

Abstract
Cross-species comparative studies are a powerful approach to understanding the epithelial Cl− channel cystic fibrosis transmembrane conductance regulator (CFTR), which is defective in the genetic disease cystic fibrosis (CF). Here, we investigate the single-channel behaviour of ovine CFTR and the impact of the most common CF mutation, F508del-CFTR, using excised inside-out membrane patches from transiently transfected CHO cells. Like human CFTR, ovine CFTR formed a weakly inwardly rectifying Cl− channel regulated by PKA-dependent phosphorylation, inhibited by the open-channel blocker glibenclamide. However, for three reasons, ovine CFTR was noticeably more active than human CFTR. First, single-channel conductance was increased. Second, open probability was augmented because the frequency and duration of channel openings were increased. Third, with enhanced affinity and efficacy, ATP more strongly stimulated ovine CFTR channel gating. Consistent with these data, the CFTR modulator phloxine B failed to potentiate ovine CFTR Cl− currents. Similar to its impact on human CFTR, the F508del mutation caused a temperature-sensitive folding defect, which disrupted ovine CFTR protein processing and reduced membrane stability. However, the F508del mutation had reduced impact on ovine CFTR channel gating in contrast to its marked effects on human CFTR. We conclude that ovine CFTR forms a regulated Cl− channel with enhanced conductance and ATP-dependent channel gating. This phylogenetic analysis of CFTR structure and function demonstrates that subtle changes in structure have pronounced effects on channel function and the consequences of the CF mutation F508del.
Key points
Malfunction of the cystic fibrosis transmembrane conductance regulator (CFTR), a gated pathway for chloride movement, causes the common life-shortening genetic disease cystic fibrosis (CF).
Towards the development of a sheep model of CF, we have investigated the function of sheep CFTR.
We found that sheep CFTR was noticeably more active than human CFTR, while the most common CF mutation, F508del, had reduced impact on sheep CFTR function.
Our results demonstrate that subtle changes in protein structure have marked effects on CFTR function and the consequences of the CF mutation F508del.
Introduction
The cystic fibrosis transmembrane conductance regulator (CFTR; ABCC7; Riordan et al. 1989) is the ATP-binding cassette (ABC) transporter that functions as a ligand-gated anion channel (Gadsby et al. 2006). Located predominantly in the apical membrane of epithelia lining ducts and tubes throughout the body, CFTR plays a pivotal role in transepithelial ion transport as its dysfunction in the genetic disease cystic fibrosis (CF) testifies (Welsh et al. 2001).
To understand better the diverse physiological roles CFTR plays in different organs and learn how CFTR dysfunction causes organ-level disease, a variety of CF animal models have been developed. Soon after the identification of the CFTR gene, CFTR knockout mice were generated and used to evaluate innovative therapies for CF (for review, see Wilke et al. 2011). However, anatomical and physiological differences between humans and mice have driven the search for new animal models of CF, leading to the development of CFTR knockout pigs, ferrets and rats (Rogers et al. 2008; Sun et al. 2010; Tuggle et al. 2014). These new animal models have provided important insight into the pathogenesis of CF lung disease (Pezzulo et al. 2012) and CF-related diabetes (Olivier et al. 2012). Nevertheless, for several reasons, sheep remain an attractive species to use to develop a large animal model of CF. First, ovine CFTR shows a high degree of sequence conservation with human CFTR (91% identity and 95% similarity at the amino acid level) with the pattern of ovine CFTR gene expression during development mirroring that of human CFTR (Tebbutt et al. 1995). Second, the sheep lung exhibits a high degree of anatomical and functional similarity with the human lung (Alcorn et al. 1981; Olver & Robinson, 1986). Third, the sheep lung has proved to be a valuable model system to evaluate experimental therapeutics for CF lung disease, including small-molecules (Coote et al. 2009) and gene therapy (McLachlan et al. 2011). Finally, the technology to successfully clone sheep from somatic cells has existed for some time (Campbell et al. 1996) and could be used to generate a CF sheep.
In this study, our goal was to establish the potential utility of ovine CFTR as a model of human CFTR. We investigated the behaviour of ovine CFTR and the consequences of the most common CF mutation, F508del-CFTR on the processing, plasma membrane stability and function of ovine CFTR. To address our aims, we assembled a full-length cDNA encoding ovine CFTR, introduced the F508del mutation by site-directed mutagenesis, transiently expressed these cDNAs in Chinese hamster ovary (CHO) cells and studied single-channels and macroscopic currents using excised inside-out membrane patches. We discovered that ovine CFTR forms a regulated Cl− channel with enhanced conductance and ATP-dependent gating compared to human CFTR. In contrast to its effects on human CFTR, the F508del mutation had reduced impact on ovine CFTR channel gating, but disrupted protein processing and membrane stability. By demonstrating that wide-ranging effects on single-channel behaviour can result from subtle changes in protein structure our results inform the phylogenetic analysis of CFTR structure and function.
Methods
Assembly of a full-length ovine CFTR cDNA
The full-length ovine CFTR cDNA was assembled in 2005 by joining the partial cDNAs α, A, B, C, D′, F and G as described in Table1 of Tebbutt et al. (1995). Each fragment was excised from the pCRII cloning vector with relevant restriction enzymes, ligated onto an adjacent fragment and cloned into pUC18, before further assembly of the construct as follows: fragments F and G were joined first and sequenced, revealing a frameshift mutation that was subsequently repaired by substitution of a 217 bp MscI-BlpI wild-type fragment; fragments C and D′ were joined and sequenced; fragments CD′ and FG were ligated and cloned into pUC18. Fragments A and B were ligated and cloned into pUC18 and then joined to fragment α in the same vector. Sequencing revealed several mutations in fragment A, which were corrected by site-directed mutagenesis with the exception of F229L, a polymorphism found in some wild-type CFTR homologues (e.g. murine). Segments αAB were then combined with CD′FG and cloned into pcDNA3.1zeo. The cryptic bacterial promoter in exon 6 of CFTR was abolished by site-directed mutagenesis (using the primer 5′ CGT CAG AAA TGA TTG AAA ACA TCC AAT CAG TTA AGGC 3′) to generate T933C (equivalent to T936C in human CFTR; Cheng et al. 1990). The F508del mutation was introduced into this construct using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA) and the following primer 5′ GAA CCA TTA AAG ATA ACA TCA TCG GTG TTT CCT ATG ATG AAT ATA G 3′.
Table 1.
Open and closed time constants of ovine CFTR Cl− channels
| Human | Ovine | |||
|---|---|---|---|---|
| Wild-type | F508del | Wild-type | F508del | |
| τO1 (ms) | — | 4.7 ± 1.9 | — | 2.2 ± 0.2 |
| τO2 (ms) | 36.6 ± 2.1 | 46.9 ± 7.6 | 55.5 ± 4.0* | 75.7 ± 5.9† |
| τC1 (ms) | 3.0 ± 0.1 | 3.9 ± 0.4 | 1.7 ± 0.2* | 1.5 ± 0.1† |
| τC3 (ms) | 109 ± 7 | 1584 ± 212 | 87 ± 7* | 239 ± 49† |
| Area under curve τO1 | — | 0.46 ± 0.06 | — | 0.24 ± 0.06 |
| Area under curve τO2 | 1 | 0.54 ± 0.06 | 1 | 0.76 ± 0.06 |
| Area under curve τC1 | 0.68 ± 0.01 | 0.68 ± 0.05 | 0.81 ± 0.02* | 0.84 ± 0.01† |
| Area under curve τC3 | 0.33 ± 0.01 | 0.32 ± 0.05 | 0.19 ± 0.02* | 0.16 ± 0.01† |
| Events per minute | 1462 ± 88 | 216 ± 44 | 1506 ± 130 | 1407 ± 147† |
| Total time (s) | 1029 | 948 | 1952 | 779 |
| n | 6 | 5 | 13 | 6 |
Time constants (τO1, fast open time constant; τO2, slow open time constant; τC1, fast closed time constant; τC3, slow closed time constant) were derived from the fitting of one- or two-component exponential functions to open and closed time histograms using the maximum likelihood method as described in the Methods. Area under curve indicates the proportion of the total open or closed time distribution that corresponds to the different time constants. Events per minute represents the number of transitions between the open and closed states within 1 min. The total time analysed for human and ovine CFTR is shown and in each patch approximately 5000 events were analysed for wild-type human and ovine CFTR, 700 events for F508del human CFTR and 3000 events for F508del ovine CFTR. Values are means ± SEM of n observations
P < 0.05 vs. human CFTR
P < 0.05 vs. F508del human CFTR. Measurements were made at 37°C in the presence of the catalytic subunit of PKA (75 nm) and ATP (1 mm) in the intracellular solution; voltage was –50 mV and there was a large Cl− concentration gradient across the membrane ([Cl−]internal = 147 mm; [Cl−]external = 10 mm). All F508del-CFTR data analysed were acquired before channel rundown commenced (Wang et al. 2014).
Cells and CFTR expression systems
Chinese hamster ovary K1 (CHO-K1) and human embryonic kidney 293 (HEK-293) cells were cultured as previously described (Graham et al. 1977; Lansdell et al. 1998a). For biochemical studies, human and ovine CFTR were transiently expressed in HEK-293 cells using calcium phosphate (Leir & Harris, 2011), whereas for single-channel studies they were transiently expressed with green fluorescent protein (GFP) in CHO-K1 cells using the Lipofectamine Plus® system (Life Technologies Ltd, Paisley, UK). Thirty-six to sixty hours after transfection, GFP-expressing CHO-K1 cells were selected for study using the patch-clamp technique. To investigate the single-channel behaviour of F508del ovine CFTR, transiently transfected CHO cells were incubated at 27°C for 24–72 h prior to use to deliver the mutant protein to the cell surface (Denning et al. 1992; Aleksandrov et al. 2012). For comparison, we studied human CFTR Cl− channels in excised inside-out membrane patches from CHO cells transiently and mouse mammary epithelial cells (C127 cells) stably expressing wild-type and F508del human CFTR. The single-channel behaviour of wild-type human CFTR in excised membrane patches from different mammalian cell lines is equivalent (Lansdell et al. 1998a; Chen et al. 2009).
Western blotting
To assess the expression of ovine CFTR protein, we used Western blotting. HEK-293 cells were lysed 72 h post-transfection using NET Buffer (150 mm NaCl, 5 mm EDTA, 50 mm Tris-HCl, 1% Triton X-100, pH 7.5) supplemented with protease inhibitors (Sigma, St. Louis, MO, USA; P8340) and 100 mm phenylmethylsulfonyl fluoride. Lysates were then passed through a 25 G needle, incubated on ice for 15 min and cleared by centrifugation (16,000 g at 4°C for 15 min). SDS loading buffer (50 mm Tris-HCl, 2% SDS, 10% glycerol, 12.5 mm EDTA, pH 6.8) with 5% β-mercaptoethanol was added to each lysate, followed by incubation at 55°C for 15 min. Lysates were then resolved on 3%/6% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels and transferred to Immobilon membranes (Millipore, Billerica, MA, USA). Membranes were blocked in Tris-buffered saline (TBS)–Tween (TBST) containing 5% milk and then probed overnight at 4°C using the mouse anti-CFTR monoclonal antibody (596), which recognizes nucleotide-binding domain 2 (NBD2) of human and ovine CFTR (Cui et al. 2007; a generous gift of T. Jensen and J. R. Riordan (University of North Carolina, USA) and Cystic Fibrosis Foundation Therapeutics (Bethesda, MD, USA)) diluted 1:2000 in TBST containing 1% milk. Membranes were washed with TBST, probed at room temperature with polyclonal goat anti-mouse-HRP (Dako, Carpinteria, CA, USA) diluted 1:10,000 in 1% milk TBST for 2 h and washed with TBST before developing using enhanced chemiluminescence (ECL) Western Blotting Substrate (Pierce, Rockford, IL, USA; 32106).
