

Abstract
Adult mammalians possess three cell systems that are activated by acute bodily hypoxia: pulmonary artery smooth muscle cells (PASMC), carotid body chemoreceptor cells (CBCC) and erythropoietin (EPO)-producing cells. In rats, chronic perinatal hyperoxia causes permanent carotid body (CB) atrophy and functional alterations of surviving CBCC. There are no studies on PASMC or EPO-producing cells. Our aim is to define possible long-lasting functional changes in PASMC or EPO-producing cells (measured as EPO plasma levels) and, further, to analyse CBCC functional alterations. We used 3- to 4-month-old rats born and reared in a normal atmosphere or exposed to perinatal hyperoxia (55–60% O2 for the last 5–6 days of pregnancy and 4 weeks after birth). Perinatal hyperoxia causes an almost complete loss of hypoxic pulmonary vasoconstriction (HPV), which was correlated with lung oxidative status in early postnatal life and prevented by antioxidant supplementation in the diet. O2-sensitivity of K+ currents in the PASMC of hyperoxic animals is normal, indicating that their inhibition is not sufficient to trigger HPV. Perinatal hyperoxia also abrogated responses elicited by hypoxia on catecholamine and cAMP metabolism in the CB. An increase in EPO plasma levels elicited by hypoxia was identical in hyperoxic and control animals, implying a normal functioning of EPO-producing cells. The loss of HPV observed in adult rats and caused by perinatal hyperoxia, comparable to oxygen therapy in premature infants, might represent a previously unrecognized complication of such a medical intervention capable of aggravating medical conditions such as regional pneumonias, atelectases or general anaesthesia in adult life.
Key points
Adult animals that have been perinatally exposed to oxygen-rich atmospheres (hyperoxia), recalling those used for oxygen therapy in infants, exhibit a loss of hypoxic pulmonary vasoconstriction, whereas vasoconstriction elicited by depolarizing agents is maintained.
Loss of pulmonary hypoxic vasoconstriction is not linked to alterations in oxygen-sensitive K+ currents in pulmonary artery smooth muscle cells.
Loss of hypoxic vasoconstriction is associated with early postnatal oxidative damage and corrected by an antioxidant diet.
Perinatal hyperoxia damages carotid body chemoreceptor cell function and the antioxidant diet does not reverse it. The hypoxia-elicited increase in erythropoietin plasma levels is not affected by perinatal hyperoxia.
The potential clinical significance of the findings in clinical situations such as pneumonia, chronic obstructive pulmonary disease or general anaesthesia is considered.
Introduction
Mammals possess three interrelated systems that work in concert to maintain an adequate supply of O2 to their organs, thus preventing or alleviating the adverse effects of hypoxia. Pulmonary artery smooth muscle cells (PASMC) sense alveolar hypoxia and respond with a Ca2+-dependent contractile response. In situations of uneven ventilation of the lungs, contraction of PASMC almost instantaneously diverts blood to well ventilated lung regions to optimize ventilation/perfusion matching and blood oxygenation (Marshall et al. 1994a,b; Sylvester et al. 2012). Carotid body (CB) chemoreceptor cells (CBCC) detect hypoxic hypoxia and respond with a Ca2+-dependent release of neurotransmitters triggering respiratory reflexes that increase alveolar ventilation, alveolar  and haemoglobin saturation (Gonzalez et al. 1994; Kumar, 2009). Erythropoietin (EPO)-producing cells (primarily interstitial fibroblasts in the kidney) sense local tissue hypoxia and, in a time scale of a few hours, respond with increased rates of EPO gene transcription and translation and increased secretion of EPO hormone; EPO augments the O2-carrying capacity of blood by stimulating erythrocyte production in the bone marrow (Jelkmann, 1992). The three O2-sensing cell systems exhibit low hypoxic thresholds (i.e. they start responding when
and haemoglobin saturation (Gonzalez et al. 1994; Kumar, 2009). Erythropoietin (EPO)-producing cells (primarily interstitial fibroblasts in the kidney) sense local tissue hypoxia and, in a time scale of a few hours, respond with increased rates of EPO gene transcription and translation and increased secretion of EPO hormone; EPO augments the O2-carrying capacity of blood by stimulating erythrocyte production in the bone marrow (Jelkmann, 1992). The three O2-sensing cell systems exhibit low hypoxic thresholds (i.e. they start responding when 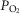 is relatively high: alveolar or arterial
is relatively high: alveolar or arterial 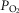 of 65–70 mmHg; O2 content >90%) and their coordinated function allows healthy humans to assure an adequate O2 content in arterial blood at barometric pressures as low as 400 mmHg or an ambient
of 65–70 mmHg; O2 content >90%) and their coordinated function allows healthy humans to assure an adequate O2 content in arterial blood at barometric pressures as low as 400 mmHg or an ambient 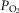 of 84 mmHg (Ward et al. 1995; Gonzalez et al. 2002). Thus, as a whole, these three cell types represent the general system that adult mammals employ to compensate for or adapt to systemic hypoxia (Gonzalez et al. 2010). The aortic and neuroepithelial bodies and, in the full-term fetus and newborn, the adrenal medulla also contribute to those adjustments.
of 84 mmHg (Ward et al. 1995; Gonzalez et al. 2002). Thus, as a whole, these three cell types represent the general system that adult mammals employ to compensate for or adapt to systemic hypoxia (Gonzalez et al. 2010). The aortic and neuroepithelial bodies and, in the full-term fetus and newborn, the adrenal medulla also contribute to those adjustments.
Perinatal exposure of rats to a hyperoxic atmosphere (60% O2, 4 weeks) produces permanent hypotrophy of the CB and a marked diminution of the response of the organ to hypoxia when assessed as the carotid sinus nerve action potential frequency (the afferent arm of the CB chemoreflex; Ling et al. 1997; Erickson et al. 1998; Fuller et al. 2002; Bisgard et al. 2003; Prieto Lloret et al. 2004; Wenninger et al. 2006; Bavis et al. 2013). However, when the CB chemoreflex is explored via phrenic nerve activity or ventilation (i.e. the efferent arm of the chemoreflex), the dysfunction produced by perinatal hyperoxia is appreciated differently. In the original study of Ling et al. (1996), it was reported that 1 month of perinatal hyperoxic treatment caused a marked decrease in hypoxic ventilation when the animals were studied up to the age of 5 months (i.e. 4 months after hyperoxic treatment); it was concluded that animals may suffer impaired chemoresponsiveness throughout their lives. By contrast, in our previous study (Prieto-Lloret et al. 2004), as well as those of Dauger et al. (2003) and Wenninger et al. (2006), no alterations in control, hypoxic and hypercapnic ventilation were noted when animals were explored at ≥ 4 months of age. In another study, Ling et al. (1998) extended their observations until the animals reached 14 months of age and they found a marked decrease in the hypoxic response that recovered with time, so that, at 14 months of age, the responses to hypoxia were normalized. Subsequently, Fuller et al. (2002) re-examined the issue and found that animals aged up to 14–15 months exhibited functional impairment in the hypoxic responses and concluded that perinatal hyperoxia causes life-long impairment of carotid chemoreceptor function; a recent review is provided by Bavis et al. (2013). Prieto-Lloret et al. (2004) and, more recently, Kim et al. (2013) located the hyperoxic damage in surviving CBCC in a step prior to cell depolarization and Ca2+ entry into the hypoxic transduction cascade (Gonzalez et al. 1992).
By contrast to the situation for CB, there are no studies exploring the effects of perinatal hyperoxia on the other two systems of oxygen-sensitive cells. Clinically, it is important to determine whether exposure to perinatal hyperoxia damages PASMC and hypoxic pulmonary vasoconstriction (HPV) because, although oxygen therapy in hypoxemic, usually premature, newborn infants is aimed at restoring a normal blood O2 content and avoiding hyperoxaemia, lung tissues are unavoidably exposed to hyperoxia. Thus, if hyperoxia deteriorates the HPV, it would imply the loss of an important homeostatic mechanism. This loss could be fatal in lung pathologies such as non-generalized pneumonias or atelectases or mild chronic obstructive pulmonary diseases, as well as in situations of general anaesthesia, particularly in thoracic surgery, where systemic arterial 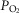 is supported by HPV (Glasser et al. 1983, Eisenkraft, 1990; Marshall et al. 1994a,b; Nagendran et al. 2006; Karzai and Schwarzkopf, 2009; Sylvester et al. 2012). Additionally, hyperoxaemia is a frequent undesirable accident in infant oxygen therapy (Tracy et al. 2004; Hartnett and Penn, 2012; Bavis et al. 2013), which not only would augment the exposure of PASMC to high
is supported by HPV (Glasser et al. 1983, Eisenkraft, 1990; Marshall et al. 1994a,b; Nagendran et al. 2006; Karzai and Schwarzkopf, 2009; Sylvester et al. 2012). Additionally, hyperoxaemia is a frequent undesirable accident in infant oxygen therapy (Tracy et al. 2004; Hartnett and Penn, 2012; Bavis et al. 2013), which not only would augment the exposure of PASMC to high 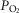 , but also would expose EPO-producing cells to abnormally high
, but also would expose EPO-producing cells to abnormally high 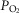 . In this context, the present study aimed to further analyse the functional alterations in the CB and to define possible alterations in the other two O2-sensing cell types. Accordingly, we used 3- to 4-month-old rats born and reared in a normal atmosphere or exposed to perinatal hyperoxia (55–60% O2 for the last 6 days of pregnancy and the initial 4 weeks after birth). Additionally, although it has been reported that perinatal hyperoxia does not cause oxidative stress as assessed via plasma carbonylated proteins (Bavis et al. 2008), it should be expected to result in an increase in reactive oxygen species (Turrens et al. 2003; Halliwell and Gutteridge 2007). We measured the levels of reduced glutathione (GSH) and oxidized glutathione (GSSG) in the liver, brain and lung, and found that the glutathione redox potential was diminished in the liver and lung in hyperoxic animals of 7 days of age. This prompted additional experiments in which mothers and litters received an antioxidant diet during hyperoxic exposure. We found that perinatal hyperoxic exposure alters the metabolism of catecholamine (CA) in the CB but causes no alteration in the ventilatory response to hypoxia or hypercapnia. Hyperoxic animals showed a normal EPO plasma levels in response to 10 h of hypoxic exposure. By contrast, we found that hyperoxic exposure caused an almost complete loss of HPV. An antioxidant diet corrected the deviations of the redox status and prevented the loss of the HPV but did not reverse the alterations in the CB. The potential mechanisms and clinical significance of the findings are discussed.
