HD, et al. The electrophysiological effects of qiliqiangxin on cardiac ventricular myocytes of rats. Evid based Complement Alternat Med. 2013;2013:213976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewin AJ, Kereiakes DJ, Chrysant SG, et al. Triple-combination treatment with olmesartan medoxomil/amlodipine/hydrochlorothiazide in Hispanic/Latino patients with hypertension: the TRINITY study. Ethn Dis. 2014;24:41–47. [PubMed] [Google Scholar]
- 14.Yilmaz S, Pekdemir M, Tural U, et al. Comparison of alprazolam versus captopril in high blood pressure: a randomized controlled trial. Blood Press. 2011;20:239–243. [DOI] [PubMed] [Google Scholar]
- 15.Chakraborty A, Chatterjee S. Convergence in findings from randomized trials and elaborately analysed observational data on mortality reduction with carvedilol in heart failure in comparison with metoprolol. Eur J Heart Fail. 2014;16:595–597. [DOI] [PubMed] [Google Scholar]
- 16.Zhou N, Li L, Wu J, et al. Mechanical stress-evoked but angiotensin II-independent activation of angiotensin II type 1 receptor induces cardiac hypertrophy through calcineurin pathway. Biochem Biophys Res Commun. 2010;397:263–269. [DOI] [PubMed] [Google Scholar]
- 17.Zou YZ, Liang YY, Gong H, et al. Ryanodine receptor type 2 is required for the development of pressure overload-induced cardiac hypertrophy. Hypertension. 2011;58:1099–U363. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Zhou N, Gong H, et al. Comparison of angiotensin II type 1-receptor blockers to regress pressure overload-induced cardiac hypertrophy in mice. Hypertens Res. 2010;33:1289–1297. [DOI] [PubMed] [Google Scholar]
- 19.Nakaya M, Chikura S, Watari K, et al. Induction of cardiac fibrosis by beta-blocker in G protein-independent and G protein-coupled receptor kinase 5/beta-arrestin2-dependent signaling pathways. J Biol Chem. 2012;287:35669–35677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diao TY, Pan H, Gu SS, et al. Effects of angiotensin-converting enzyme inhibitor, captopril, on bone of mice with streptozotocin-induced type 1 diabetes. J Bone Miner Metab. 2014;32:261–270. [DOI] [PubMed] [Google Scholar]
- 21.Sano M, Minamino T, Toko H, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. [DOI] [PubMed] [Google Scholar]
- 22.Lin L, Tang CY, Xu JF, et al. Mechanical stress triggers cardiomyocyte autophagy through angiotensin II type 1 receptor-mediated p38MAP kinase independently of angiotensin II. PLoS One. 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teiger E, Than VD, Richard L, et al. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest. 1996;97:2891–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mei Y, Thompson MD, Cohen RA, et al. Autophagy and oxidative stress in cardiovascular diseases. Biochim Biophys Acta. 2014;185:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou YZ, Akazawa H, Qin YJ, et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. [DOI] [PubMed] [Google Scholar]
- 26.Tutor AS, Penela P, Mayor F., Jr Anti-beta1-adrenergic receptor autoantibodies are potent stimulators of the ERK1/2 pathway in cardiac cells. Cardiovasc Res. 2007;76:51–60. [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, Singh VP, Baker KM. The intracellular renin-angiotensin system in the heart. Curr Hypertens Rep. 2009;11:104–110. [DOI] [PubMed] [Google Scholar]
- 28.Kurdi M, De Mello WC, Booz GW. Working outside the system: an update on the unconventional behavior of the renin-angiotensin system components. Int J Biochem Cell Biol. 2005;37:1357–1367. [DOI] [PubMed] [Google Scholar]
- 29.Hall JL, O'Connell TD, Francis GS. Promising small molecule for heart failure targeting adrenal catecholamine release and beta-adrenergic receptor signaling in the heart. J Am Coll Cardiol. 2014;63:2558–2559. [DOI] [PubMed] [Google Scholar]
- 30.Katragadda S, Arora RR. Role of angiotensin-converting enzyme inhibitors in vascular modulation: beyond the hypertensive effects. Am J Ther. 2010;17:e11–23. [DOI] [PubMed] [Google Scholar]
- 31.Israili ZH, Velasco M, Bermudez V. Direct renin inhibitors as antihypertensive agents. Am J Ther. 2010;17:237–254. [DOI] [PubMed] [Google Scholar]
- 32.Siragy HM. Comparing angiotensin II receptor blockers on benefits beyond blood pressure. Adv Ther. 2010;27:257–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMurray JJ, Young JB, Dunlap ME, et al. Relationship of dose of background angiotensin-converting enzyme inhibitor to the benefits of candesartan in the Candesartan in Heart failure: assessment of Reduction in Mortality and morbidity (CHARM)-Added trial. Am Heart J. 2006;151:985–991. [DOI] [PubMed] [Google Scholar]
- 34.Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372:547–553. [DOI] [PubMed] [Google Scholar]
- 35.Amanfu RK, Saucerman JJ. Modeling the effects of beta1-adrenergic receptor blockers and polymorphisms on cardiac myocyte Ca2+ handling. Mol Pharmacol. 2014;86:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wikstrand J, Wedel H, Castagno D, et al. The large-scale placebo-controlled beta-blocker studies in systolic heart failure revisited: results from CIBIS-II, COPERNICUS and SENIORS-SHF compared with stratified subsets from MERIT-HF. J Intern Med. 2014;275:134–143. [DOI] [PubMed] [Google Scholar]
- 37.Choi YH, Cowan DB, Moran AM, et al. Myocyte apoptosis occurs early during the development of pressure-overload hypertrophy in infant myocardium. J Thorac Cardiovasc Surg. 2009;137:1356–1362. 62 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang ZV, Rothermel BA, Hill JA. Autophagy in hypertensive heart disease. J Biol Chem. 2010;285:8509–8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song Z, An L, Ye Y, et al. Essential role for UVRAG in autophagy and maintenance of cardiac function. Cardiovasc Res. 2014;101:48–56. [DOI] [PubMed] [Google Scholar]
- 40.Amaravadi RK, Yu DN, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booth LA, Tavallai S, Hamed HA, et al. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisenberg-Lerner A, Bialik S, Simon HU, et al. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. [DOI] [PubMed] [Google Scholar]
- 43.Kim TJ, Sun J, Lu S, et al. The regulation of beta-adrenergic receptor-mediated PKA activation by substrate stiffness via microtubule dynamics in human MSCs. Biomaterials. 2014;35:8348–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bersell K, Arab S, Haring B, et al. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. [DOI] [PubMed] [Google Scholar]
- 45.Bostrom P, Mann N, Wu J, et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuller SJ, Sivarajah K, Sugden PH. ErbB receptors, their ligands, and the consequences of their activation and inhibition in the myocardium. J Mol Cell Cardiol. 2008;44:831–854. [DOI] [PubMed] [Google Scholar]
- 47.Sebastian T, Johnson PF. Stop and go: anti-proliferative and mitogenic functions of the transcription factor C/EBPbeta. Cell Cycle. 2006;5:953–957. [DOI] [PubMed] [Google Scholar]