Patch-clamp experiments
CFTR Cl− channels were recorded in excised inside-out membrane patches using an Axopatch 200B patch-clamp amplifier and pCLAMP software (versions 6.0 and 9.2) (all from Molecular Devices, Union City, CA, USA) as described previously (Sheppard & Robinson, 1997). The pipette (extracellular) solution contained (mm): 140 N-methyl-d-glucamine (NMDG), 140 aspartic acid, 5 CaCl2, 2 MgSO4 and 10 N-tris[hydroxymethyl]methyl-2-aminoethanesulphonic acid (Tes), adjusted to pH 7.3 with Tris ([Cl−], 10 mm). The bath (intracellular) solution contained (mm): 140 NMDG, 3 MgCl2, 1 CsEGTA and 10 Tes, adjusted to pH 7.3 with HCl ([Cl−], 147 mm; free [Ca2+], <10−8 m) and was maintained at 37°C.
Within 5 min of membrane patch excision, we added the catalytic subunit of protein kinase A (PKA, 75 nm) and ATP (1 mm) to the intracellular solution to activate CFTR Cl− channels. To minimize channel rundown, we added PKA to all intracellular solutions, maintained the ATP concentration at ≥0.3 mm and clamped voltage at −50 mV. The effects of CFTR modulators on ovine CFTR Cl− channels were tested by addition to the intracellular solution in the continuous presence of ATP (1 mm) and PKA (75 nm). Because the effects of the CFTR potentiator phloxine B are only partially reversible (Cai & Sheppard, 2002), specific interventions were compared with the pre-intervention control period made with the same concentration of ATP and PKA, but without the test CFTR modulator.
For single-channel studies, we used membrane patches containing ≤5 active channels (human CFTR, number of active channels (N) ≤4; F508del human CFTR, N = 1; ovine CFTR, N ≤ 4; F508del ovine CFTR, N ≤ 5); for all other studies, membrane patches contained large numbers of active channels. To determine channel number, we used the maximum number of simultaneous channel openings observed during the course of an experiment (for further information, see Cai et al. 2006). Using voltage ramp protocols, we acquired macroscopic current–voltage (I–V) relationships (Cai et al. 2003). To evaluate the plasma membrane stability of F508del ovine CFTR at 37°C, we calculated open probability (Po) values in 20 s intervals over a 10–15 min period, commencing Po measurements only after channels were fully activated.
Single-channel currents were initially recorded on digital audiotape using a digital tape recorder (model DTR-1204, Bio-Logic Science Instruments; Intracel Ltd., Royston, UK) at a bandwidth of 10 kHz. On playback, records were filtered with an eight-pole Bessel filter (model F-900C/9L8L, Frequency Devices Inc., Ottawa, IL, USA) at a corner frequency (fc) of 500 Hz and acquired using a DigiData1320A interface (Molecular Devices, CA, USA) and pCLAMP software at sampling rates of 1.0 kHz (voltage ramp protocols), 2.5 kHz (time course studies) or 5 kHz (single-channel studies).
To measure single-channel current amplitude (i), Gaussian distributions were fitted to current amplitude histograms; chord conductance was calculated as described by Cai et al. (2003). For Po and kinetic analyses, lists of open and closed times were created using a half-amplitude crossing criterion for event detection and dwell-time histograms constructed as previously described (Sheppard & Robinson, 1997); transitions <1 ms were excluded from the analysis (eight-pole Bessel filter rise time (T10–90) ∼0.73 ms at fc = 500 Hz). Histograms were fitted with one or more component exponential functions using the maximum likelihood method. To determine which component function fitted best, the log-likelihood ratio test was used and considered statistically significant at a value of 2.0 or greater (Winter et al. 1994). For wild-type human CFTR, time constant values were unaffected when data were filtered at fc = 1 kHz, digitized at 10 kHz and transitions <0.5 ms excluded from the analysis (T10–90 ∼0.34 ms) (data not shown). Burst analysis was performed as described by Cai et al. (2006) using a tc (the time that separates interburst closures from intraburst closures determined from closed time histograms (e.g. human CFTR, tc = 13.6 ± 0.4 ms (n = 6); ovine CFTR, tc = 8.9 ± 0.7 ms (n = 11) in the presence of 1 mm ATP). The mean interburst interval (TIBI) was calculated using the equation (Cai et al. 2003):
| 1 |
where, Tb = (mean burst duration) × (open probability within a burst). Mean burst duration (TMBD) and open probability within a burst (Po(burst)) were determined directly from experimental data using pCLAMP software. Only membrane patches which contained a single active CFTR Cl− channel were used for burst and kinetic analyses.
To calculate the voltage-dependent dissociation constant (Kd) for CFTR inhibition by glibenclamide, we used the equation:
| 2 |
where, Kd(V) is the voltage-dependent dissociation constant at voltage V, and I and Io are current values in the presence and absence of drug, respectively. To determine the electrical distance across the membrane sensed by blocking ions, we used the equation (Woodhull, 1973):
| 3 |
where z′ is the apparent valency of the blocking ion (defined as the actual valency of the blocking ion (z) multiplied by the electrical distance across the membrane experienced by the blocking ion (δ)) and F, R and T are the Faraday constant, gas constant and absolute temperature, respectively. For glibenclamide inhibition of CFTR, we assume a valency of −1 and a single binding site (Sheppard & Robinson, 1997).
Some electrophysiology data for wild-type human CFTR in this paper are reproduced from previously published work (Sheppard & Robinson, 1997; Cai & Sheppard, 2002; Cai et al. 2003; Scott-Ward et al. 2007). In Figs 6C and 7E, the wild-type human CFTR data are directly reproduced. In the remainder of the electrophysiology figures, the wild-type human CFTR data are a combination of new and old data. The contribution of previously published data to the total data set is indicated in the relevant figure legends.
Figure 6.
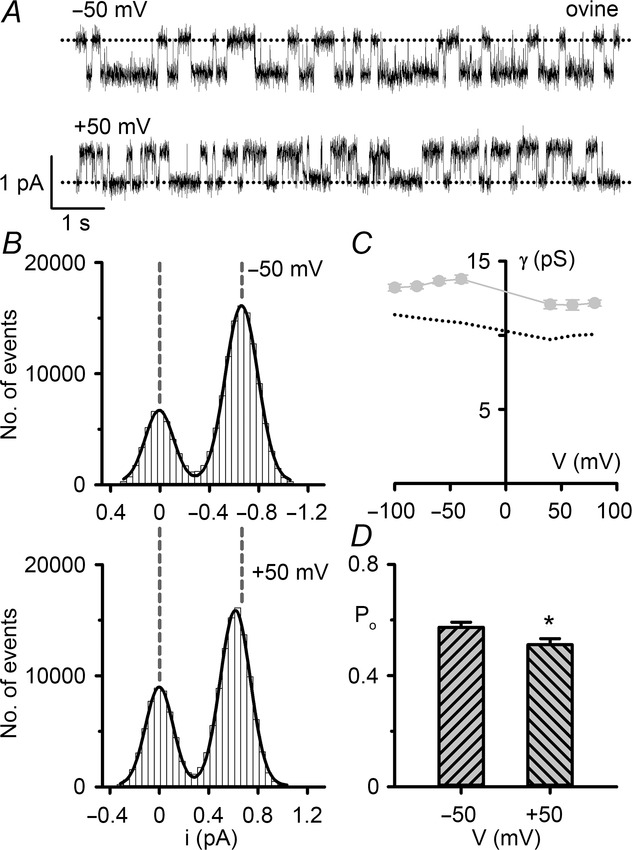
The single-channel behaviour of ovine CFTR in symmetrical Cl−-rich solutions
A, representative recordings of a single ovine CFTR Cl− channel at –50 mV (top) and +50 mV (bottom). The excised inside-out membrane patch from a CHO cell transiently expressing ovine CFTR was bathed in symmetrical 147 mm NMDGCl solutions and ATP (1 mm) and PKA (75 nm) were continuously present in the intracellular solution. Dotted lines indicate the closed channel state, while downward deflections at –50 mV and upward deflections at +50 mV correspond to channel openings. B, current amplitude histograms of the single ovine CFTR Cl− channel shown in A; the closed channel amplitudes are shown on the left. The continuous lines represent the fit of Gaussian distributions to the data and the vertical dashed lines indicate the position of the open and closed channel levels at –50 mV. C, relationship between chord conductance and voltage for ovine CFTR. Data are means ± SEM (ovine, n = 6–11). For comparison, equivalent data for human CFTR (©2003 Cai et al. Journal of General Physiology. 122:605–620. doi:10.1085/jgp.200308921) are shown as a dotted line. Chord conductance was calculated as described in the Methods. D, single-channel open probability (Po) of ovine CFTR at –50 and +50 mV. Data are means ± SEM (n = 9); *P < 0.05 vs. ovine CFTR value at –50 mV. For comparison, Po values of human CFTR Cl− channels at –50 and +50 mV were 0.54 ± 0.03 and 0.53 ± 0.02, respectively (n = 6); P = 0.71.
Figure 7.
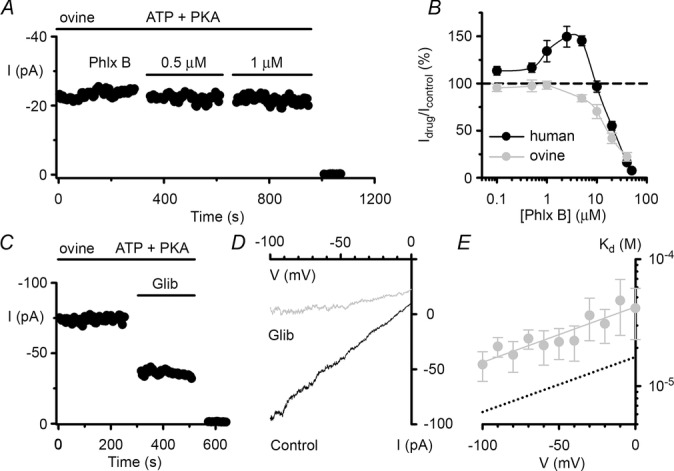
Effects of phloxine B and glibenclamide on ovine CFTR Cl− currents
A and C, representative time courses of ovine CFTR Cl− currents in excised inside-out membrane patches from CHO cells expressing ovine CFTR; voltage was –50 mV and there was a large Cl− concentration gradient across the membrane patch ([Cl−]internal = 147 mm; [Cl−]external = 10 mm). During the periods indicated by the bars, ATP (1 mm), PKA (75 nm), phloxine B (Phlx B, 0.5 and 1 μm) and glibenclamide (Glib, 50 μm) were present in the intracellular solution. For the purpose of illustration, the time courses have been inverted so that upward deflections represent inward current. B, effects of phloxine B concentration (0.1–50 μm) on human and ovine CFTR. Values above the dashed line indicate CFTR potentiation, whereas values below the line denote CFTR inhibition. D, representative I–V relationships of ovine CFTR Cl− currents recorded in the absence and presence of glibenclamide (50 μm) using the experimental conditions described in C. E, relationship between the voltage-dependent dissociation constant (Kd) and voltage for ovine CFTR inhibition by glibenclamide. In B and E, data are means ± SEM (phloxine B, n = 5–11; glibenclamide, n = 5); the continuous line in E is the fit of a first order regression to Kd values over the range –100 to 0 mV (r2 = 0.84). In B, some human CFTR data were originally published in Cai & Sheppard (2002), copyright the American Society for Biochemistry and Molecular Biology (n = 4–7); other human CFTR data are newly acquired using C127 cells stably expressing human CFTR. In E, equivalent human CFTR data were originally published in Sheppard & Robinson (1997) and shown as a dotted line.
Reagents
PKA purified from bovine heart was purchased from Calbiochem (Merck Chemicals Ltd., Nottingham, UK). All other chemicals were of reagent grade and supplied by the Sigma-Aldrich Company Ltd. (Gillingham, UK). ATP was dissolved in intracellular solution directly before each experiment. All other reagents were dissolved in DMSO and stored at −20°C. Immediately before use, stock solutions were diluted to achieve final concentrations and, where necessary, the pH of the intracellular solution was re-adjusted to pH 7.3 to avoid pH-dependent changes in CFTR function (Chen et al. 2009). DMSO did not affect CFTR activity (Sheppard & Robinson, 1997).