. In this context, the present study aimed to further analyse the functional alterations in the CB and to define possible alterations in the other two O2-sensing cell types. Accordingly, we used 3- to 4-month-old rats born and reared in a normal atmosphere or exposed to perinatal hyperoxia (55–60% O2 for the last 6 days of pregnancy and the initial 4 weeks after birth). Additionally, although it has been reported that perinatal hyperoxia does not cause oxidative stress as assessed via plasma carbonylated proteins (Bavis et al. 2008), it should be expected to result in an increase in reactive oxygen species (Turrens et al. 2003; Halliwell and Gutteridge 2007). We measured the levels of reduced glutathione (GSH) and oxidized glutathione (GSSG) in the liver, brain and lung, and found that the glutathione redox potential was diminished in the liver and lung in hyperoxic animals of 7 days of age. This prompted additional experiments in which mothers and litters received an antioxidant diet during hyperoxic exposure. We found that perinatal hyperoxic exposure alters the metabolism of catecholamine (CA) in the CB but causes no alteration in the ventilatory response to hypoxia or hypercapnia. Hyperoxic animals showed a normal EPO plasma levels in response to 10 h of hypoxic exposure. By contrast, we found that hyperoxic exposure caused an almost complete loss of HPV. An antioxidant diet corrected the deviations of the redox status and prevented the loss of the HPV but did not reverse the alterations in the CB. The potential mechanisms and clinical significance of the findings are discussed.
Methods
Animals
The experiments were performed in Wistar rats of both sexes born and reared in the normal room air atmosphere for their entire life (controls) or in an atmosphere of 55–60% O2 for the last 5–7 days of intrauterine life and for 4 weeks after birth (hyperoxic); thereafter, the pups were brought to the normal atmosphere of the vivarium until use (rats age 90–120 days). Groups of seven rats at days 14–15 of pregnancy were individually caged and placed in a big glass cage with inlets and outlets for gases and orifices for a temperature probe. The chamber was continuously flushed with a gas mixture of 55–60% O2/balance N2. The continuous gas flow secured washing out of CO2, heat and vapour water; temperature was stable at 22–25ºC. Every 4 days, animals were cleaned and the food was replaced; immediately after cleaning, the rat cages were brought back to the big cage and a high gas flow was applied to achieve 55–60% O2 in 15–20 min. The control animals were maintained in the same room for the entire period. At 28 days of postnatal age, control and experimental mothers and litters were transferred to the general vivarium until the experiments (Prieto-Lloret et al. 2004).
One additional group of experiments was performed in identically hyperoxic animals, for which, during the entire exposure to an O2-rich atmosphere, mothers (and offspring) were fed with a diet supplemented with vitamin E (α-tocopherol, 300 mg kg−1 of regular rodent pellet) and R-lipoic acid (1 g kg−1 of pellet); the diet was provided by Panlab, Barcelona, Spain). Body weight of the animals was 332 ± 19, 302 ± 20 and 367 ± 14 g in control, hyperoxic and hyperoxic supplemented groups, respectively.
Handling of the animals was conducted in accordance with the European Community Council directive of 24 of November 1986 (86/609/EEC) for the Care and Use of Laboratory Animals. The Institutional Committee of the University of Valladolid for Animal Care and Use approved the protocols. Animals were killed by an intracardiac injection (250 mg kg−1) of sodium pentobarbital.
Recording of pulmonary arterial pressure (PAP) and HPV
Isolated in situ lung preparation
In an initial series of experiments, we used a blood perfused in situ isolated preparation of rat lungs (Hauge, 1968; Tamayo et al. 1997) to register HPV. Rats of both sexes were anaesthetized with ketamine (50 mg; i.p.), tracheostomized and pump ventilated (CL Palmer, London, UK) with a gas mixture (20% O2, 5% CO2/balance N2, normoxia; or 2% O2, 5% CO2/balance N2, hypoxia) at a positive end-expiratory pressure of 2 cmH2O and a frequency of 40 breaths min−1. Tidal volume was adjusted in every animal to achieve a peak inspiratory pressure of 10 cmH2O. In each experiment, a donor rat was heparinized and its blood withdrawn through the carotid arteries; the blood from the donor was placed in a reservoir (38ºC) and, when combined with the blood of the rat under study, was used to fill the perfusion circuit (20 ml), as described in detail in Tamayo et al. (1997); for surgical procedures, see also Liu et al. (1991).
The pressure transducer connected to the pulmonary artery was fed to a polygraph (Dynograph Recorder R 612; Beckman Coulter, Fullerton, CA, USA) and PAP was continuously monitored. The recorded signal was filtered with a 30 Hz low-pass filter. The reservoir was filled with blood to appproximately one-third of its volume, and the atmosphere above during the experiment had the same composition as the ventilating gas. The tubing of the pump driving the blood to the lung circulation was placed at the bottom of the reservoir. In all experiments, the reservoir itself was maintained at a fixed altitude to yield a constant pressure of 2 mmHg in the left atrium. Blood was perfused at a constant rate of 0.03 ml g−1 min−1. This perfusion rate produced a PAP between 10 and 15 mmHg (mean ± SD: 13.2 ± 1.1 mmHg; n = 48) at the onset of the experiment (20–30 min after surgical procedures). With the perfusion rate maintained constant, any change recorded in the perfusion pressure reflected changes in the pulmonary vascular resistance. Blood samples taken from the bottom of the reservoir and from the left atrial cannula yield the same values for blood gas analysis (Automatic Blood Gas System, AVL 990; AVL Biochemical Instruments, Graz, Austria). Blood pH was maintained at between 7.36 and 7.44 by injection (as required) of small boluses of isotonic NaCO3H into the reservoir.
PAP recordings started after ventilating the lungs with the normoxic gas mixture and perfusing the lungs with blood equilibrated with the normoxic gas for a few minutes. After achieving a stable normoxic baseline (20% O2, 5% CO2, 75% N2), the ventilation of the lungs and the equilibration of the blood in the reservoir was switched to a hypoxic gas mixture (2% O2, 5% CO2, 93% N2) for 7–8 min and, after a new period of normoxia (9–10 min), a second hypoxic test was made. In other experiments, after a hypoxic test and under in normoxic conditions, 4-aminopyridines were added to the reservoir to reach a final concentration of 10 mm or N-methyl-l-arginine (NMLA; final concentrations in the circuit of 5 × 10−5 m. Changes in PAP (ΔPAP) were determined as the difference between peak PAP recorded during the stimuli and the previous control baseline PAP.
In vivo recording of PAP and HPV
In a different group of experiments, PAP was recorded from intact animals. Ketamine anaesthetized rats were tracheostomized and pump ventilated with room air (CL Palmer) (60 cycles min–1 and a positive end-expiratory pressure of 2 cmH2O) to measure systemic arterial pressure (AP) and PAP when the ventilating pump was fed with air or with 10% O2. Systemic and pulmonary arterial pressures were continuously monitored with catheters inserted in the carotid and pulmonary arteries, respectively. To reach the pulmonary artery, a minimal thoracotomy was performed, a catheter was inserted into the right ventricle and, under oscilloscopic control, driven to the pulmonary artery. As in the case of the systemic arterial pressure, the catheter was connected to a pressure transducer, with signals being stored for subsquent analysis. In both cases (i.e. systemic and pulmonary arterial pressure), mean arterial pressures (MAP) are plotted. They were calculated according to the expression: MAP = DP + 1/3 (SP – DP), where DP is diastolic pressure and SP is systolic pressure.
Immunostaining and morphometric analysis of the muscle cell layer of the pulmonary arteries
Lungs from three control and three hyperoxic rats were removed and their arterial trees were dissected under a microscope (Tamayo et al. 1997) and fixed during 6 h by immersion in 4% paraformaldehyde in 0.1 m phosphate buffer (PB) (pH 7.40) at 4ºC. Thereafter, the arterial trees transferred to 30% sucrose in PB for cryoprotection. After embedding in Tissue-Tek (Sakura Finetek USA, Inc., Torrance, CA, USA), the arteries were frozen at –20ºC, serial sectioned (10 μm thick) in a Leitz Cryostat (1720) (Leica Microsystems, Wetzlar, Germany) and collected in glass slides coated with 3-aminopropyltriethoxy-silane (Sigma, Madrid, Spain). The sections were washed in phosphate-buffered saline (PBS) at room temperature (5 min), and incubated in PBS containing 0.1% Triton X-100 and 2% non-immunized goat serum (permeabilizing-blocking solution) for 30 min. The incubation with the primary antibody (mouse anti-smooth muscle actin; Sigma; dilution 1:500 in permeabilizing solution) was made overnight at 4ºC. Thereafter, sections were washed in PBS (3 × 10 min) and incubated for 1 h at room temperature with the secondary antibody (rabbit anti-mouse-horseradish peroxidase; Abcam, Cambridge, MA, USA; dilution 1:300 in permeabilizing solution). The specific labelling was developed with diaminobenzidine. Finally, sections were rinsed with distilled water, dehydrated, washed with xylene and mounted in Eukitt (Kindler GMBH and Co., Hannover, Germany). Negative controls were similarly incubated but in the absence of primary antibody. Sections were examined using an Axioscop 2 (mot plus) microscope (Carl Zeiss, Oberkochen, Germany) with Nomarski optics and were images captured with a digital camera (CoolSnap; Photometrics, Tucson, AZ, USA). In the captured images (10×; Phan-Neofluor) the thickness of the muscular layer was measured and the perimeter of the artery was obtained directly using MetaMorph, version 6.3 (Molecular Devices, Sunnyvale, CA, USA) for image analysis.
Electrophysiological recordings
All of the procedures used to dissociate PASMC from small calibre resistance arteries (200–500 μm in diameter) have been described previously (Cogolludo et al. 2003; Frazziano et al. 2011). In brief, small pulmonary arteries (300–500 μm of internal diameter) were dissected into Ca2+-free physiological saline (in mm): 130 NaCl, 5 KCl, 1.2 MgCl2, 0 CaCl2, 10 glucose, 10 Hepes; pH 7.3 with NaOH, and then endothelium denuded and cut into small rings (2 mm in length). Cell dissociation was carried out by incubation in the same saline containing 1 mg ml–1 papain, 0.8 mg ml–1 dithiothreitol and 0.7 mg ml–1 albumin; cells were used within 8 h. Membrane currents were recorded with an Axopatch 200B and a Digidata 1322A (Axon Instruments, Burlingame, CA, USA) using the whole-cell configuration of the patch-clamp technique, normalized for cell capacitance and expressed in pA/pF. For optimal IKv recording, cells were superfused with the same Ca2+-free PSS and the internal solution was (in mm): 110 KCl, 1.2 MgCl2, 5 Na2ATP, 10 Hepes and 10 EGTA (pH 7.3). Currents were elicited by 200 ms depolarizing pulses from –60 mV to +60 mV in incrementsof 10 mV. All experiments were performed at room temperature (22–24ºC).
Whole body plethysmography
Ventilation was measured in conscious freely-moving rats by whole body plethysmography. The system (Emka Technologies, Paris, France) consists of 5 litre metacrylate chambers continuously fluxed (2 l min−1) with the desired gas mixtures. Temperature was maintained in the chamber within the thermo-neutral range (22–24ºC). Tidal volume (TV; ml kg–1), respiratory frequency (breaths min–1) and minute ventilation (ml min–1 kg–1) were measured. Briefly, the rats were placed in the plethysmographic chamber and breathed room air for at least 30 min until they were adapted to the ambience of the chamber and acquired a standard resting behaviour. Subsequently, we started recording ventilatory parameters during 20 min, followed by fluxing the chamber with a gas mixture containing 12%, 10% or 7% O2 (rest N2; 2 l min–1) and 5% CO2 in air for 10 min. Each hypoxic or hypercapnic exposure was followed by a 10–20 min recovery period in air. The pressure changes within the chamber reflecting TV were measured with a high-gain differential pressure transducer. Ideally, the frequency of pressure fluctuations is identical to the breathing movements; spurious fluctuations of the pressure as a result of animal movements were rejected electronically. The amplitude of the pressure oscillations is proportionally related to TV; a calibration of the system by injections of 2–5 ml of air into the chamber allowed the direct estimation of TV. Pressure signals were fed to a computer for visualization and storage, allowing offline analysis with EMKA software (emka Technologies SAS, Paris, France).
Exposure to hypoxia
The control and hyperoxic rats used for the measurement of plasma EPO and content of CA in the CB were exposed for 10 h at an atmosphere of 10% O2, which was achieved by flushing a plastic cage capable of holding two smaller rat cages containing five rats each. During this period (08.00–18.00 h), the animals had water and food available ad libitum. In some of the initial experiments, the hypoxic exposure lasted only 5 h (08.00–13.00 h).
Measurement of endogenous CA in the CB and the superior cervical ganglion (SCG)
After cleaning the surrounding tissues under a microscope, the CBs and the SGC were transferred to cold Eppendorf tubes containing, respectively, 75 and 250 μl of 0.1 n perchloric acid and 0.1 mm EDTA, weighed and usually frozen at -80ºC until HPLC analysis. Regardless of whether they were immediately processed or frozen, the tissues were glass-to-glass homogenized at 0–4ºC and the homogenates, plus an additional aliquot of 50 μl of the perchloric acid-EDTA solution, as used to quantitatively collect the tissue homogenate, were centrifuged (10 min; 0–4ºC) in a microfuge (Beckmann, Madrid, Spain). Aliquots (10-50 μl) of supernatants were directly injected into the HPLC system, composed of a programmable Waters 600 pump, a Waters 717 injector (Waters, Milford, MA, USA ) and an electrochemical detector BAS LC-4C (Bioanalytical Systems, Inc., West Lafayette, IN, USA). The mobile phase was a PB (25 mm) containing 0.65 mm of octanesulfonic sodium salt, 0.1 mm EDTA and 6% methanol (final pH was 3.46). The sensitivity of the detector and the volume of the injected samples were adjusted to obtain chromatographic peaks of adequate height. Signals from the detector were fed to an analogue-to-digital converter controlled by Peak Sample Chromatography System Software (Buck Scientific, East Northwalk, CT, USA). These chromatographic conditions very satisfactorily resolve norepinephrine (NE), epinephrine, dihydroxyphenyl acetic acid (the main catabolite of dopamine) and dopamine (DA) (Ramirez et al. 2012). The identification and quantification of endogenous CA in tissue samples were verified using internal and external standards.
Measurement of GSH and GSSG
The assay of GSH and GSSG has been described in detail by Gonzalez et al. (2004); see also Quintero et al. (2013). In brief, the upper half right lung, liver lobe or whole brain (without cerebellum) were cleaned of surrounding tissues, dry-blotted by touching on filter paper, weighed and transferred to assay tubes containing a solution of 5-sulfosalicylic acid (SSA) at 5% and 0.25 mm EDTA; the final volume SSA solution was adjusted to × 5 tissue weight. Tissues were homogenized at 0–4ºC, centrifuged (12,000 g at 4ºC for 10 min) and the supernatant used to assay GSH/GSSG. The assay can be performed immediately or the supernatant stored –80ºC until assay. Glutahione redox potentials (EGSH), which approximate the overall redox potential of the cells (Schafer and Buettner, 2001), were calculated by the Nernst equation: EGSH = EºGSH – RT/zF × Ln [GSH]2/[GSSG] and a value for Eº’GSH of –240 mV.
Measurement of 3H-CA release
The experimental protocols and analytical procedures have been described in detail previously (Vicario et al. 2000a; Prieto Lloret et al. 2004). In brief, isolated CBs were incubated for 2 h with 3H-tyrosine of high specific activity (40–50 Ci mmol−1), which is the natural precursor of CA. Once the CA stores were labelled, organs were transferred to vials containing precursor-free Tyrode's solution (in mM): 116 NaCl, 5 KCl, 2 CaCl2, 1.1 MgCl2, 5.5 glucose, 24 NaCO3H and 10 Hepes. The solution was equilibrated with gas mixtures containing 5% CO2 and different percentages of O2 (maintaining pH 7.40) (see Results). In other experiments, we incubated using K+-rich solutions and the osmolarity was maintained by removing an equimolar amount of NaCl. Incubating solutions were collected and their content with respect to 3H-CA was measured by scintillation spectrometry.
Measurement of cAMP
The measurement of cAMP was made in accordance with previously described protocols (Cachero et al. 1996; Conde et al. 2008). In brief, after isolation, CBs were pre-incubated (15 min for 37ºC) in Tyrode's solution equilibrated with 95% O2–5% CO2. Thereafter, the solution was renewed with an incubating solution containing 500 μm isobutylmethylxanthine (Sigma), a phosphodiesterase inhibitor. Incubating solutions were equilibrated with either 95% O2–5% CO2 (basal) or 7% O2–5% CO2–88%N2 (hypoxia); incubations lasted 30 min. CBs were homogenized in ice-cold 6% trichloroacetic acid and centrifuged (12,000 g for 10 min at 4ºC). Supernatants were extracted with water-saturated diethyl ether (3×), the aqueous phase lyophilized and dried samples stored at –20ºC until cAMP was assayed. We used a commercial enzyme immunoassay kit in accordance with the manufacturer's instructions (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). cAMP levels are expressed as pmol mg–1 tissue.
Determination of EPO in plasma
Control male and female rats breathing room air were used to measure baseline plasma EPO levels. To measure the effect of hypoxia on EPO plasma levels, separate groups of control animals (male and female) were exposed to hypoxia for 5 or 10 h. Finally, a third group, also comprising male and female rats perinatally exposed to hyperoxia, was exposed to hypoxia for 10 h. In all the cases, rats anaesthetized with sodium pentobarbital (60 mg kg−1; i.p.) were punctured in the left ventricle through the thoracic wall and 1 ml of blood was withdrawn and added to standard haematological test tubes containing lithium heparin and centrifuged at 3500 g (10 min) at room temperature; 100 μl of plasma was used for the automated immunometric assay of EPO in accordance with the manufacturer's instructions (Immulite-EPO; Diagnostic Products Corporation, Los Angeles, CA, USA). Standard curves constructed with known amounts of EPO are linear from 0 to 200 mU ml−1. EPO levels in samples are read by interpolation.
Statistical analysis
Aside from some sample recording, data are presented as the mean ± SD and the 95% confidence interval (CI). Comparison of data obtained in each experimental group was made by a paired or unpaired two-tailed Student's t test, and by one-way ANOVA, two-way ANOVA and two-way ANOVA for repeated measures, as required. P < 0.05 was considered statistically significant.
Results
Hypoxic pulmonary vasoconstriction
Isolated in situ preparation and smooth muscle actin immunohistochemistry
In a first set of experiments, hypoxia (lung ventilation and blood equilibration with 2% O2) augmented PAP in control rats by 5.5 ± 2.7 mmHg (n = 8) (Fig.1A and C). In hyperoxic animals, the same hypoxia produced no changes (Fig.1B) or only minimal changes in PAP; mean ± SD ΔPAP observed in nine hyperoxic rats amounted to 1.04 ± 1.08 mmHg (P < 0.001 vs. control animals; unpaired Student's t test) (Fig.1C). In a second set of experiments, we tested the possibility that the reduced response to hypoxia observed in hyperoxic rats could be the result of an increased production of NO, a potent vasodilator of the pulmonary arterial tree. Indeed, Yeh et al. (2006) have reported that a short-lasting protocol of exposure to hyperoxia (>90% FIO2 for 60 h) in adult rats increased baseline NO production and blunted the HPV response during hypoxic challenges compared to normoxic rats. As shown in Fig.1D, the NO synthase inhibitor NMLA (5 × 10−5 m) augmented the hypoxic response in control rats from ΔPAP of 6.2 ± 2.4 mmHg (95% CI = 3.7–8.7) to 12.2 ± 5.5 mmHg (95% CI = 6.4–17.9; n = 6 in both cases; P < 0.01, Student's paired t test) (Fig.1F). In hyperoxic animals, ΔPAP induced by hypoxia was 1.0 ± 0.7 mmHg (95% CI = 0.23–1.75) in the absence of the NO synthase inhibitor and 1.5 ± 1.0 mmHg in its presence (95% CI = 0.44–2.52; n = 6 in both cases; P = 0.3490, Student's paired t test) (Fig. 1E and F). By contrast to expectations, the lack of effect of NMLA is compatible with a deficient or no expression of endothelial nitric oxide synthase (eNOS) in hyperoxic animals; this observation led us to determine the level of expression of eNOS and inducible nitric oxide synthase (iNOS) (western blot) in pulmonary artery homogenates of control, hyperoxic and lipopolysaccaride (LPS)-treated rats. The data (Fig. 2A and B) indicate that the amount of eNOS protein was not different in control, hyperoxic and LPS-treated rats; basal expression of iNOS in control and hyperoxic animals was almost absent; LPS administration markedly induced this enzyme (Fig.2C).
Figure 1.

Effects of hypoxia (2% O2, 8 min) on PAP recorded in an in situ isolated preparation
A and B, sample recordings obtained, respectively, in a control and in a perinatally hyperoxic rat. C, mean increases in PAP under the effects of hypoxia in both groups. Data are the mean ± SD of eight and nine individual values. ***P < 0.001. D–F, effects of the NOS inhibitor NMLA (5 × 10−5 m) on the response elicited by hypoxia in a control (D) and a perinatally hyperoxic rat (E). Mean ±SD of the responses obtained in six individual values in both groups (C). *P < 0.05.