Modelling
Like that of human CFTR (Mornon et al. 2008), the three-dimensional model of the membrane-spanning domains (MSDs) and NBDs of ovine CFTR protein was constructed using as a template the Sav1866 three-dimensional structure in an outward-facing conformation (pdb code: 2hyd; Dawson & Locher, 2006). Based on new three-dimensional structures of ABC exporters (pdb codes: 3g5u, 3g60, 4ksb, 4f4c, 3wme, 3wmf, 3zdq, 4ayt, 4ayx, 4ayw, 4myc, 4mrn, 4mrp, 4mrr, 4mrs, 4mrv and 3qf4), the alignment of the MSDs described in Mornon et al. (2008) was modified slightly (Mornon et al. 2015).
The three-dimensional model of the MSD:NBD assembly of ovine CFTR was built using Modeller 9.10 (Martí-Renom et al. 2000). It includes amino acids 65–648 (MSD1:NBD1), with the regulatory insertion (404–434), and amino acids 843–1448 (MSD2:NBD2), with the linker insertion (1183–1203). The overall stereo-chemical quality of the model was assessed using PROCHECK (Laskowski et al. 1993).
Statistics
Results are expressed as means ± SEM of n observations. To test for differences between groups of data, we used Student's t test. Differences were considered statistically significant when P < 0.05. All tests were performed using SigmaStat™ (version 3.5; Systat Software Inc., Richmond, CA, USA).
Results
Ovine CFTR has augmented conductance and gating compared with human CFTR
In this study, we investigated the single-channel behaviour of ovine CFTR in excised inside-out membrane patches from transiently transfected CHO cells. Figure1A shows representative single-channel recordings of human and ovine CFTR following phosphorylation with PKA. Two important differences are apparent from visual inspection of these traces. First, the single-channel current amplitude (i) of ovine CFTR was larger than that of human CFTR (Fig.1A and B). To quantify this difference, we measured single-channel current amplitude at negative voltages (Fig.2A), where the i–V relationship is linear in the presence of a large Cl− concentration gradient (Fig.2B) (Sheppard et al. 1993). At negative voltages, the single-channel conductance of ovine CFTR (9.89 ± 0.14 pS; n = 13) was significantly greater than that of human CFTR (8.59 ± 0.08; n = 10; P < 0.05) (Fig.2C).
Figure 1.
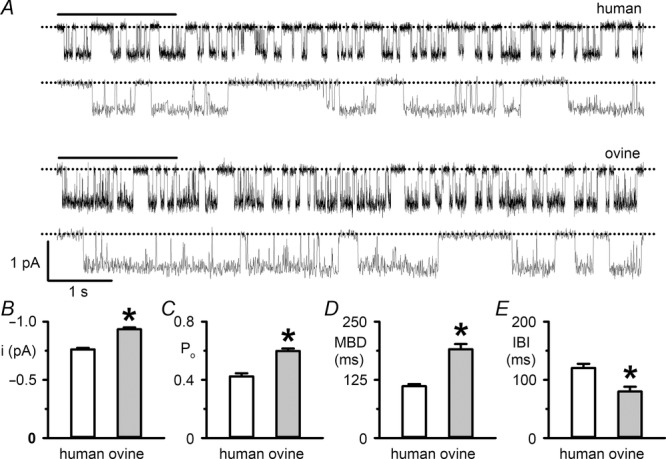
The single-channel behaviour of ovine CFTR
A, representative recordings show the single-channel activity of human and ovine CFTR in excised inside-out membrane patches from transiently transfected CHO cells. ATP (1 mm) and PKA (75 nm) were continuously present in the intracellular solution. Dotted lines indicate the closed channel state and downward deflections correspond to channel openings. Beneath each prolonged 10 s recording, the 2 s portions indicated by bars are shown on an expanded time scale. Unless otherwise indicated in this and subsequent figures, excised inside-out membrane patches were voltage clamped at –50 mV and there was a large Cl− concentration gradient across the membrane patch ([Cl−]internal = 147 mm; [Cl−]external = 10 mm). For the purpose of illustration, single-channel records were filtered at 500 Hz and digitized at 5 kHz before file size was compressed by 5-fold data reduction. B–E, single-channel current amplitude (i), open probability (Po), mean burst duration (MBD) and interburst interval (IBI) of human and ovine CFTR for the experimental conditions described in A. Data are means ± SEM (human, i and Po, n = 10, MBD and IBI, n = 6; ovine, i and Po, n = 24, MBD and IBI, n = 13); *P < 0.05 vs. human CFTR. In B–E, some human CFTR data were originally published in Scott-Ward et al. (2007), copyright (2007) National Academy of Sciences, USA (i and Po, n = 5; MBD and IBI, n = 5); other human CFTR data are newly acquired using CHO cells expressing human CFTR.
Figure 2.
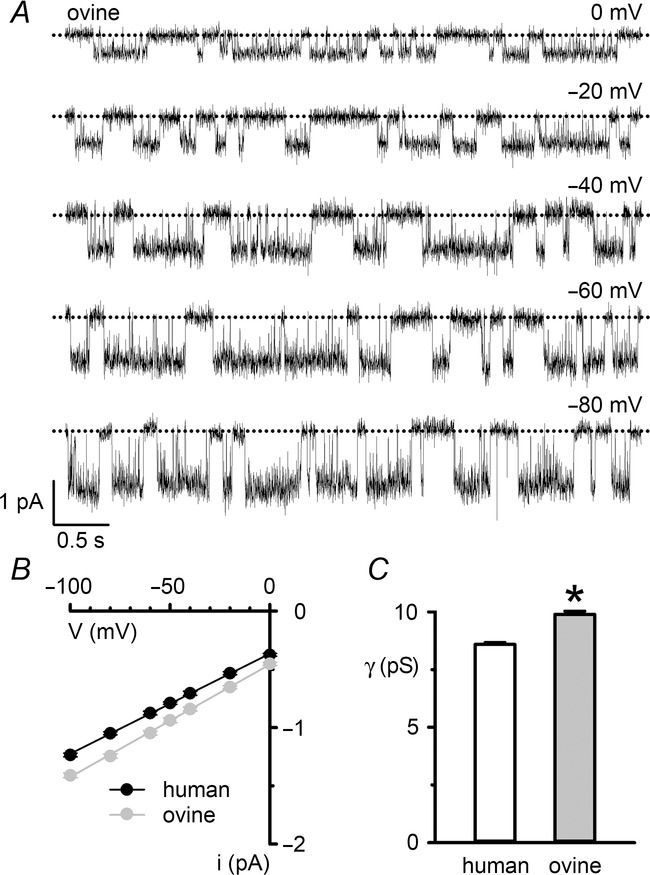
Single-channel conductance of ovine CFTR
A, representative recordings show the single-channel activity of ovine CFTR in an excised inside-out membrane patch from a CHO cell at the indicated voltages. ATP (1 mm) and PKA (75 nm) were continuously present in the intracellular solution. Dotted lines indicate the closed channel state and downward deflections correspond to channel openings. B, single-channel current–voltage (i–V) relationships of human and ovine CFTR. Data are means ± SEM (human, n = 10; ovine, n = 13); error bars are smaller than symbol size except where shown. The continuous lines are the fit of first order linear regression functions to mean data (r2 > 0.99). C, single-channel slope conductance (γ) of human and ovine CFTR determined from the slope of the i–V relationships in B; *P < 0.05 vs. human CFTR. In B and C, some human CFTR data were originally published in Scott-Ward et al. (2007), copyright (2007) National Academy of Sciences, USA (n = 5); other human CFTR data are newly acquired using CHO cells expressing human CFTR.
Second, there was a difference in the pattern of channel gating. The gating behaviour of human CFTR is characterized by bursts of channel openings interrupted by brief flickery closures, separated by longer closures between bursts. Like human CFTR, the ovine CFTR Cl− channel exhibited a bursting pattern of channel activity (Fig.1A). However, the mean burst duration (MBD) of ovine CFTR was 71% longer than that of human CFTR, while its interburst interval (IBI) was 34% shorter (Fig.1D and E). As a result, the Po of ovine CFTR was 41% greater than that of human CFTR (Fig.1C).
To explore further the gating behaviour of ovine CFTR, we investigated gating kinetics using membrane patches that contained only a single active CFTR Cl− channel. For both human and ovine CFTR, open and closed time histograms were best fitted with one- and two-component exponential functions, respectively (Fig.3A and B and Table1). The two populations of channel closures, described by fast (τC1) and slow (τC3) closed time constants, represent the brief, flickery closures that interrupt channel openings and the prolonged closures, which separate one channel opening from the next. Consistent with the burst analysis (Fig.1D and E), Fig.3A and B and Table1 suggest that the augmented Po of ovine CFTR is a consequence of a 51% lengthening of the open time constant (τO2) and a 20% reduction in the slow closed time constant (τC3). Figure 3A and B and Table1 also reveal that the fast closed time constant (τC1) of ovine CFTR was 45% shorter than that of human CFTR. However, its share of the closed time distribution was 19% larger than that of human CFTR (Table1). Thus, ovine CFTR has augmented conductance and gating compared to that of human CFTR.
Figure 3.
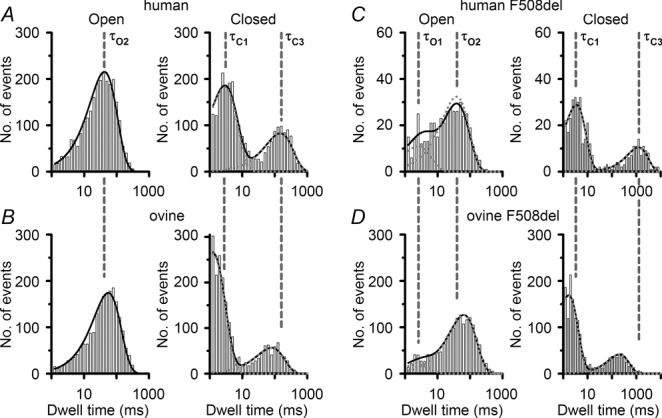
Dwell time histograms of ovine CFTR
Representative dwell time histograms of human and ovine CFTR in the absence (A and B) and presence (C and D) of the F508del mutation. Data are from experiments in which the excised inside-out membrane patch from transiently transfected CHO cells (or stably transfected C127 cells expressing F508del human CFTR) contained only one active channel, studied in the presence of ATP (1 mm) and PKA (75 nm). The continuous lines are the fit of one- or two-component exponential functions to the data and the dotted lines show the individual components of these functions. The vertical dashed lines indicate the mean values of the open (τO1, τO2) and closed (τC1, τC3) time constants of human CFTR (A and B) and F508del human CFTR (C and D). Logarithmic x-axes with 10 bins decade−1 were used for dwell time histograms; note the changes in abscissa (C and D) and ordinate (C).
ATP gates ovine CFTR with greater affinity and efficacy than human CFTR
CFTR channel gating is tightly controlled by cycles of ATP binding and hydrolysis at the NBDs (Gadsby et al. 2006). Therefore, we speculated that the enhanced channel gating of ovine CFTR might reflect altered ATP affinity and/or efficacy. To test this idea, we examined the ATP dependence of channel gating using membrane patches containing ≤4 active channels.
Figure 4A shows the effects of increasing the intracellular ATP concentration from 0.03 to 3 mM in the continuous presence of PKA (75 nm) on ovine CFTR, while Fig.4B plots the relationship between ATP concentration and Po for human and ovine CFTR. By fitting Michaelis–Menten functions to mean data (Fig.4B), we calculated values of KD (the ATP concentration required for half-maximal activity, which describes the apparent affinity of CFTR for ATP) and Po(max) (the maximum Po). For human CFTR, KD = 230 μm and Po(max) = 0.66 (r2 = 0.95), while for ovine CFTR, KD = 77 μm and Po(max) = 0.70 (r2 = 0.93). These data suggest that ATP gates ovine CFTR with enhanced affinity and efficacy compared to human CFTR.