Figure 2.
Level of expression of eNOS and iNOS in pulmonary arteries of control, hyperoxic and LPS-treated rats
A, sample western blot analysis obtained in the different experimental conditions. Note that the level of eNOS expression in every condition is comparable; note also that iNOS was only visible in the lane corresponding to a tissue sample obtained form a LPS-treated rat. B, mean densities of the eNOS bands in the three experimental conditions, showing that they are comparable. C, by contrast, iNOS band density was minimal, barely above background in control and hyperoxic tissues, and it was very intense in the tissue of LPS-treated animals.
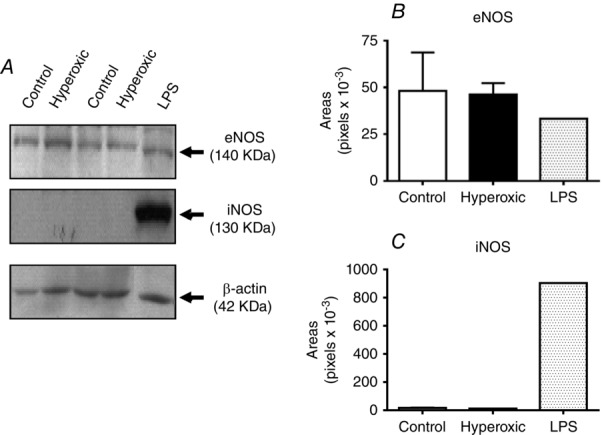
As shown in Fig.1, perinatal hyperoxia blunted HPV and produced an atrophy of the CB (Erickson et al. 1998; Prieto Lloret et al. 2004). Accordingly, in two different ways, we explored the possibility that the reduced response of PAP to hypoxia was the result of atrophy of the muscle layer of pulmonary vessels. In a first experiment, we tested the ability of the pulmonary vessels to contract in response to the depolarizing agent 4-aminopyridine (4AP). Figure 3A and C shows that, in control rats, 10 mm 4AP produced an increase in the PAP of 7.3 ± 4.6 mmHg comparable to that produced by hypoxia 5.6 ± 4.1 mmHg (n = 12). In hyperoxic rats, the hypoxic ΔPAP was minimal (1.2 ± 0.7 mmHg; n = 8; P < 0.05 vs. control; one-way ANOVA followed by Dunnet's multiple comparison test), although the response to 4AP was statistically identical to that obtained in control rats (7.4 ± 2.8 mmHg; n = 8) (Fig. 3B and C). In the second experiment, we measured the thickness of the muscular layer in lung resistance arteries. Figure 3D shows sections of small arteries taken from a control (D1) and a hyperoxic (D2) rats immunocytochemically stained for smooth muscle actin. As indicated, the thickness of the muscle layer was measured at different points and the mean of those measurements was taken to represent the thickness of that particular artery. The diameter of the artery was directly obtained with the appropriate software (MetaMorph v.6.3, Molecular Devices, Sunny Valley, CA, USA). Figure 3E shows mean thickness in small pulmonary arteries, which are the vessels responsible for most of the circulatory resistances, demonstrating that they are essentially identical in control and hyperoxic animals. Figure 3D shows that a section incubated in the absence of primary antibody provides a negative control, demonstrating the specificity of the immunostaining. The specificity of the primary antibody was not tested. Thus, both functional and morphological criteria indicate that the muscular of the pulmonary arteries has a normal capacity to generate tension, in turn indicating the specificity of the loss of their property to respond to hypoxia.
Figure 3.
Comparison of the effects of hypoxia (2% O2) and 4AP (10 mm) on PAP in control and perinatally hyperoxic rats
A, representative recording obtained in a control rat. B, representative recording obtained in a perinatally hyperoxic rat. C, mean ± SD of the increase in PAP elicited by hypoxia and 4AP in control (n = 12) and perinatally hyperoxic rats (n = 8). **P < 0.01. D, representative immunostaining of a section of the small arteries belonging to control and perinatally hyperoxic rats and a blank obtained by elimination of the primary antibody. D, procedure used to calculate the thickness of the muscular layer as the mean of five (or more) individual thickness measured on the images. E, mean thickness in eight control and eight hyperoxic small arteries of 200–300 μm [17.3 ± 1.9 (95% CI = 15.72–18.93) vs. 16.9 ± 1.2 (95% CI = 15.86–17.89)] and four control and nine hyperoxic arteries of 100–200 μm [13.4 ± 2.6 (95% CI = 9.29–17.45) vs. 13.1 ± 0.9 (95% CI = 12.40–13.82)] (As shown in the drawing, thickness is expressed in micrometers in all instances).
In vivo experiments
Using intact anaesthetized rats, we obtained results almost identical to those for the isolated in situ preparation (Fig.4). As shown in Fig.4A and B, in control animals, there was a clear increase in PAP on switching the respirator from air to 10% O2 reservoir, whereas, in hyperoxic animals, there was no noticeable change. In control animals, mean ± SD PAP immediately prior to starting breathing 10% O2 was 11.4 ± 3.2 mmHg (95% CI = 9.40–13.44) and this rose to 14.5 ± 4.6 mmHg (95% CI = 11.6–17.37) during the hypoxic episode (n = 12; P < 0.001); on returning to air breathing (recovery), PAP fell to 11.4 ± 3.1 mmHg (95% CI = 9.37–13.35) (Fig.4C). In fourteen hyperoxic animals, mean ± SD basal PAP was 12.3 ± 4.3 mmHg (95% CI = 9.63–15.05), rising to 13.07 ± 4.9 mmHg (95% CI = 9.93-16.27) during hypoxia and subsequently falling to 12.80 ± 4.8 mmHg (Fig.4D). The mean ± SD ΔPAP (ΔPAP = [(air+R)/2] – 10%) in control animals was 3.1 ± 2.0 mmHg (95% CI = 1.77–4.37) and, in hyperoxic animals, 0.5 ± 1.0 (95% CI –0.09 to 1.14) (P < 0.001) (Fig.4E). As we will see in a later figure, this difference in the response of the pulmonary circulation cannot be attributed to differences in systemic AP responses to hypoxia.
Figure 4.
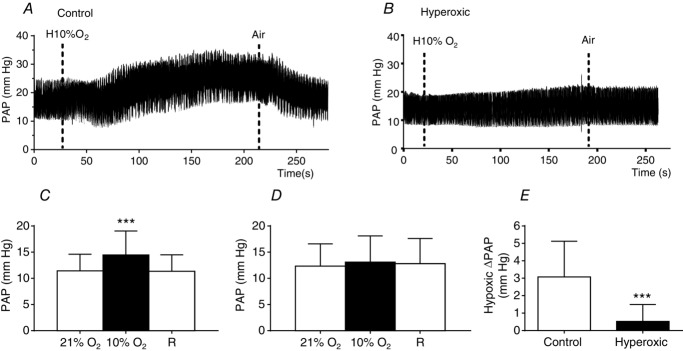
Hypoxic pulmonary vasoconstriction as recorded in intact anaesthetized control and hyperoxic rats
A and B, sample recordings, respectively, from control and perinatally hyperoxic rats. Hypoxia (10% O2) was administered as labelled for a period of ∼3 min. C, mean PAP before (20% O2), during (10% O2) and after the hypoxic episode (recovery, R) in control rats. D, as in C, but with data belonging to hyperoxic rats. E, mean ΔPAP during hypoxia obtained as [10% O2 PAP – (20% O2 PAP + R PAP)/2]. Data are the mean ± SD (n = 12–14). ***P < 0.001 (paired t test in C and unpaired t test in E).
Some basic electrophysiological properties of PASMC in control and hyperoxic animals
In an attempt to disclose possible mechanisms damaged by the perinatal hyperoxic exposure, we investigated some basic properties of smooth muscle cells isolated from resistance pulmonary arteries, including membrane potential (Em), membrane capacitance and K+ currents density, and their sensitivity to hypoxia. Figure 5 shows that Em was quite similar in cells from control and hyperoxic rats –37.3 ± 8.8 mV (95% CI = –59.28 to –15.38; n = 6) and –35.08 ± 4.3 mV (95% CI = –39.64 to –30.53; n = 6), respectively (P > 0.05). Membrane capacitance also was comparable 14.4 ± 1.7 pF (n = 6) in controls and 13.05 ± 3.0 pF (n = 8) in hyperoxic rats, suggesting that control and hyperoxic cells had similar sizes (Fig.5B). The IK+ density was not different (one-way ANOVA, followed by Tukey's multiple comparisons test) (Fig.5C). Figure 6 shows the changes induced by hypoxia on the K+ currents and the changes in Em during hypoxic exposure. Sample recordings and I–V curves in control and hyperoxic cells also demonstrate that both groups of cells exhibit a comparable response to the hypoxic exposure (no statistically significant differences were seen after applying a one-way ANOVA with Tukey's post hoc test). If the data are plotted (Fig.6C), it can be appreciated that, in control and hyperoxic cells, hypoxia (6 min) inhibited K+ currents by ∼25% at –30 mV. Similarly, the depolarization observed in both groups of cells was comparable 2.5 ± 0.7 mV vs. 3.8 ± 0.3 mV (n = 3), in control and hyperoxic cells, respectively (Fig.6D).
Figure 5.
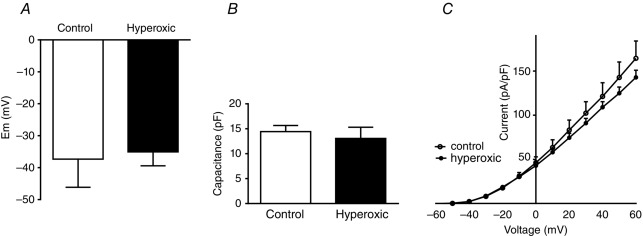
Basic electrophysiological properties of pulmonary artery smooth muscle cells
A,Membrane potential, B, membrane capacitance and C, K+ currents I–V curves in pulmonary artery smooth muscle cells obtained from resistance arteries (<500 μm of diameter) of control and perinatally hyperoxic rats. Data are the mean ± SD (n = 5–6 cells obtained from three animals).
Figure 6.
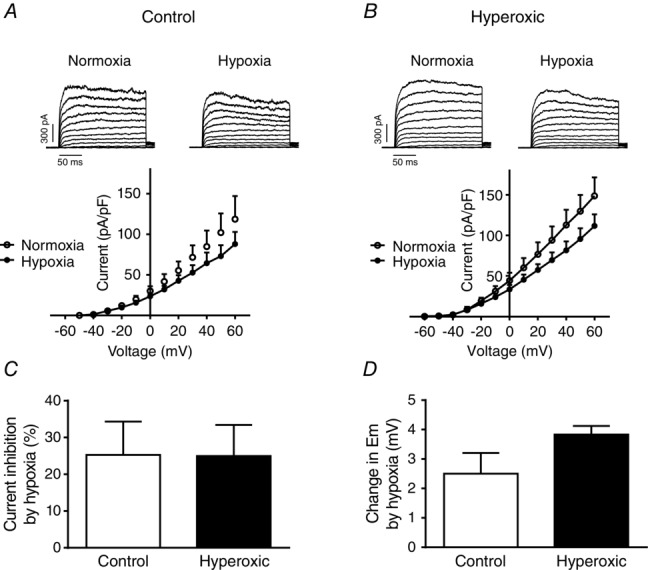
Changes induced by hypoxia in the density of K+ currents and in Em
A, sample recordings and I–V curves obtained in pulmonary artery smooth muscle cells isolated from control rats and superfused with a normoxic solution (air-equilibrated) and a hypoxic (N2-equilibrated) solution (n = 5 cells from three animals). B, as in A, but cells were obtained from a perinatally hyperoxic rats (n = 5 cells from two animals). C, percentage IK inhibition at a membrane potential of +30 mV (n = 5 cells from two animals). D, magnitude of the hypoxia-induced depolarization in cells from control and hyperoxic rats (n = 3 cells from two animals).
Effects of hyperoxic exposure on ventilation
In a previous study (Prieto-Lloret et al. 2004), we evaluated ventilation based on measurement of the breathing frequency. The present plethysmographic recordings demonstrated that perinatal hyperoxic exposure did not alter breathing frequency or TV in any of the atmospheres assessed. Computed minute ventilation indicated that the effects of different atmospheres on ventilation follow the same trend in control and perinatal hyperoxic animals (Table1).
Table 1.
Breathing parameters in control and hyperoxic animals
| Breathing atmosphere | 20% O2, air | 12% O2 | 10% O2 | 7% O2 | 5% CO2 in air | |
|---|---|---|---|---|---|---|
| Control animals | ||||||
| Breathing frequency (breaths min–1) | 69.0 ± 4.8 | 97.3 ± 9.7*** | 129.2 ± 8.6*** | 113.6 ± 17.6*** | 107.1 ± 9.7*** | |
| Tidal volume (ml kg–1) | 8.9 ± 1.5 | 8.6 ± 1.3 | 8.94 ± 1.1 | 11.5 ± 1.5** | 11.9 ± 1.4** | |
| Minute ventilation (ml min–1 kg–1) | 630.6 ± 122 | 900.1 ± 204.7* | 1135.2 ± 164.3*** | 1289.3 ±319.8*** | 1268.6 ± 159.3*** | |
| Hyperoxic animals | ||||||
| Breathing frequency (breaths min–1) | 70.7 ± 4.8 | 98.3 ± 11.3*** | 116.7 ± 10.3*** | 115.3 ± 11.5*** | 115.4 ± 29.3*** | |
| Tidal volume (ml kg–1) | 9.2 ± 2.1 | 8.4 ± 1.9 | 9.5 ± 2.3 | 12.1 ± 2.6** | 13.0 ± 1.2*** | |
| Minute ventilation (ml min–1 kg–1) | 672.8 ±166.3 | 818.8 ± 185.2 | 1072.9 ± 300.6*** | 1380.8 ± 308.1*** | 1474 ± 405.4*** | |
Data are the mean ± SD of seven to 12 individual values.
P < 0.05
P < 0.01
P < 0.001 vs. 20% O2. No differences were observed between control and hyperoxic animals. The P value for interaction term was 0.1993, indicating that the effects of different atmospheres on ventilation follow the same trend (two-way ANOVA for repeated measures. Bonferroni's post hoc test).
Effect of hypoxia on CB CA content
In control rats, hypoxia (10 h; 10% O2; barometric pressure 710 mmHg) augmented DA and, more moderately, NE levels in the CB, demonstrating the activation chemoreceptor cells and short-term regulation and induction of tyrosine hydroxylase (Fig. 7A and B). Thus, DA levels in normoxic conditions amounted to 256.2 ± 132.6 pmole/mg tissue (95% CI = 203.7–308.6) and, after the hypoxic episode, increased to 497.0 ± 172.2 pmole/mg tissue (95% CI = 439.6–554.4; P < 0.001; one-way ANOVA followed by Tukey's multiple comparisons test). By contrast, in hyperoxic rats, which have augmented normoxic levels of DA per unit of tissue weight (466.8 ± 229.2 pmole/mg tissue 95% CI = 344.7–588.9; Prieto-Lloret et al. 2004), the hypoxic episode markedly decreased DA levels to 248.7 ± 299.7 pmole/mg tissue (95% CI = 144.1–353.3; P < 0.001; one-way ANOVA followed by Tukey's multiple comparisons test). NE followed a comparable behaviour (Fig. 7A and B). These findings reveal the incapacity of the biosynthetic machinery with respect to coping with the increased utilization of CA in the CB of hyperoxic animals under hypoxia.
Figure 7.
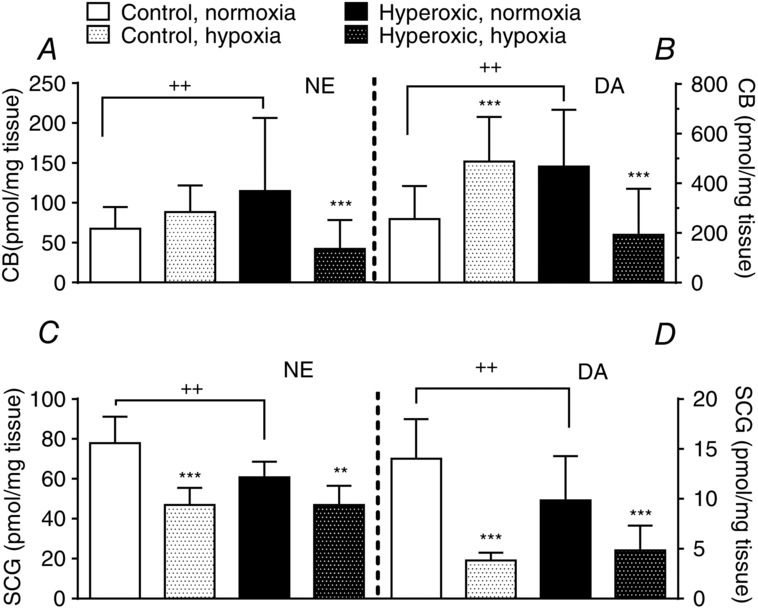
Effects of a hypoxic episode lasting ten hours on the levels of endogenous catecholamines, NE and DA, in the carotid body (A and B) and the superior cervical ganglion (C and D) of standard control rats and of animals perinatally exposed to hyperoxia. Data are expressed as the mean ± SD (n = 11 to 14). **P < 0.01; ***P < 0.001 vs. correspondent normoxic animals not exposed to the 10 h hypoxic episode. ++P < 0.01 control vs. hyperoxic animals in normoxic conditions as indicated.
However, the evolution of NE in chemoreceptor cells during hypoxia is difficult to trace because a significant amount of NE is present in sympathetic endings (Mir et al. 1982). To obtain additional insight, we measured CA in the SCG (Fig. 7C and D). The per unit weight steady-state levels of both NE and DA tend to be smaller in the hyperoxic animals than in animals reared in a standard atmosphere. The 10 h hypoxic episode reduced NE content in the SCG by 31% in control rats and by only 13% in hyperoxic animals; the hypoxic episode diminished DA levels by almost 70% in control rats and by 40% in hyperoxic rats. Thus, the regulation of the storing/utilization of NE and DA in the SCG during the hypoxia exhibits a fundamental difference compared to the CB in control animals but, in hyperoxic animals, there is a comparable trend.
Glutathione levels and redox status in lung, liver, and brain in hyperoxic animals: effects of supplementation with an antioxidant diet
Although, based on plasma measurements of carbonylated proteins, it has been reported that perinatal hyperoxia does not cause oxidative stress (Bavis et al. 2008), we aimed to re-examine this aspect by measuring the levels of GSH and GSSG, the main determinant of the redox status in several tissues. The extension of the measurement of glutathione to these three tissues relies on the observation that intermittent and sustained hypoxia cause tissue-specific oxidative damage, with some tissues showing altered redox status, whereas others maintain this (Singh et al. 2001; Jun et al. 2008; Quintero et al. 2013). Figure 8 shows that, at 7 days of age, control levels of total glutathione (GSH + GSSG) approached 1 μmol g–1 tissue in the lung and brain, being approximately tripled in liver. Glutathione levels did not increase with age in the lung or brain, whereas, in the liver, total glutathione almost doubled, reaching levels close to 6 μmol g–1 tissue in adult animals. Hyperoxic exposure caused a significant decrease in GSH levels in the three tissues at 7 days of age, whereas, in the lung and liver, GSSG levels increased but decreased in the brain. The net result was that the EGSH was very significantly decreased in lung from –192.9 ± 9.3 mV (95% CI = –198.8 to –187) to -170.7 ± 11.7 mV (95% CI = −176.9 to −164.4) and in the liver from −224.9 ± 2.9 mV (95% CI = −226.9 to −222.9) to −206.3 ± 9.8 mV (95% CI = −211.3 −201.3) (i.e. there was an increased oxidative status in these tissues) being normal in the brain −211.4 ± 3.6 mV (95% CI = −214.1 to −208.7) vs. −207.8 ± 7.5 mV (95% CI = −211.8 to −203.8). At 90 days, hyperoxic animals had normal GSH, GSSG and EGSH. Supplementation of the diet with antioxidants reversed the increased oxidative status in the lung and liver at 7 days of age, which became normal and remained normal or even improved at 90 days of age. Again, supplementation with antioxidants resulted in a somewhat paradoxical effect in the brain because it produced a diminution of the EGSH at 7 days, remaining low at 90 days.
Figure 8.

Levels of GSH and GSSG (A and B, respectively) and EGSH (C) in lung (top), liver (middle) and brain (bottom) of control, hyperoxic and antioxidant diet supplemented hyperoxic rats of 7 and ≥ 90 days of age. Data are the mean ± SD of 12–16 individual values. *P < 0.05; **P < 0.01; ***P < 0.001 vs. controls of the same age. Two-way ANOVA, followed by Tukey's multiple comparison test.
Effects of supplementation with an antioxidant diet on ventilation: CB responses and HPV
The results outlined above prompted us to investigate whether supplementation with antioxidants restored functional responses. Figure 9A–C shows the effect of supplementation on HPV. An antioxidant diet restored the sensitivity-response of PASMC to hypoxia so that the ΔPAP produced by the hypoxic test (3 min, 10% O2) amounted to 3.00 ± 0.93 mmHg (95% CI = 2.43–3.56), which is identical to that found in control animals 3.1 ± 2.0 mmHg (95% CI = 1.77–4.37). At the systemic level, the hypoxic test in hyperoxic supplemented rats caused a hypotensive response similar to that encountered in control animals (Fig.9D).