Figure 4.
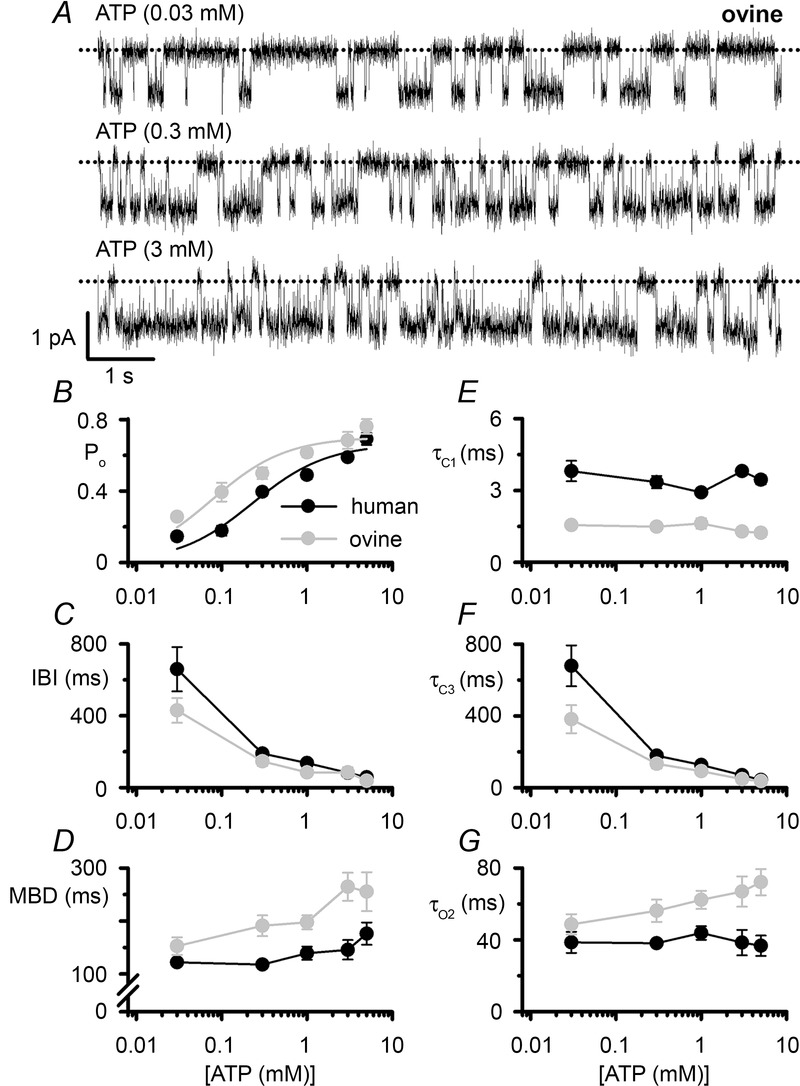
The ATP dependence of ovine CFTR
A, representative recordings show the activity of a single ovine CFTR Cl− channel in an excised inside-out membrane patch from a CHO cell at the indicated intracellular ATP concentrations. PKA (75 nm) was continuously present in the intracellular solution. Dotted lines indicate the closed channel state and downward deflections correspond to channel openings. B–G, relationship between ATP concentration and open probability (Po), interburst interval (IBI), mean burst duration (MBD), fast closed time constant (τC1), slow time constant (τC3) and open time constant (τO2) of human and ovine CFTR plotted using a logarithmic x-axis. For ovine CFTR, data are from transiently transfected CHO cells, whereas human CFTR data are pooled from CHO cells transiently and C127 cells stably expressing human CFTR. Data are means ± SEM (Po, human, n = 4–16; ovine, n = 5–13; MBD, IBI, τC1, τC3 and τO2, human, n = 5–14; ovine, n = 4–12) from experiments where ≥2 (ovine) or ≥3 (human) ATP concentrations were tested in each membrane patch. In B, continuous lines are the fits of Michaelis–Menten functions to mean data. In B–G, some human CFTR data were originally published in Scott-Ward et al. (2007), copyright (2007) National Academy of Sciences, USA and Cai & Sheppard (2002), copyright the American Society for Biochemistry and Molecular Biology (n = 4); other human CFTR data are newly acquired using C127 cells stably expressing human CFTR.
To understand further how ATP stimulates ovine CFTR channel gating, we investigated gating kinetics. For human CFTR, ATP modulates the duration of long closures separating bursts of channel openings, but has little or no effect on channel openings or the short closures that interrupt them (Winter et al. 1994). Like those of human CFTR, the long closures of ovine CFTR were strongly ATP dependent, whereas the short closures were ATP independent (Fig.4C, E and F). Figure 4D and G demonstrates that ATP had little or no effect on channel openings of human CFTR, but increased the duration of those of ovine CFTR. As a result, at 3 mm ATP, the MBD and open time constant (τO2) of ovine CFTR were 82% and 79% longer than those of human CFTR (Fig.4D and G). Taken together, the data demonstrate that ATP stimulates human CFTR by increasing the rate of channel opening only, whereas it stimulates ovine CFTR both by increasing the rate of channel opening and by slowing the rate of channel closure.
Voltage dependence of ovine CFTR
Human CFTR has a pseudo-linear I–V relationship, exhibiting weak inward rectification at large positive voltages (Cai et al. 2003). To investigate whether ovine CFTR exhibits similar behaviour, we bathed excised inside-out membrane patches in symmetrical 147 mm Cl− solutions and recorded membrane currents over the voltage range ±100 mV using a voltage-ramp protocol (Fig.5A). Under basal conditions, membrane currents were tiny (Fig.5A). However, following the activation of ovine CFTR by PKA-dependent phosphorylation large membrane currents were observed (Fig.5A). Figure5B and C demonstrates that at negative voltages, the I–V relationship of ovine CFTR was linear, whereas at positive voltages, it exhibited weak inward rectification (Fig.5C). The data also suggest that rectification is weaker for ovine CFTR than human CFTR (current at +100 mV: human, 74 ± 2% of that at −100 mV (n = 10) (Cai et al. 2003); ovine, 84 ± 2% of that at −100 mV (n = 6); P < 0.01).
Figure 5.
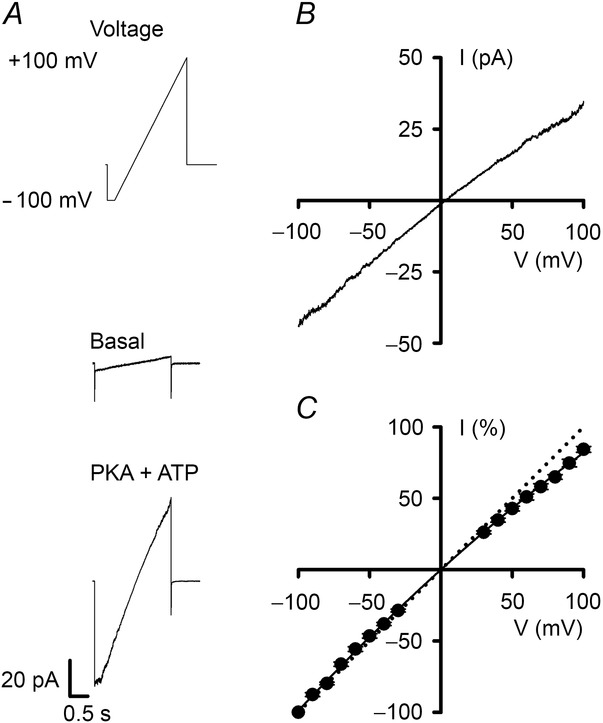
The I–V relationship of ovine CFTR in the presence of symmetrical Cl−-rich solutions
A, current traces from an excised inside-out membrane patch from a CHO cell expressing ovine CFTR. The recordings were made in the absence (middle) and presence (bottom) of ATP (1 mm) and PKA (75 nm) in the intracellular solution. The basal recording is the current in response to a single ramp of voltage with no active CFTR Cl− channels, whereas the recording in the presence of PKA and ATP is the average of 11 ramps of voltage. Holding voltage was –50 mV and the membrane patch was bathed in symmetrical 147 mm NMDGCl solutions. The voltage ramp protocol used is shown above the recordings and currents were acquired at a sampling rate of 1 kHz as described in Cai et al. (2003). B, I–V relationship constructed by subtracting the basal trace from the PKA and ATP trace shown in A. C, I–V relationship of ovine CFTR Cl− currents. Data are means ± SEM (n = 6) at each voltage calculated by expressing individual current values measured from –100 to +100 mV in 10 mV increments as a percentage of the current value at –100 mV. Error bars are smaller than symbol size. The continuous line is the fit of a second order regression function to the data. The dotted line shows the predicted ohmic I–V relationship.
To investigate further the inward rectification of ovine CFTR, we studied single Cl− channels. Figure6A and B compares the activity of a single ovine CFTR Cl− channel at −50 mV and +50 mV using identical conditions to those employed to study ovine CFTR Cl− currents, while Fig.6C and D quantifies changes in single-channel chord conductance and Po. Similar to the behaviour of human CFTR, the chord conductance of ovine CFTR decreased with membrane depolarization (e.g. −50 mV, γ = 13.76 ± 0.26 pS; +50 mV, γ = 11.66 ± 0.29 pS; n = 11; P < 0.05) (Fig.6C). However, the decrease in Po of ovine CFTR at +50 mV, while small, was greater than that of human CFTR (human, 2%; ovine 13%; P = 0.03) (Fig.6D). Reminiscent of human CFTR (Cai et al. 2003), visual inspection of single-channel records suggested that at +50 mV the duration of both bursts of channel openings and the intervening long closures were decreased in length (Fig.6A). Thus, rectification of both human and ovine CFTR is caused by a reduction in i and changes in channel gating.
Pharmacology of ovine CFTR
Previous studies have highlighted differences in the response of CFTR homologues to small-molecule CFTR modulators (e.g. Lansdell et al. 1998a; Stahl et al. 2012). We were therefore keen to explore the pharmacological profile of ovine CFTR, selecting for study the fluorescein derivative phloxine B, which potentiates robustly human CFTR (Cai & Sheppard, 2002), and the sulphonylurea drug glibenclamide, a widely-used open-channel blocker of human CFTR (Schultz et al. 1996; Sheppard & Robinson, 1997).
Figure 7A and B shows the effects of phloxine B on ovine CFTR Cl− currents. In contrast to its potentiation of human CFTR, low micromolar concentrations of phloxine B (0.1–5 μm) were without effect or inhibited weakly ovine CFTR Cl− currents (Fig.7B). However, elevated concentrations of phloxine B (≥10 μM) inhibited strongly both human and ovine CFTR (Fig.7B).
Figure 7C demonstrates that ovine CFTR Cl− currents were inhibited by glibenclamide (50 μm), albeit with reduced efficacy compared with human CFTR (human, Idrug/Icontrol = 31 ± 3% (n = 8); ovine, Idrug/Icontrol = 44 ± 2% (n = 9); P < 0.05). To investigate further glibenclamide inhibition of ovine CFTR, we examined the voltage dependence of channel block (Fig.7D and E). Although glibenclamide inhibition of ovine CFTR was weaker than that of human CFTR (human, Kd(0 mV) = 16 ± 2 μm; ovine CFTR, Kd(0 mV) = 42 ± 12 μm (n = 5); P < 0.05), there was no difference in the electrical distance across the membrane sensed by glibenclamide between human and ovine CFTR (human CFTR, z′ = 0.25 ± 0.05; ovine CFTR, z′ = 0.31 ± 0.05 (n = 5) measured from the inside of the membrane; P < 0.44). Thus, the pharmacological profile of ovine CFTR shows similarities to, but also differences from, that of human CFTR.