Figure 9.
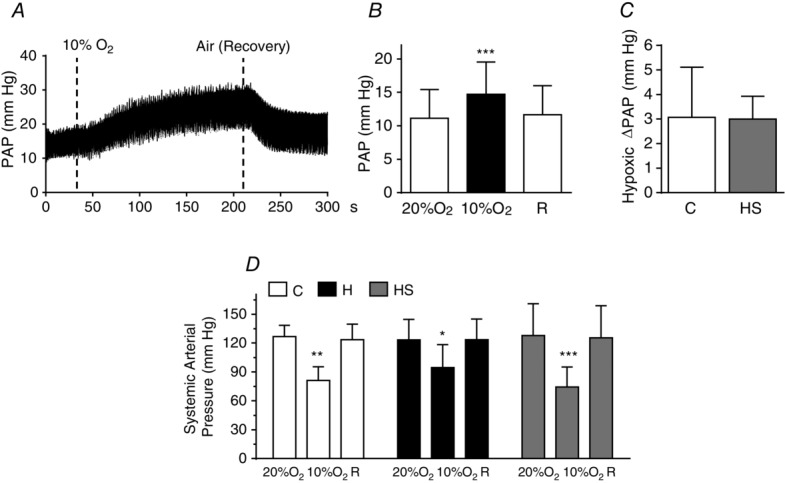
Hypoxic pulmonary vasoconstriction recorded in anaesthetized rats perinatally exposed to hyperoxia and fed with an antioxidant diet
A, sample recording in a hyperoxic supplemented rat. B, mean pulmonary arterial pressure in normoxia (air), in hypoxia (10% O2) and during recovery (air, R) in hyperoxic supplemented rats. C, ΔPAP induced by hypoxia in control and hyperoxic supplemented rats (data for control group are the same as those plotted in Fig. 6E). D, systemic arterial pressure in rats of the three groups (control, hyperoxic and hyperoxic supplemented animals) recorded when breathing air or 10% O2. Data are the mean ± SD of 12–13 individual values. *P < 0.05; **P < 0.01; ***P < 0.001 vs. normoxia.
Restoration of the HPV with the antioxidant diet prompted an evaluation of whether it also restored CB-related functions. Figure 10A shows minute ventilation in control, hyperoxic and hyperoxic diet-supplemented animals expressed as percentage of normoxic ventilation (20% O2), which, in all cases, was taken as 100% and amounted, respectively, to 620.4 ± 175.9 ml min−1 kg−1 (95% CI = 526.72–714.17; n = 16), 642.5 ± 156.6 mlmin−1 kg−1 (95% CI = 559.0–725.92; n = 16) and 584.5 ± 82.5 ml min−1 kg−1 (95% CI = 532.11–636.93; n = 12). There was a non-significant tendency for ventilation to decrease in a 12% O2 atmosphere in the hyperoxic supplemented animals. However, neither hyperoxia, nor hyperoxia plus diet supplementation altered normoxic, CB-mediated hypoxic ventilation or hypercapnic ventilation, which is mediated both by CB and the brain stem (two-way ANOVA for repeated measures followed by Bonferroni's post hoc test). Figure 10B shows that the CB response to hypoxia, measured as 3H-CA release, was markedly diminished in both hyperoxic and hyperoxic supplemented animals, whereas the response to high external K+ was not different from controls. Figure 10C shows that, in control CBs, a 10 min incubation period in a hypoxic solution augmented the levels of cAMP from 7.0 ± 5.6 (95% CI = 3.62–10.42) to 15.8 ± 12.1 pmole/mg tissue (95% CI = 6.48–25.17) (P < 0.01 two-way ANOVA with a Bonferroni's multiple comparison test), whereas, in both hyperoxic groups, it did not increase the levels of this second messenger.
Figure 10.
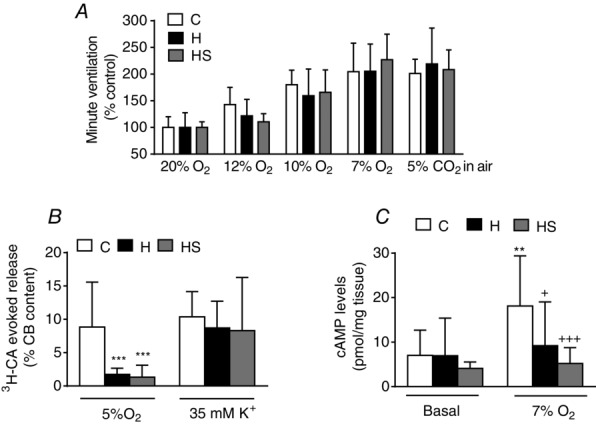
Ventilation, release of 3H-CA and cAMP in control, hyperoxic and hyperoxic diet supplemented hyperoxic rats
A, minute ventilation in different atmospheres (as drawn) in control, hyperoxic and hyperoxic supplemented rats. B, release of 3H-CA from the CBs of rats of same experimental groups elicited by hypoxia and high external K+. C, normoxic and hypoxic (10 min of incubation with 7% O2 equilibrated atmosphere) cyclic AMP levels in the CBs of rats of the same three groups. Data are the mean ± SD of 12–16 individual values. In every case, statistical comparisons have been made comparing data obtained in CBs from the two different experimental groups (hyperoxic and hyperoxic supplemented) with the control group. **P < 0.01; ***P < 0.001. C, +P < 0.05 and +++P < 0.001 vs. control hypoxic.
Erythropoietin production
Normoxic baseline values of EPO plasma levels in the control rats yield values above the threshold level of detection, and the mean ± SD EPO levels measured in seven rats amounted to 0.4 ± 0.9 mU ml−1 of plasma. A separate group of seven animals exposed to hypoxia (10% O2) for 5 h showed EPO levels of 3.24 ± 2.77 mU ml−1 and, in an additional group of 14 animals exposed to 10 h of hypoxia, EPO levels reached 23.11 ± 26.56 mU ml−1 (n = 14) (Fig.11A). This temporal pattern of EPO production in response to hypoxia reported in the present study is similar to that originally described by Eckardt et al. (1990). In these initial experiments, we noted the existence of a marked sexual dimorphism with respect to EPO production in response to this level of hypoxia. This dimorphism is responsible for the high variability of the data shown in Fig.11A and lead us to separate males and females in subsequent experiments. Both control and perinatally hyperoxic male and female rats had mean basal EPO levels below 1 mU ml−1 of plasma (0.84 ± 0.25 mU ml−1; 95% CI = 0.77–0.92; n = 44) (Fig.11B). A 10 h period of exposure to hypoxia increased EPO levels, albeit these were markedly different according to sex. Thus, in control and hyperoxic male rats, EPO levels reached, respectively, 41.8 ± 29.5 mU ml−1 (n = 30; 95% CI = 30.7–52.8) and 43.8 ± 31.4 mU ml−1 (n = 11: 95% CI = 22.7–64.9) (P < 0.001 vs. normoxia); in female control and hyperoxic rats, EPO levels reached, respectively, 6.9 ± 8.4 mU ml−1 (95% CI = 2.67–11.28; n = 17) and 9.9 ± 17.7 mU ml−1 (95% CI = 1.68–18.27; n = 20) (n.s. vs. normoxia; two-way ANOVA followed by Bonferroni's multiple comparison test). As shown in Fig.11B, the hypoxic EPO levels in males vs. females were statistically different both in control and in hyperoxic animals.
Figure 11.
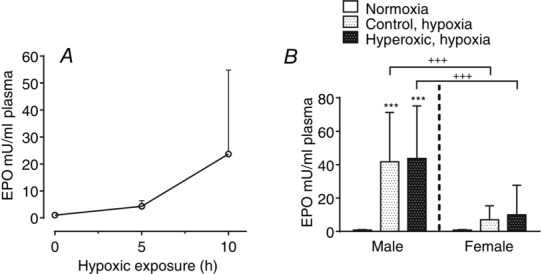
Effects of a hypoxic episode lasting five or ten hours on the levels of erythropoietin in plasma of standard control rats and of animals perinatally exposed to hyperoxia
A, time course of plasma EPO levels change in control animals. Mean ± SD of n = 7 (0 h), n = 7 (5 h) and n = 14 (10 h) rats; the data were obtained from an approximately equal number of males and females. B, normoxic and hypoxic (10 h in a 10% O2 atmosphere) plasma levels of EPO in control and perinatally hyperoxic male and female rats. Data are the mean ± SD of 30 and 11 male control and perinatally hyperoxic rats, respectively, exposed to a 10 h hypoxic episode; in female rats, n = 17 (control in hypoxia) and n = 20 (perinatally hyperoxic rats in hypoxia). ***P < 0.001 vs. normoxia; +++P < 0.001 males vs. females.
Discussion
The most novel and important finding of present study is that perinatal hyperoxia causes a loss of HPV in adult animals, which is associated with an early postnatal oxidative status in the lung, is reversed by supplementation with an antioxidant diet and, in dissociated cells, is not linked to alterations in the O2-sensitive K+ currents. In the discussion that follows, we first deal with the findings and their mechanisms, and follow-up by discussing their potential clinical significance. Additionally, we confirm that perinatal hyperoxia damages the CB functionally, whereas hypoxic and hypercapnic ventilatory responses are maintained. We also find that exposure to perinatal hyperoxia does not alter the increase in EPO plasma levels produced by hypoxia, suggesting a normal responsive to hypoxia by EPO-producing cells. The data also show a marked sexual dimorphism, with males attaining hypoxic plasma EPO levels almost four times larger than that of females.
At the outset, we want to justify the experimental model used. In the present study, as in previous studies conducted with CBs (Ling et al. 1997; Erickson et al. 1998; Fuller et al. 2002; Bisgard et al. 2003; Prieto Lloret et al. 2004; Wenninger et al. 2006; see Bavis et al. 2013), the model used recalls perinatal oxygen therapy in humans. However, we emphasize that the model does not exactly mimic oxygen therapy in humans: in infants, the aim of oxygen therapy is to attain normal O2 levels in blood, whereas, in our model, a higher than normal  should be expected and therefore PASMC, CBCC and kidney EPO-producing cells are exposed to a hyperoxic milieu in our animals. Nonetheless, we would suggest that, in infants subjected to oxygen therapy, a certain level of hyperoxia would occur in pulmonary vessels along the O2 diffusion pathway from the alveoli to erythrocytes. It should be noted that infants requiring oxygen therapy comprise low weight preterm babies most frequently born from eclamptic or pre-eclamptic mothers kept under oxygen therapy (commonly 55–60% O2). It has been shown that oxygen therapy in mothers lowers the perinatal mortality rate (Say et al. 2003) because extra days of intrauterine life allow the weight gain of the fetus. In this context, we also note that our model is not a model of bronchopulmonary dysplasia, a condition that might represent the outcome of up to 25% preterm-low weight O2-treated surviving infants (Saugstad et al. 2014) and is characterized separately from a great number of pathologic lung lesions, including anomalous lung vasculature development and pulmonary hypertension (Baker and Alvira, 2014); instead, the model used in the present study would represent the remaining 75% preterm infants who reach adulthood without apparent pathologies. In sum, our model deals with long-term silent and not previously recognized complications of perinatal oxygen therapy. The normality of blood gases (Ling et al. 1996), blood cell counts (Prieto-Lloret et al. 2004) and basal pulmonary arterial pressure observed in animals in the present study excludes bronchopulmonary dysplasia. The present experimental model was not designed to investigate the subacute effects of perinatal hyperoxia immediately after exposure to an O2-rich atmosphere (Bland et al. 2000).
should be expected and therefore PASMC, CBCC and kidney EPO-producing cells are exposed to a hyperoxic milieu in our animals. Nonetheless, we would suggest that, in infants subjected to oxygen therapy, a certain level of hyperoxia would occur in pulmonary vessels along the O2 diffusion pathway from the alveoli to erythrocytes. It should be noted that infants requiring oxygen therapy comprise low weight preterm babies most frequently born from eclamptic or pre-eclamptic mothers kept under oxygen therapy (commonly 55–60% O2). It has been shown that oxygen therapy in mothers lowers the perinatal mortality rate (Say et al. 2003) because extra days of intrauterine life allow the weight gain of the fetus. In this context, we also note that our model is not a model of bronchopulmonary dysplasia, a condition that might represent the outcome of up to 25% preterm-low weight O2-treated surviving infants (Saugstad et al. 2014) and is characterized separately from a great number of pathologic lung lesions, including anomalous lung vasculature development and pulmonary hypertension (Baker and Alvira, 2014); instead, the model used in the present study would represent the remaining 75% preterm infants who reach adulthood without apparent pathologies. In sum, our model deals with long-term silent and not previously recognized complications of perinatal oxygen therapy. The normality of blood gases (Ling et al. 1996), blood cell counts (Prieto-Lloret et al. 2004) and basal pulmonary arterial pressure observed in animals in the present study excludes bronchopulmonary dysplasia. The present experimental model was not designed to investigate the subacute effects of perinatal hyperoxia immediately after exposure to an O2-rich atmosphere (Bland et al. 2000).
Our findings demonstrate that hyperoxic exposure abrogates the expression of some element of the hypoxic transduction cascade indispensable for developing HPV. As in the case of CBCC (Prieto-Lloret et al. 2004; Kim et al. 2013), the data shown in Figs 1 to 4 indicate that the unexpressed element in PASMC of hyperoxic animals is upstream of depolarization and Ca2+ entry via voltage-operated Ca2+ channels. Thus, 4-AP, a depolarizing agent that would almost completely inhibit Kv currents, leading to cell depolarization and activation of voltage-operated Ca2+ channels, Ca2+ entry and activation of the contraction (Olschewski et al. 2002; Cogolludo et al. 2005), generates an increase in PAP that is almost identical in control and hyperoxic rats (Figs 3A–C). These observations, along with the immunohistochemical demonstration of the normal thickness of the muscular layer in resistance arterioles (Fig. 3D and E), would indicate a normal development of lung vasculature. If voltage-operated Ca2+ channels and contractile machinery function with normality, and even further, if IK+ and the O2-sensitive IK+ component are normal (Figs 5 and 6), what might represent the element damaged by hyperoxia? The O2-sensing property appears to depend on an O2-sensor specifically expressed in PASMC and in CBCC (Plathosyn et al. 2006; Gonzalez et al. 2009; Sylvester et al. 2012). On the other hand, and in contrast to CBCC (Vicario et al. 2000b) intracellular Ca2+ stores are necessary for the expression of the oxygen dependent contraction in PASMC because emptying of the Ca2+ stores almost abolishes the hypoxic response (Dipp et al. 2001; Dipp and Evans, 2001). However, Ca2+ entry in PASMC is necessary to generate a sustained response because, in Ca2+-free solutions, hypoxia produces a short-lasting Ca2+ transient and a near ablation of the HPV; suppression of the expression of the store-operated Ca2+ entry complex also almost abolishes the Ca2+ increase and PASMC contraction elicited by hypoxia (Dipp et al. 2001; Wang et al. 2005; Lu et al. 2009; Ng et al. 2010). These findings suggest that O2-sensing machinery generates a signal that leads to empty Ca2+ stores, which in turn leads to activation of the store-operated Ca2+ entry complex, entry of Ca2+ and genesis of the sustained response in PASMC (Sylvester et al. 2012; Swenson, 2013). From this perspective, postnatal oxidative stress produced by hyperoxia (Figs 8 and 9) either damages or silences the expression of the mechanism linking O2-sensor with Ca2+ stores releasing machinery or, alternatively, it silences the store-operated Ca2+ entry complex (stromal interaction molecule and Orai-1 channels; STIM). Because STIM and Orai are overexpressed in pulmonary hypertension (Song et al. 2011; Ogawa et al. 2012), and oxidative stress can regulate the abundance of STIM and Orai (Patterson et al. 2012, Nunes and Demaurex, 2014), we would propose that hyperoxic stress causes a suppression of their expression. We omit a possible role for the vascular endothelium because our measurement of eNOS expression is normal and the hypoxia-induced contraction of PASMC is an intrinsic property of these cells (Murray et al. 1990).
The findings shown in Fig.7A confirm that, per unit weight, hyperoxic CBs have higher NE and DA content compared to controls (Prieto Lloret et al. 2004), in addition to showing that the regulation of CA metabolism in chemoreceptor cells is profoundly altered in hyperoxic animals. Because hypoxia causes an increase in the release of CA, particularly of DA, the increased levels of DA after 10 h of hypoxia in control animals indicate that the activation of CBCC causes an increase in the activity of tyrosine hydroxylase (the limiting enzyme in CA synthesis), which is capable of coping with the augmented release and even causing an overfilling of CA stores. The mechanisms leading to activation of tyrosine hydroxylase would include an initial reduction of the feedback inhibition by product, as well as phosphorylation and induction of the enzyme (Gonzalez et al. 1979; Fidone et al. 1982; Czyzyk-Krzeska et al. 1992). Figure 7A and B indicates that the overall activation of DA synthesis in response to hypoxia in the CB of control animals is stronger than that of NE synthesis, probably as a result of a very significant part of NE (>50%; Mir et al. 1982) being stored in sympathetic fibre endings (DA is stored exclusively in CBCC) whose biosynthetic machinery is less strongly activated by hypoxia (Gonzalez et al. 1979); consistent with this interpretation, the levels of NE and DA in the SCG of control animals decreased as a result of the hypoxic stimulation (Fig.7C and D). In hyperoxic animals, the 10 h hypoxic episode caused a marked decrease of both NE and DA, indicating that, even if the release of both CA induced by hypoxia is smaller than in control (Prieto-Lloret et al. 2004) (Fig.10B), the CA biosynthetic machinery is unable to maintain the CA stores. As shown in Fig.10C, perinatal hyperoxic exposure causes a loss of the capacity of hypoxia to increase cAMP levels in the CB (Perez-Garcia et al. 1990) and therefore the covalent regulation by phosphorylation and the cAMP response element-binding-mediated induction (Lim et al. 2000) are probably lost. Additionally, cAMP augments the release of CA from the CB in response to hypoxia and does not affect the release induced by high external K+ (Perez-Garcia et al. 1991) and the findings of the present study also show that hyperoxia lessens the release induced by hypoxia without affecting the release induced by high K+. Both observations combined would suggest that the incapacity of CBCC to release CA in response to hypoxia is a result, at least in part, of an incapacity to generate cAMP.
Despite these diminished functions of the CB in hyperoxic animals (i.e. diminished capacity to synthesize and release CA during hypoxia, as well as diminished sensory chemoreceptor activity in response to hypoxia), the redundancy of the chemoreceptor activity reaching the brain stem (Prieto-Lloret et al. 2004) is capable of maintaining basal and hypoxic ventilation (Table1). Additionally, the normal capacity of animals with respect to responding to hypercapnia (a response mediated by ∼60–70% by central chemoreceptors; Gonzalez et al. 1994) would indicate that central mechanisms controlling respiration are not affected by the hyperoxic exposure. It cannot be excluded that the smaller increase in minute ventilation with the very moderate hypoxic stimulation (12% O2) observed in hyperoxic animals represents a lower sensitivity to this very mild level of hypoxia (Bavis et al. 2011).
Hyperoxic exposure does not alter the hypoxia triggered EPO production that is known to be mediated by HIF-1α. However, our experiments unveil a very marked sexual dimorphism in the response to hypoxia. It has also been shown that male rats contain 3-fold greater levels of EPO in the kidney than females after 4 h of exposure to hypoxia (Fried et al. 1982). Because oestradiol greatly attenuates the activity/expression of HIF-1α and the EPO production induced by hypoxia in HEP3B cells (Mukundan et al. 2004), this would implicate sexual hormones in the diminished EPO plasma levels found in female rats. We have not observed any sexual dimorphism in the ventilatory response to hypoxia and hypercapnia. It should be noted that studies conducted in humans and laboratory animals in this regard have generated conflicting results (Behan et al. 2003). However, Gassman et al. (2009) have reported that the effects of EPO on ventilation exhibit a clear sexual dimorphism. At this point, we want to explicitly state that, if our interpretation has been correct, perinatal oxidative damage would be specific for pulmonary vessels because the antioxidant diet did not reverse the alterations induced by hyperoxia in the CB and EPO plasma levels were not affected by hyperoxia.
In sum, our observations would suggest that antioxidants may be beneficial to human preterm infants. Certainly, the idiosyncratic responses of brain tissue to hyperoxia and the antioxidant supplementation imposes a word of caution because it appears that a long-lasting oxidative status might be present in brain tissue. However, we should state that, behaviourally, nothing abnormal (i.e. sleep rhythm, general motor activity, exploratory behaviour) was detected in hyperoxic rats and the responses measured that involve central nervous system integration (ventilation, systemic blood pressure) were normal (Fig. 10 and Table1). Additionally, we would recommend that, when facing lung diseases or thoracic surgery, a patient's medical history should be meticulously examined with respect to perinatal O2 administration.