Impact of the F508del mutation on the ovine CFTR Cl− channel
The most common CF mutation F508del is a temperature-sensitive folding mutant that prevents CFTR processing and intracellular transport, reduces CFTR stability at the plasma membrane and disrupts channel gating (Cheng et al. 1990; Dalemans et al. 1991; Denning et al. 1992; Lukacs et al. 1993). Because the ovine CFTR Cl− channel had greater activity than human CFTR, we were interested to learn the consequences of the F508del-CFTR mutation on ovine CFTR expression and function.
Consistent with previous results (Aleksandrov et al. 2012), when expressed in HEK-293 cells, ovine CFTR generated band C, the mature, fully glycosylated form of CFTR protein, whereas F508del-ovine CFTR produced reduced levels of band C (Fig.8). Based on these and other data (Aleksandrov et al. 2012), we incubated CHO cells expressing ovine F508del-CFTR at 27°C for 24–72 h before investigating its single-channel behaviour in excised inside-out membrane patches at 37°C. In marked contrast to its effects on human CFTR, the F508del mutation had less impact on ovine CFTR channel gating, but caused a small, albeit significant (P < 0.05), reduction in i (Fig.9A and B). Figure9C demonstrates that the F508del mutation reduced the Po of human CFTR by 89%, but that of ovine CFTR by only 32% to approximate the Po of wild-type human CFTR. To explain these results, we analysed gating kinetics. Figure3C and D and Table1 demonstrate that the F508del mutation introduced two populations of open times described by fast (τO1) and slow (τO2) open time constants in contrast to the single population of open times described by τO2 in human and ovine CFTR. For both human and ovine CFTR, the F508del mutation prolonged τO2 values (Table1). However, the τO2 of F508del ovine CFTR was 61% longer than that of F508del human CFTR and its share of the open time distribution was 76%, whereas that of F508del human CFTR was only 54% (Table1). These results probably explain the different effects of the F508del mutation on the MBD of human and ovine CFTR, which was reduced by 36% and 12%, respectively (Fig.9D). Figure 9E demonstrates that the F508del mutation had markedly different effects on IBI, increasing that of human CFTR 9-fold, but that of ovine CFTR only 2-fold. Consistent with these results, the slow closed time constant (τC3) of F508del ovine CFTR was 85% shorter than that of F508del human CFTR, but its share of the closed time distribution was half that of F508del human CFTR (Fig.3C and D and Table1). Thus, different impacts on the rate of channel opening predominantly explain the varying severity of the F508del mutation on the gating behaviour of human and ovine CFTR.
Figure 8.
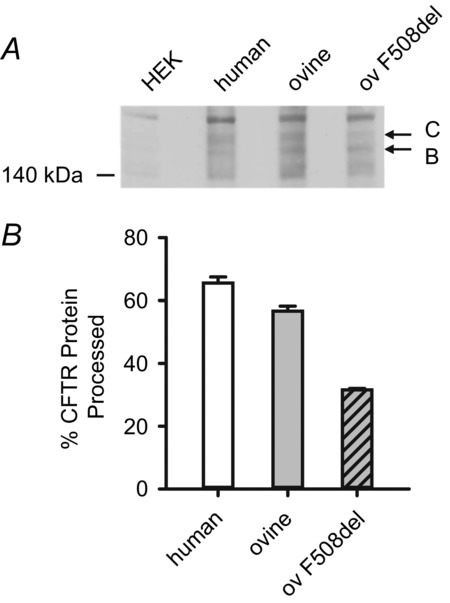
Processing of wild-type and F508del ovine CFTR
A, representative Western blot of total protein extracts from HEK-293 cells transiently expressing the indicated CFTR constructs (ov F508del, F508del ovine CFTR) lysed 72 h post-transfection. Lysates from untransfected HEK-293 cells (HEK) were used as a control. CFTR was detected with the mouse anti-CFTR monoclonal antibody (596), which recognizes a region of NBD2 conserved between human and ovine CFTR (amino acids 1204–1211) (Cui et al. 2007). Arrows indicate the positions of the band B (immature) and C (mature) forms of CFTR and the position of molecular weight markers is indicated. B, quantification of CFTR expression. The amount of CFTR protein present in the mature form (band C) is expressed as a percentage of total CFTR protein (%CFTR protein processed = [band C/(bands B + C)] × 100). Data are means ± SD (n = 2).
Figure 9.
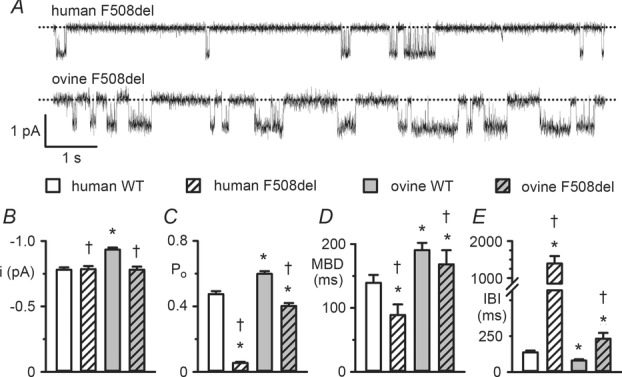
The single-channel activity of F508del ovine CFTR
A, representative single-channel recordings of F508del human CFTR and F508del ovine CFTR in excised inside-out membrane patches from C127 and CHO cells. ATP (1 mm) and PKA (75 nm) were continuously present in the intracellular solution; temperature was 37°C. Dotted lines indicate the closed channel state and downward deflections correspond to channel openings. B–E, single-channel current amplitude (i), open probability (Po), mean burst duration (MBD) and interburst interval (IBI) of wild-type and F508del human and ovine CFTR Cl− channels. For ovine CFTR, data are from CHO cells transiently expressing CFTR constructs, whereas for wild-type human CFTR results are pooled from both transiently expressing CHO and stably expressing C127 cells (cf. human CFTR data in Fig.1), and for F508del human CFTR data are from stably expressing C127 cells. Data are means ± SEM (wild-type human, i and Po, n = 23, MBD and IBI, n = 14; F508del human, i and Po, n = 5, MBD and IBI, n = 5; wild-type ovine, i and Po, n = 24, MBD and IBI, n = 13; F508del ovine, i and Po, n = 8, MBD and IBI, n = 6); *P < 0.05 vs. wild-type human CFTR; †P < 0.05 vs. wild-type ovine CFTR. In B–E, some wild-type human CFTR data were originally published in Scott-Ward et al. (2007), copyright (2007) National Academy of Sciences, USA and Cai & Sheppard (2002), copyright the American Society for Biochemistry and Molecular Biology (n = 12); other human CFTR data are newly acquired using CHO and C127 cells; none of the F508del human CFTR data have previously been published.
Accelerated channel rundown at 37°C is indicative of the reduced plasma membrane stability of F508del human CFTR (e.g. Schultz et al. 1999). However, the observation that F508del chicken CFTR is stable at 37°C (Aleksandrov et al. 2012) suggests that there are cross-species differences in the impact of the F508del mutation on CFTR plasma membrane stability. To test the effects of the F508del mutation on the plasma membrane stability of ovine CFTR, we analysed the duration of F508del ovine CFTR Cl− channel activity in excised inside-out membrane patches at 37°C in the continuous presence of PKA (75 nm) and ATP (1 mm). Figure10 shows a representative prolonged recording of F508del ovine CFTR commenced following complete channel activation and its corresponding Po time course. Over the 14 min recording, channel activity was progressively lost, with Po declining from ∼0.4 to ∼0.1 (Fig.10). Taken together, the data suggest that the F508del mutation alters the processing, plasma membrane stability and channel gating of ovine CFTR, but that its impact is less severe than on human CFTR.
Figure 10.
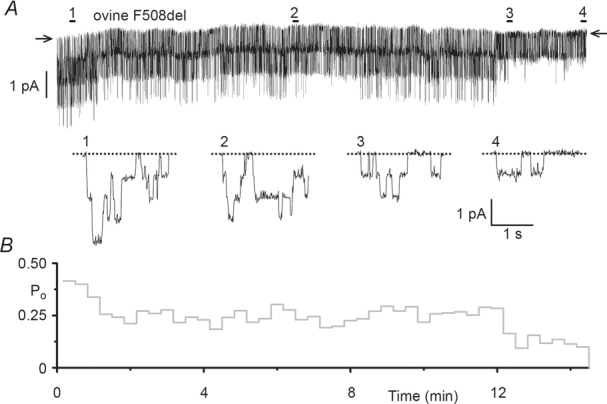
The single-channel activity of F508del ovine CFTR exhibits thermo-instability
A and B, representative single-channel recording (A) and corresponding open probability (Po) time course (B) of F508del ovine CFTR in an excised inside-out membrane patch from a CHO cell commenced following full channel activation. ATP (1 mm) and PKA (75 nm) were continuously present in the intracellular solution; temperature was 37°C. In A, the 2 s long recordings labelled 1–4 indicated by the bars are displayed on an expanded time scale beneath the current recording. Arrows and dotted lines indicate the closed channel state and downward deflections correspond to channel openings. For the purpose of illustration, the single-channel record was filtered at 500 Hz and digitized at 5 kHz before file size was compressed by 50-fold data reduction. In B, Po values were calculated for each 20 s time interval. Similar results were observed in 7 other experiments.
Discussion
In this study, we investigated the single-channel behaviour of ovine CFTR and the impact of the most common CF mutation F508del. Our data demonstrate that ovine CFTR is a more active Cl− channel than human CFTR because it has enhanced conductance and ATP-dependent channel gating. They also show that the F508del mutation has modest effects on ovine CFTR channel gating, but disrupts protein processing and membrane stability. Given the high degree of amino acid similarity between human and ovine CFTR, the magnitude of these functional differences is surprising.
In previous work, Aleksandrov et al. (2012) demonstrated that recombinant ovine CFTR forms a regulated Cl− channel with conductance and Po similar to human CFTR when reconstituted into planar phospholipid bilayers. The reasons for the differences between the data acquired by Aleksandrov et al. (2012) and ourselves are not known. However, it is important to emphasize that the two studies used different experimental techniques and conditions. Moreover, our ovine CFTR construct contained the polymorphism F229L, whereas that used by Aleksandrov et al. (2012) had green fluorescent protein fused to its C-terminus. Irrespective of these differences, both studies draw important conclusions about the function of CFTR homologues. Below, we compare and contrast the behaviour of ovine CFTR with that of human CFTR.
Using the relationship ICFTR = N × i × Po (where ICFTR is the macroscopic CFTR Cl− current, N is the number of CFTR Cl− channels in the plasma membrane, i is the current that flows through an individual CFTR Cl− channel, and Po is the probability that an individual CFTR channel is open) and the data in Fig.1B and C, we can calculate the amount of Cl− current that passes through a single CFTR Cl− channel in 1 s (Valkenier et al. 2014). For wild-type human CFTR, Cl− transport per second is 2.0 × 106, whereas for wild-type ovine CFTR it is 3.5 × 106. These data suggest that ovine CFTR transports 1.5 times more Cl− per second than human CFTR. While it remains plausible that this difference is a consequence of heterologous expression of recombinant ovine CFTR, the present data and other results (Price et al. 1996; Scott-Ward et al. 2007; Dong et al. 2012) argue that amino acid sequence differences are responsible for the functional differences observed between CFTR homologues. Thus, our data raise the possibility that native ovine CFTR Cl− channels in ovine epithelial cells might be more active than their human CFTR counterparts. Perhaps this might reflect metabolic differences between humans and sheep. Alternatively, the demands of ruminant digestion might necessitate that ovine CFTR is more active than human CFTR. Future studies should investigate the behaviour of native ovine CFTR Cl− channels and their physiological roles.
The single-channel behaviour of several recombinant CFTR homologues has been investigated. In all cases, CFTR expression generated Cl−-selective channels regulated by cAMP-dependent phosphorylation (Hanrahan et al. 1993; Price et al. 1996; Lansdell et al. 1998a; Ostedgaard et al. 2007; Demmers et al. 2010; Aleksandrov et al. 2012; present study). Allowing for experimental differences, single-channel conductance varies between species, decreasing in the rank order chicken >> rabbit > ovine ≥ Xenopus > human = possum > porcine > murine ≥ shark (Hanrahan et al. 1993; Price et al. 1996; Lansdell et al. 1998a; Ostedgaard et al. 2007; Demmers et al. 2010; Aleksandrov et al. 2012; present study). These data raise the possibility that metabolic differences might directly or indirectly influence single-channel conductance.
Previous work demonstrates that arginine residues act as fixed positive charges within the intra- and extracellular vestibules of the CFTR pore, playing an important role in maximizing the rate of Cl− flow (Smith et al. 2001; Gong & Linsdell, 2003; St Aubin & Linsdell, 2006; Zhou et al. 2010). To understand better why ovine CFTR has a higher single-channel conductance than human CFTR, we generated a molecular model of ovine CFTR based on the atomic-resolution structure of the ABC transporter Sav1866 in an outward-facing configuration and previous molecular models of human CFTR (Dawson & Locher, 2006; Serohijos et al. 2008; Mornon et al. 2008) (Fig.11). Analysis of this molecular model suggests that there are no clear differences in the architecture of the CFTR pore between human and ovine CFTR (Fig.11). First, it is doubtful whether A872V (seventh transmembrane segment; M7) and I982V (M9), the only two amino acids that are different between human and ovine CFTR within the pore-lining sequence, substantially affect pore properties. Second, the string of amino acid changes in the fourth extracellular loop (human, L884GNTP888; ovine, F884PKIL888) is probably too far from the pore to alter directly single-channel conductance (see also Zhou & Linsdell, 2009). Third, the model suggests that the sequence polymorphism F229L (M4) is unlikely to affect single-channel conductance because it is located in a transmembrane segment distant from the CFTR pore (Mornon et al. 2009; Norimatsu et al. 2012a) (Fig.11). Based on recent studies of human CFTR (Mornon et al. 2015), future studies using molecular dynamics might elucidate the structural basis of the different pore properties exhibited by human and ovine CFTR.
Figure 11.
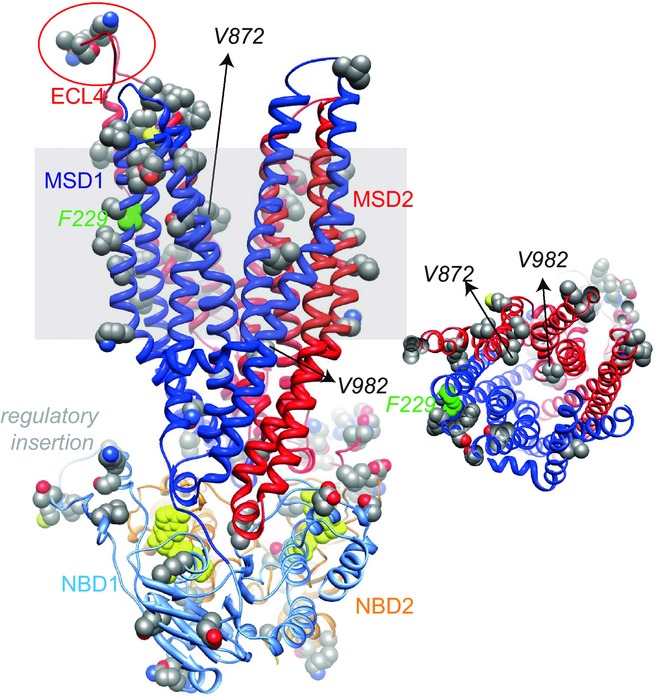
Molecular model of ovine CFTR
Left, ribbon representation of the ovine CFTR MSD:NBD assembly modelled using as a template Sav1866 in an outward-facing conformation (pdb code: 2hyd). Atoms where amino acids differ between human and ovine CFTR are shown with spheres and the ATP molecules are coloured yellow. The grey shading represents the position of the membrane bilayer. Positions discussed in the text are highlighted. Right, orthogonal view of the CFTR pore, viewed from the extracellular side of the membrane.
The gating behaviour of a few recombinant CFTR homologues has been examined. Interestingly, a common characteristic of ovine, porcine and the sub-conductance state (O1) of murine CFTR is the prolonged duration of open-channel bursts compared with that of human CFTR (Lansdell et al. 1998b; Scott-Ward et al. 2007; Ostedgaard et al. 2007; present study). Visual inspection of single-channel recordings suggests that Xenopus and shark CFTR have brief channel openings, whereas those of chicken CFTR are markedly prolonged (Hanrahan et al. 1993; Price et al. 1996; Aleksandrov et al. 2012). Phylogenetic analysis of these data suggests that with the exception of human CFTR, CFTRs from eutherian mammals have prolonged channel openings compared with other species.
The present results demonstrate that ATP more strongly stimulates ovine CFTR than human CFTR. The data suggest that ovine CFTR has a higher apparent ATP affinity and ATP more strongly stimulates ovine CFTR by both increasing the opening rate and decreasing the closing rate. Using the ATP-driven NBD dimerization model of CFTR channel gating (Vergani et al. 2005; Csanády et al. 2010), the reduced IBI of ovine CFTR suggests that the energy barrier for NBD dimerization in ovine CFTR is less than that of human CFTR. Moreover, once the ovine CFTR NBD1:NBD2 dimer forms, it has greater stability than that of human CFTR. Increased stability of the ovine CFTR NBD1:NBD2 dimer might result from tighter binding or slower hydrolysis of ATP at the canonical ATP binding site (site 2), where ATP binding initiates channel opening and ATP hydrolysis leads promptly to channel closure (Gadsby et al. 2006). Alternatively, using the energetic coupling model of CFTR channel gating (Jih et al. 2012), the prolonged open time of ovine CFTR might be explained by the stabilization of a posthydrolytic open state leading to decoupling of the gating cycle from the ATP hydrolysis cycle.
As for the CFTR pore, analysis of the molecular model of ovine CFTR identified no clear differences between human and ovine CFTR within the critical motifs that form the two ATP-binding sites (Figs11 and 12). Instead, the amino acid differences between human and ovine CFTR (9%) are mostly concentrated in sequence fragments that play (or are likely to play) regulatory roles. These regions include the N-terminus, the regulatory insertion of NBD1, the regulatory domain, the linker region between MSD2 and NBD2 and the C-terminus (Fig.12). Interestingly, in several of these regions, the differences lead to the presence of additional serine and threonine residues in ovine CFTR.
Figure 12.
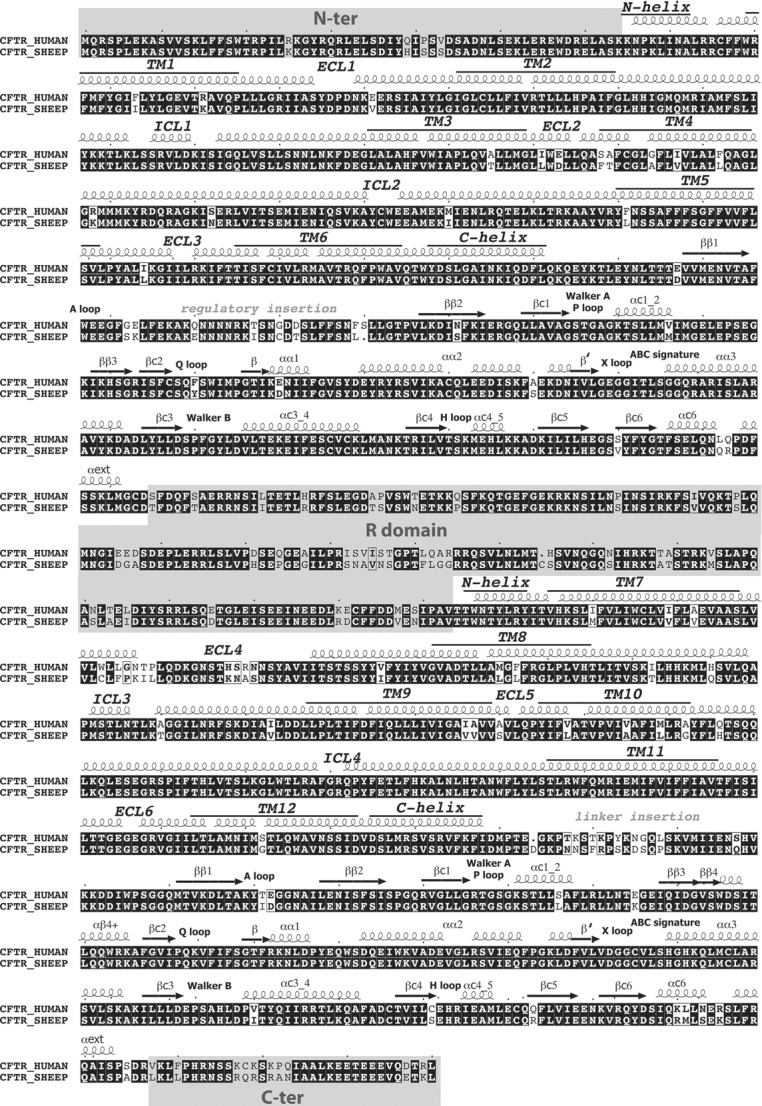
Alignment of the sequences of human and ovine CFTR
Amino acid sequence alignments of human (Riordan et al. 1989) and ovine (Tebbutt et al. 1995) CFTR are shown. Above the sequence alignments are the predicted secondary structures of CFTR based on the alignment of its sequence with that of Sav1866 (Mornon et al. 2008), whose three-dimensional structure (Dawson & Locher, 2006) was used to construct the models of CFTR's MSD:NBD assembly. Amino acids that are identical between human and ovine CFTR are shown as white letters on a black background. Regions which are not included in the ovine CFTR model, because of the absence of suitable templates, are shaded grey (N-terminus, regulatory (R) domain and C-terminus).
Cross-species differences in the molecular pharmacology of CFTR have important implications for drug development. Our observation that low micromolar concentrations of the fluorescein derivative phloxine B, an efficacious CFTR potentiator (Cai & Sheppard, 2002; Cai et al. 2006), were without effect on ovine CFTR Cl− currents suggests that ovine CFTR might be insensitive to CFTR potentiators. These data are reminiscent of the action of CFTR potentiators on murine CFTR, which is unresponsive to pyrophosphate (Lansdell et al. 1998a), phloxine B (Z. Cai & D. N. Sheppard, unpublished observations) and ivacaftor (Van Goor et al. 2009). Building on these data, we previously demonstrated that transfer of both NBDs of murine CFTR to human CFTR renders chimeric CFTR Cl− channels insensitive to potentiation by pyrophosphate (Scott-Ward et al. 2007). Given that both murine and ovine CFTR have prolonged channel openings and both are insensitive to potentiation by phloxine B, we speculate that sequence differences in the NBDs between human and ovine CFTR are responsible for the insensitivity of ovine CFTR to potentiation by phloxine B.
The sulphonylurea drug glibenclamide, a widely used open-channel blocker of CFTR, inhibited ovine CFTR Cl− currents, albeit with reduced efficacy compared to human CFTR. Interestingly, previous work has identified species-dependent differences in the action of glibenclamide, the thiazolidinone, CFTRinh-172 (Ma et al. 2002; Kopeikin et al. 2010) and the glycine hydrazide, GlyH-101 (Muanprasat et al. 2004; Norimatsu et al. 2012b). When tested on recombinant CFTR homologues expressed in Xenopus oocytes (Stahl et al. 2012) or airway epithelia expressing endogenous CFTR (Liu et al. 2007), GlyH-101 inhibited all homologues tested with the exception of murine CFTR, shark CFTR was insensitive to CFTRinh-172, and porcine CFTR was unaffected by glibenclamide (Liu et al. 2007; Stahl et al. 2012). To explain these differences in channel block, Stahl et al. (2012) speculated that subtle differences in the three-dimensional structure of the CFTR pore and local environment (e.g. hydrophobicity and charge) in the vicinity of drug-binding sites might be responsible. Based on the interaction of glibenclamide with molecular models of CFTR (Dalton et al. 2012) and our own modelling of ovine CFTR, the same explanation probably applies for differences in glibenclamide inhibition between human and ovine CFTR.
In marked contrast to the impact of the F508del mutation on human CFTR (Cheng et al. 1990), Ostedgaard et al. (2007) found that the F508del-CFTR processing defect was less severe in porcine and murine CFTR. Subsequently, Aleksandrov et al. (2012) demonstrated that the F508del mutation disrupts to varying degrees the processing of mammalian CFTR homologues, whereas in non-mammalian species (e.g. frog and chicken) there was substantial maturation of F508del-CFTR protein at 37°C. Aleksandrov et al. (2012) further demonstrated that F508del chicken CFTR exhibits marked thermostability, remaining active at physiological temperatures, unlike F508del human CFTR, which deactivates very rapidly. The present results demonstrate that F508del ovine CFTR exhibits plasma membrane instability, characterized by time-dependent loss of channel activity in excised membrane patches at 37°C. However, the retention of F508del ovine CFTR channel activity beyond 10 min at 37°C suggests that it has less severe thermal instability than F508del human CFTR (Wang et al. 2011; Liu et al. 2012; Wang et al. 2014).
Hypothesizing that structural differences between human and chicken CFTR account for the thermostability of F508del chicken CFTR, Aleksandrov et al. (2012) identified the F508del revertant mutation I539T (deCarvalho et al. 2002) and four proline residues at key positions within NBD1 (S422P, S434P, S492P, A534P). Of note, introducing the I539T revertant and the four proline mutations into F508del human CFTR restored protein processing, plasma membrane stability and channel function (Aleksandrov et al. 2012), suggesting that correction of NBD1 structure is sufficient to overcome the F508del mutation. By contrast, Ostedgaard et al. (2007) interpreted the species-dependent effects of F508del-CFTR on CFTR processing and channel function to suggest that these defects have different causes. This conclusion is supported by studies of human–murine CFTR chimeras which revealed that maturation of F508del-CFTR requires NBD1, whereas rescue of F508del-CFTR channel gating requires NBD1–MSD2 interactions (Dong et al. 2012). Consistent with these ideas, other studies (e.g. Mendoza et al. 2012; Rabeh et al. 2012) demonstrate that correction of NBD1 folding and the interaction of NBD1 with the fourth intracellular loop (ICL4) are required to suppress fully the processing and functional defects of F508del-CFTR.
Similar to its effects on murine and porcine CFTR Cl− channels (Ostedgaard et al. 2007), the impact of the F508del mutation on the gating behaviour of ovine CFTR was modest. In all species tested, the F508del mutation prolonged the duration of long closures separating bursts of channel openings (Ostedgaard et al. 2007; present study), arguing that the mutation disrupts the formation of the NBD1:NBD2 dimer. Building on these data, Jih et al. (2011) demonstrated that F508del-CFTR destabilizes both the full and partial NBD1:NBD2 dimer configurations during channel gating. We speculate that our observation that F508 deletion generates two populations of open times is further evidence that the F508del mutation destabilizes CFTR channel gating.
A notable feature of the present results is the reduction of i caused by the F508del mutation in ovine CFTR. Previous studies of F508del-CFTR and other CF mutations in the NBDs have found little or no evidence for effects of mutations on current flow through the CFTR pore unlike those in the MSDs (e.g. Sheppard et al. ,). Given the location of F229 (M4, a transmembrane segment predicted not to line the CFTR pore; Fig.1), we consider it unlikely that the F229L polymorphism is responsible for this change in conductance. Instead, the location of F508 at the interface between NBD1 and ICL4 raises the interesting possibility that under some circumstances F508del might destabilize the MSDs, reducing current flow through the CFTR pore.
In conclusion, ovine CFTR has a high degree of amino acid sequence identity with human CFTR. However, for several reasons, ovine CFTR formed a Cl− channel with noticeably greater activity than that of human CFTR. First, the single-channel conductance was increased. Second, ovine CFTR had a higher Po because the frequency and duration of channel openings were augmented. Third, with enhanced affinity and efficacy, ATP more strongly stimulated ovine CFTR channel gating. Similar to its impact on human CFTR, the F508del mutation disrupted the processing of ovine CFTR protein and reduced its plasma membrane stability. However, the mutation had less effect on ovine CFTR channel gating. These results have important implications for phylogenetic analysis of CFTR structure and function, arguing that subtle changes in structure have marked effects on channel function and the action of CF mutations.
Acknowledgments
We thank T. C. Hwang, P. Lehn and our departmental colleagues, especially J.-P. Mornon and M. K. A. Al Salmani, for valuable discussions and assistance. We are very grateful to J. R. Riordan and T. Jensen (University of North Carolina) and Cystic Fibrosis Foundation Therapeutics (Bethesda, MD, USA) for the generous gift of anti-CFTR antibodies.
Glossary
- ABC transporter
ATP-binding cassette transporter
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- i
single-channel current amplitude
- IBI
interburst interval
- ICL
intracellular loop
- M
transmembrane segment
- MBD
mean burst duration
- MSD
membrane-spanning domain
- NBD
nucleotide-binding domain
- Po
open probability
- PKA
protein kinase A
Additional information
Competing interests
None declared.
Author contributions
Z.C., A.H. and D.N.S. conceived and designed the experiments; Z.C., T.P.-P., P.K., M.J.M., C.B., B.L. and T.S.S.-W. performed the research; Z.C., T.P.-P., P.K., M.J.M., C.B., B.L., T.S.S.-W., I.C., A.H. and D.N.S. analysed and interpreted the data; Z.C., I.C., A.H. and D.N.S. drafted the article or revised it critically for important intellectual content. All authors approved the final version of the manuscript. The ovine CFTR cDNA was assembled in Professor Harris's Laboratory formerly at the Weatherall Institute of Molecular Medicine, University of Oxford, while the ovine F508del-CFTR construct was generated in her laboratory at Lurie Children's Research Center, Northwestern University Feinberg School of Medicine, where the CFTR processing experiments were also performed. Electrophysiological experiments were undertaken at the University of Bristol. Molecular modelling was carried out at the University of Pierre and Marie Curie.
Funding
This work was supported by the Cystic Fibrosis Foundation, Cystic Fibrosis Trust, the French Cystic Fibrosis Association Vaincre la Mucoviscidose and the National Institutes of Health (R01 HL094585); C.B. was supported by the Université de Poitiers and P.K. by the Strategic Scholarships Fellowships Frontier Research Networks, Office of the Higher Education Commission of Thailand.
Authors’ present addresses
T. Palmai-Pallag: School of Life Sciences, University of Lincoln, Brayford Pool, Lincoln LN6 7TS, UK.
P. Khuituan: Department of Physiology, Faculty of Science, Prince of Songkla University, Hat Yai, 90112 Thailand.
C. Boinot: Department of Physiology, School of Medicine, The Johns Hopkins University, Baltimore, MD 21205, USA.
T. S. Scott-Ward: Human Development and Health, Center for Human Development, Stem Cells and Regeneration, Faculty of Medicine, University of Southampton, Southampton SO16 6YD, UK.
References
- Alcorn DG, Adamson TM, Maloney JE. Robinson PM. A morphologic and morphometric analysis of fetal lung development in the sheep. Anat Rec. 1981;201:655–667. doi: 10.1002/ar.1092010410. [DOI] [PubMed] [Google Scholar]
- Aleksandrov AA, Kota P, Cui L, Jensen T, Alekseev AE, Reyes S, He L, Gentzsch M, Aleksandrov LA, Dokholyan NV. Riordan JR. Allosteric modulation balances thermodynamic stability and restores function of ΔF508 CFTR. J Mol Biol. 2012;419:41–60. doi: 10.1016/j.jmb.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Scott-Ward TS. Sheppard DN. Voltage-dependent gating of the cystic fibrosis transmembrane conductance regulator Cl− channel. J Gen Physiol. 2003;122:605–620. doi: 10.1085/jgp.200308921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z. Sheppard DN. Phloxine B interacts with the cystic fibrosis transmembrane conductance regulator at multiple sites to modulate channel activity. J Biol Chem. 2002;277:19546–19553. doi: 10.1074/jbc.M108023200. [DOI] [PubMed] [Google Scholar]
- Cai Z, Taddei A. Sheppard DN. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem. 2006;281:1970–1977. doi: 10.1074/jbc.M510576200. [DOI] [PubMed] [Google Scholar]
- Campbell KHS, McWhir J, Ritchie WA. Wilmut I. Sheep cloned by nuclear transfer from a cultured cell line. Nature. 1996;380:64–66. doi: 10.1038/380064a0. [DOI] [PubMed] [Google Scholar]
- Chen J-H, Cai Z. Sheppard DN. Direct sensing of intracellular pH by the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem. 2009;284:35495–35506. doi: 10.1074/jbc.M109.072678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR. Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Coote K, Atherton-Watson HC, Sugar R, Young A, MacKenzie-Beevor A, Gosling M, Bhalay G, Bloomfield G, Dunstan A, Bridges RJ, Sabater JR, Abraham WM, Tully D, Pacoma R, Schumacher A, Harris J. Danahay H. Camostat attenuates airway epithelial sodium channel function in vivo through the inhibition of a channel-activating protease. J Pharmacol Exp Ther. 2009;329:764–774. doi: 10.1124/jpet.108.148155. [DOI] [PubMed] [Google Scholar]
- Csanády L, Vergani P. Gadsby DC. Strict coupling between CFTR's catalytic cycle and gating of its Cl− ion pore revealed by distributions of open channel burst durations. Proc Natl Acad Sci USA. 2010;107:1241–1246. doi: 10.1073/pnas.0911061107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Aleksandrov L, Chang X-B, Hou Y-X, He L, Hegedus T, Gentzsch M, Aleksandrov A, Balch WE. Riordan JR. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007;365:981–994. doi: 10.1016/j.jmb.2006.10.086. [DOI] [PubMed] [Google Scholar]
- Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq J-P. Lazdunski M. Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- Dalton J, Kalid O, Schushan M, Ben-Tal N. Villà-Freixa J. New model of cystic fibrosis transmembrane conductance regulator proposes active channel-like conformation. J Chem Inf Model. 2012;52:1842–1853. doi: 10.1021/ci2005884. [DOI] [PubMed] [Google Scholar]
- Dawson RJP. Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- deCarvalho ACV, Gansheroff LJ. Teem JL. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ΔF508. J Biol Chem. 2002;277:35896–35905. doi: 10.1074/jbc.M205644200. [DOI] [PubMed] [Google Scholar]
- Demmers KJ, Carter D, Fan S, Mao P, Maqbool NJ, McLeod BJ, Bartolo R. Butt AG. Molecular and functional characterization of the cystic fibrosis transmembrane conductance regulator from the Australian common brushtail possum, Trichosurus vulpecula. J Comp Physiol B. 2010;180:545–561. doi: 10.1007/s00360-009-0433-6. [DOI] [PubMed] [Google Scholar]
- Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE. Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- Dong Q, Ostedgaard LS, Rogers C, Vermeer DW, Zhang Y. Welsh MJ. Human-mouse cystic fibrosis transmembrane conductance regulator (CFTR) chimeras identify regions that partially rescue CFTR-ΔF508 processing and alter its gating defect. Proc Natl Acad Sci USA. 2012;109:917–922. doi: 10.1073/pnas.1120065109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P. Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X. Linsdell P. Molecular determinants and role of an anion binding site in the external mouth of the CFTR chloride channel pore. J Physiol. 2003;549:387–397. doi: 10.1113/jphysiol.2002.038232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC. Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Hanrahan JW, Duguay F, Sansom S, Alon N, Jensen T, Riordan JR. Grzelczak Z. Low-conductance chloride channel activated by cAMP in the rectal gland of the shark Squalus acanthias and in cells heterologously expressing shark CFTR. Bull Mount Desert Island Biol Lab. 1993;32:48–49. [Google Scholar]
- Jih K-Y, Li M, Hwang T-C. Bompadre SG. The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. J Physiol. 2011;589:2719–2731. doi: 10.1113/jphysiol.2010.202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jih K-Y, Sohma Y. Hwang T-C. Nonintegral stoichiometry in CFTR gating revealed by a pore-lining mutation. J Gen Physiol. 2012;140:347–359. doi: 10.1085/jgp.201210834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikin Z, Sohma Y, Li M. Hwang T-C. On the mechanism of CFTR inhibition by a thiazolidinone derivative. J Gen Physiol. 2010;136:659–671. doi: 10.1085/jgp.201010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Delaney SJ, Lunn DP, Thomson SA, Sheppard DN. Wainwright BJ. a Comparison of the gating behaviour of human and murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in mammalian cells. J Physiol. 1998;508:379–392. doi: 10.1111/j.1469-7793.1998.379bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Kidd JF, Delaney SJ, Wainwright BJ. Sheppard DN. b Regulation of murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in Chinese hamster ovary cells. J Physiol. 1998;512:751–764. doi: 10.1111/j.1469-7793.1998.751bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS. Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- Leir S-H. Harris A. MUC6 mucin expression inhibits tumor cell invasion. Exp Cell Res. 2011;317:2408–2419. doi: 10.1016/j.yexcr.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Linsdell P, Tabcharani JA, Rommens JM, Hou Y-X, Chang X-B, Tsui L-C, Riordan JR. Hanrahan JW. Permeability of wild-type and mutant cystic fibrosis transmembrane conductance regulator chloride channels to polyatomic anions. J Gen Physiol. 1997;110:355–364. doi: 10.1085/jgp.110.4.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Luo M, Zhang L, Ding W, Yan Z. Engelhardt JF. Bioelectric properties of chloride channels in human, pig, ferret, and mouse airway epithelia. Am J Respir Cell Mol Biol. 2007;36:313–323. doi: 10.1165/rcmb.2006-0286OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, O'Donnell N, Landstrom A, Skach WR. Dawson DC. Thermal instability of ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) channel function: protection by single suppressor mutations and inhibiting channel activity. Biochemistry. 2012;51:5113–5124. doi: 10.1021/bi300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Chang X-B, Bear C, Kartner N, Mohamed A, Riordan JR. Grinstein S. The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane: determination of functional half-lives on transfected cells. J Biol Chem. 1993;268:21592–21598. [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJV. Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan G, Davidson H, Holder E, Davies LA, Pringle IA, Sumner-Jones SG, Baker A, Tennant P, Gordon C, Vrettou C, Blundell R, Hyndman L, Stevenson B, Wilson A, Doherty A, Shaw DJ, Coles RL, Painter H, Cheng SH, Scheule RK, Davies JC, Innes JA, Hyde SC, Griesenbach U, Alton EWFW, Boyd AC, Porteous DJ, Gill DR. Collie DDS. Pre-clinical evaluation of three non-viral gene transfer agents for cystic fibrosis after aerosol delivery to the ovine lung. Gene Ther. 2011;18:996–1005. doi: 10.1038/gt.2011.55. [DOI] [PubMed] [Google Scholar]
- Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F. Šali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA. Thomas PJ. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148:164–174. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon J-P, Hoffman B, Jonic S, Lehn P. Callebaut I. Full-open and closed CFTR channels, with lateral tunnels from the cytoplasm and an alternative position of the F508 region, as revealed by molecular dynamics. Cell Mol Life Sci. 2015;72:1377–1403. doi: 10.1007/s00018-014-1749-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon J-P, Lehn P. Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci. 2008;65:2594–2612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mornon J-P, Lehn P. Callebaut I. Molecular models of the open and closed states of the whole human CFTR protein. Cell Mol Life Sci. 2009;66:3469–3486. doi: 10.1007/s00018-009-0133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJV. Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis and in vivo efficacy. J Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norimatsu Y, Ivetac A, Alexander C, Kirkham J, O'Donnell N, Dawson DC. Sansom MSP. a Cystic fibrosis transmembrane conductance regulator: a molecular model defines the architecture of the anion conduction path and locates a ‘bottleneck’ in the pore. Biochemistry. 2012;51:2199–2212. doi: 10.1021/bi201888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norimatsu Y, Ivetac A, Alexander C, O'Donnell N, Frye L, Sansom MSP. Dawson DC. b Locating a plausible binding site for an open-channel blocker, GlyH-101, in the pore of the cystic fibrosis transmembrane conductance regulator. Mol Pharmacol. 2012;82:1042–1055. doi: 10.1124/mol.112.080267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, Xie W, Fisher JT, Keiser NW, Lei D, Zhou W, Yan Z, Li G, Evans TIA, Meyerholz DK, Wang K, Stewart ZA, Norris AW. Engelhardt JF. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Invest. 2012;122:3755–3768. doi: 10.1172/JCI60610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olver RE. Robinson EJ. Sodium and chloride transport by the tracheal epithelium of fetal, new-born and adult sheep. J Physiol. 1986;375:377–390. doi: 10.1113/jphysiol.1986.sp016122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostedgaard LS, Rogers CS, Dong Q, Randak CO, Vermeer DW, Rokhlina T, Karp PH. Welsh MJ. Processing and function of CFTR-ΔF508 are species-dependent. Proc Natl Acad Sci U S A. 2007;104:15370–15375. doi: 10.1073/pnas.0706974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzulo AA, Tang XX, Hoegger MJ, Alaiwa MHA, Ramachandran S, Moninger TO, Karp PH, Wohlford-Lenane CL, Haagsman HP, van Eijk M, Bánfi B, Horswill AR, Stoltz DA, McCray PB, Jr, Welsh MJ. Zabner J. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487:109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MP, Ishihara H, Sheppard DN. Welsh MJ. Function of Xenopus cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channels and use of human-Xenopus chimeras to investigate the pore properties of CFTR. J Biol Chem. 1996;271:25184–25191. doi: 10.1074/jbc.271.41.25184. [DOI] [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A. Lukacs GL. Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell. 2012;148:150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm ML, Iannuzzi MC, Collins FS. Tsui L-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB, Jr, Zabner J, Prather RS. Welsh MJ. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. 2008;321:1837–1841. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz BD, DeRoos ADG, Venglarik CJ, Singh AK, Frizzell RA. Bridges RJ. Glibenclamide blockade of CFTR chloride channels. Am J Physiol Lung Cell Mol Physiol. 1996;271:L192–L200. doi: 10.1152/ajplung.1996.271.2.L192. [DOI] [PubMed] [Google Scholar]
- Schultz BD, Frizzell RA. Bridges RJ. Rescue of dysfunctional ΔF508-CFTR chloride channel activity by IBMX. J Membr Biol. 1999;170:51–66. doi: 10.1007/s002329900537. [DOI] [PubMed] [Google Scholar]
- Scott-Ward TS, Cai Z, Dawson ES, Doherty A, Da Paula AC, Davidson H, Porteous DJ, Wainwright BJ, Amaral MD, Sheppard DN. Boyd AC. Chimeric constructs endow the human CFTR Cl− channel with the gating behavior of murine CFTR. Proc Natl Acad Sci USA. 2007;104:16365–16370. doi: 10.1073/pnas.0701562104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serohijos AWR, Hegedús T, Aleksandrov AA, He L, Cui L, Dokholyan NV. Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Ostedgaard LS, Winter MC. Welsh MJ. Mechanism of dysfunction of two nucleotide binding domain mutations in cystic fibrosis transmembrane conductance regulator that are associated with pancreatic sufficiency. EMBO J. 1995;14:876–883. doi: 10.1002/j.1460-2075.1995.tb07069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Rich DP, Ostedgaard LS, Gregory RJ, Smith AE. Welsh MJ. Mutations in CFTR associated with mild-disease-form Cl− channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- Sheppard DN. Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J Physiol. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SS, Liu X, Zhang Z-R, Sun F, Kriewall TE, McCarty NA. Dawson DC. CFTR: covalent and noncovalent modification suggests a role for fixed charges in anion conduction. J Gen Physiol. 2001;118:407–431. doi: 10.1085/jgp.118.4.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Aubin CN. Linsdell P. Positive charges at the intracellular mouth of the pore regulate anion conduction in the CFTR chloride channel. J Gen Physiol. 2006;128:535–545. doi: 10.1085/jgp.200609516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, Stahl K, Brubacher MB. Forrest JN., Jr Divergent CFTR orthologs respond differently to the channel inhibitors CFTRinh-172, glibenclamide, and GlyH-101. Am J Physiol Cell Physiol. 2012;302:C67–C76. doi: 10.1152/ajpcell.00225.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Sui H, Fisher JT, Yan Z, Liu X, Cho H-J, Joo NS, Zhang Y, Zhou W, Yi Y, Kinyon JM, Lei-Butters DC, Griffin MA, Naumann P, Luo M, Ascher J, Wang K, Frana T, Wine JJ, Meyerholz DK. Engelhardt JF. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbutt SJ, Wardle CJC, Hill DF. Harris A. Molecular analysis of the ovine cystic fibrosis transmembrane conductance regulator gene. Proc Natl Acad Sci USA. 1995;92:2293–2297. doi: 10.1073/pnas.92.6.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuggle KL, Birket SE, Cui X, Hong J, Warren J, Reid L, Chambers A, Ji D, Gamber K, Chu KK, Tearney G, Tang LP, Fortenberry JA, Du M, Cadillac JM, Bedwell DM, Rowe SM, Sorscher EJ. Fanucchi MV. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS One. 2014;9:e91253. doi: 10.1371/journal.pone.0091253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkenier H, Judd LW, Li H, Hussian S, Sheppard DN. Davis AP. Preorganized bis-thioureas as powerful anion carriers: chloride transport by single molecules in large unilamellar vesicles. J Am Chem Soc. 2014;136:12507–12512. doi: 10.1021/ja507551z. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PDJ, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M. Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC. Gadsby DC. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433:876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Okeyo GO, Tao B, Hong JS. Kirk KL. Thermally unstable gating of the most common cystic fibrosis mutant channel (ΔF508): ‘rescue’ by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. J Biol Chem. 2011;286:41937–41948. doi: 10.1074/jbc.M111.296061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu J, Loizidou A, Bugeja LA, Warner R, Hawley BR, Cai Z, Toye AM, Sheppard DN. Li H. CFTR potentiators partially restore channel function to A561E-CFTR, a cystic fibrosis mutant with a similar mechanism of dysfunction as F508del-CFTR. Br J Pharmacol. 2014;171:4490–4503. doi: 10.1111/bph.12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh MJ, Ramsey BW, Accurso F. Cutting GR. Cystic fibrosis. In: Valle D, editor; Scriver CR, Beaudet AL, Sly WS, editors. The Metabolic and Molecular Basis of Inherited Disease. 8th edn. New York: McGraw-Hill Inc; 2001. pp. 5121–5188. [Google Scholar]
- Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, Bot A, Jorna H, de Jonge HR. Scholte BJ. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros. 2011;10(Suppl. 2):S152–S171. doi: 10.1016/S1569-1993(11)60020-9. [DOI] [PubMed] [Google Scholar]
- Winter MC, Sheppard DN, Carson MR. Welsh MJ. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophys J. 1994;66:1398–1403. doi: 10.1016/S0006-3495(94)80930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J-J, Li M-S, Qi J. Linsdell P. Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J Gen Physiol. 2010;135:229–245. doi: 10.1085/jgp.200910327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J-J. Linsdell P. Evidence that extracellular anions interact with a site outside the CFTR chloride channel pore to modify channel properties. Can J Physiol Pharmacol. 2009;87:387–395. doi: 10.1139/y09-023. [DOI] [PubMed] [Google Scholar]