Acknowledgments
This work was supported by Grants BFU2012-37459 from the Ministry of Economy and Competitiveness (Spain) and Grant CIBER CB06/06/0050 from the Institute of Health Carlos III (Spain) to C. G. Also supported by Grants SAF2011-28150 to F.P-V and SAF2010-22066-C02-02 to AC from the Ministry of Economy and Competitiveness (Spain). Experiments were performed in Valladolid except for the electrophysiological recordings that were performed in Madrid.
Glossary
- 4-AP
4-aminopyridine
- AP
arterial pressure
- CA
catecholamine
- CB
carotid body
- CBCC
carotid body chemoreceptor cells
- CI
confidence interval
- DA
dopamine
- DP
diastolic pressure
- eNOS
endothelial nitric oxide synthase
- EPO
erythropoietin
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- HPV
hypoxic pulmonary vasoconstriction
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MAP
mean arterial pressure
- NE
norepinephrine
- NMLA
N-methyl-l-arginine
- NOS
nitric oxide synthase
- PASMC
pulmonary artery smooth muscle cells
- PAP
pulmonary arterial pressure
- PB
phosphate buffer
- PBS
phosphate-buffered saline
- SCG
superior cervical ganglion
- STIM
stromal interaction molecule
- TV
tidal volume
Additional information
Competing interests
The authors declare that they have no competing interests.
References
- Baker CD. Alvira CM. Disrupted lung development and bronchopulmonary dysplasia: opportunities for lung repair and regeneration. Curr Opin Pediatr. 2014;26:306–314. doi: 10.1097/MOP.0000000000000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Dmitrieff EF, Young KM. Piro SE. Hypoxic ventilatory response of adult rats and mice after developmental hyperoxia. Respir Physiol Neurobiol. 2011;177:342–346. doi: 10.1016/j.resp.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Fallon SC. Dmitrieff EF. Chronic hyperoxia and the development of the carotid body. Respir Physiol Neurobiol. 2013;185:94–104. doi: 10.1016/j.resp.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Wenninger JM, Miller BM, Dmitrieff EF, Olson EB, Jr, Mitchell GS. Bisgard GE. Respiratory plasticity after perinatal hyperoxia is not prevented by antioxidant supplementation. Respir Physiol Neurobiol. 2008;160:301–312. doi: 10.1016/j.resp.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan M1, Zabka AG, Thomas CF. Mitchell GS. Sex steroid hormones and the neural control of breathing. Respir Physiol Neurobiol. 2003;136:249–263. doi: 10.1016/s1569-9048(03)00086-7. [DOI] [PubMed] [Google Scholar]
- Bisgard GE, Olson EB, Jr, Wang ZY, Bavis RW, Fuller DD. Mitchell GS. Adult carotid chemoafferent responses to hypoxia after 1, 2, and 4 wk of postnatal hyperoxia. J Appl Physiol. 2003;95:946–52. doi: 10.1152/japplphysiol.00985.2002. [DOI] [PubMed] [Google Scholar]
- Bland RD, Albertine KH, Carlton DP, Kullama L, Davis P, Cho SC, Kim BI, Dahl M. Tabatabaei N. Chronic lung injury in preterm lambs: abnormalities of the pulmonary circulation and lung fluid balance. Pediatr Res. 2000;48:64–74. doi: 10.1203/00006450-200007000-00013. [DOI] [PubMed] [Google Scholar]
- Cachero TG, Rigual R, Rocher A. Gonzalez C. Cholera and pertussis toxins reveal multiple regulation of cAMP levels in the rabbit carotid body. Eur J Neurosci. 1996;8:2320–2327. doi: 10.1111/j.1460-9568.1996.tb01195.x. [DOI] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Bosca L, Tamargo J. Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93:656–63. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Lodi F, Tamargo J. Perez-Vizcaino F. Postnatal maturational shift from PKCzeta and voltage-gated K+ channels to RhoA/Rho kinase in pulmonary vasoconstriction. Cardiovasc Res. 2005;66:84–93. doi: 10.1016/j.cardiores.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Conde SV, Gonzalez C, Batuca JR, Monteiro EC. Obeso A. An antagonistic interaction between A2B adenosine and D2 dopamine receptors modulates the function of rat carotid body chemoreceptor cells. J Neurochem. 2008;107:1369–1381. doi: 10.1111/j.1471-4159.2008.05704.x. [DOI] [PubMed] [Google Scholar]
- Czyzyk-Krzeska MF, Bayliss DA, Lawson EE. Millhorn DE. Regulation of tyrosine hydroxylase gene expression in the rat carotid body by hypoxia. J Neurochem. 1992;58:1538–1546. doi: 10.1111/j.1471-4159.1992.tb11376.x. [DOI] [PubMed] [Google Scholar]
- Dauger S, Ferkdadji L, Saumon G, Vardon G, Peuchmaur M, Gaultier C. Gallego J. Neonatal exposure to 65% oxygen durably impairs lung architecture and breathing pattern in adult mice. Chest. 2003;123:530–538. doi: 10.1378/chest.123.2.530. [DOI] [PubMed] [Google Scholar]
- Dipp M. Evans AM. Cyclic ADP-ribose is the primary trigger for hypoxic pulmonary vasoconstriction in the rat lung in situ. Circ Res. 2001;89:77–83. doi: 10.1161/hh1301.093616. [DOI] [PubMed] [Google Scholar]
- Dipp M, Nye PC. Evans AM. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2001;281:L318–L325. doi: 10.1152/ajplung.2001.281.2.L318. [DOI] [PubMed] [Google Scholar]
- Eckardt KU, Dittmer J, Neumann R, Bauer C. Kurtz A. Decline of erythropoietin formation at continuous hypoxia is not due to feedback inhibition. Am J Physiol Renal Physiol. 1990;258:F1432–F1437. doi: 10.1152/ajprenal.1990.258.5.F1432. [DOI] [PubMed] [Google Scholar]
- Eisenkraft JB. Effects of anaesthetics on the pulmonary circulation. Br J Anaesth. 1990;65:63–78. doi: 10.1093/bja/65.1.63. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Mayer C, Jawa A, Ling L, Olson EB, Jr, Vidruk EH, Mitchell GS. Katz DM. Chemoafferent degeneration and carotid body hypoplasia following chronic hyperoxia in newborn rats. J Physiol. 1998;509:519–26. doi: 10.1111/j.1469-7793.1998.519bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidone S, Gonzalez C. Yoshizaki K. Effects of hypoxia on catecholamine synthesis in rabbit carotid body in vitro. J Physiol. 1982;333:81–91. doi: 10.1113/jphysiol.1982.sp014440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazziano G, Moreno L, Moral-Sanz J, Menendez C, Escolano L, Gonzalez C, Villamor E, Alvarez-Sala JL, Cogolludo AL. Perez-Vizcaino F. Neutral sphingomyelinase, NADPH oxidase and reactive oxygen species. Role in acute hypoxic pulmonary vasoconstriction. J Cell Physiol. 2011;226:2633–2640. doi: 10.1002/jcp.22611. [DOI] [PubMed] [Google Scholar]
- Fried W, Barone-Varelas J. Barone T. The influence of age and sex on erythropoietin titers in the plasma and tissue homogenates of hypoxic rats. Exp Hematol. 1982;10:472–477. [PubMed] [Google Scholar]
- Fuller DD, Bavis RW, Vidruk EH, Wang ZY, Olson EB, Jr, Bisgard GE. Mitchell GS. Life-long impairment of hypoxic phrenic responses in rats following 1 month of developmental hyperoxia. J Physiol. 2002;538:947–955. doi: 10.1113/jphysiol.2001.012908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M, Tissot van Patot M. Soliz J. The neuronal control of hypoxic ventilation: erythropoietin and sexual dimorphism. Ann NY Acad Sci. 2009;1177:151–161. doi: 10.1111/j.1749-6632.2009.05028.x. [DOI] [PubMed] [Google Scholar]
- Glasser SA, Domino KB, Lindgren L, Parcella P, Marshall C. Marshall BE. Pulmonary blood pressure and flow during atelectasis in the dog. Anesthesiology. 1983;58:225–231. doi: 10.1097/00000542-198303000-00004. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Agapito MT, Rocher A, Gomez-Niño A, Rigual R, Castañeda J, Conde SV. Obeso A. A revisit to O2 sensing and transduction in the carotid body chemoreceptors in the context of reactive oxygen species biology. Respir Physiol Neurobiol. 2010;174:317–330. doi: 10.1016/j.resp.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A. Rigual R. Oxygen and acid chemoreception in the carotid body chemoreceptors. Trends Neurosci. 1992;15:146–153. doi: 10.1016/0166-2236(92)90357-e. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A. Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Kwok Y, Gibb J. Fidone S. Effects of hypoxia on tyrosine hydroxylase activity in rat carotid body. J Neurochem. 1979;33:713–719. doi: 10.1111/j.1471-4159.1979.tb05216.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sanz-Alfayate G, Agapito MT, Gomez-Niño A, Rocher A. Obeso A. Significance of ROS in oxygen sensing in cell systems with sensitivity to physiological hypoxia. Respir Physiol Neurobiol. 2002;132:17–41. doi: 10.1016/s1569-9048(02)00047-2. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sanz-Alfayate G, Obeso A. Agapito MT. Role of glutathione redox state in oxygen sensing by carotid body chemoreceptor cells. Methods Enzymol. 2004;381:40–71. doi: 10.1016/S0076-6879(04)81003-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Vaquero LM, Lopez-Lopez JR. Perez-Garcia MT. Oxygen-sensitive potassium channels in chemoreceptor cell physiology: making a virtue of necessity. Ann NY Acad Sci. 2009;1177:82–88. doi: 10.1111/j.1749-6632.2009.05037.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Gutteridge JMC. Free Radicals in Biology and Medicine, 4th edn. Oxford: Oxford University Press; 2007. [Google Scholar]
- Hartnett ME. Penn JS. Mechanisms and management of retinopathy of prematurity. N Engl J Med. 2012;367:2515–2526. doi: 10.1056/NEJMra1208129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge A. Conditions governing the pressor response to ventilation hypoxia in isolated perfused rat lungs. Acta Physiol Scand. 1968;72:33–44. doi: 10.1111/j.1748-1716.1968.tb03823.x. [DOI] [PubMed] [Google Scholar]
- Hayes P. Knaus UG. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. Antioxid Redox Signal. 2013;18:1937–1945. doi: 10.1089/ars.2012.4895. [DOI] [PubMed] [Google Scholar]
- Jelkmann W. Erythropoietin: structure, control of production, and function. Physiol Rev. 1992;72:449–489. doi: 10.1152/physrev.1992.72.2.449. [DOI] [PubMed] [Google Scholar]
- Jun J, Savransky V, Nanayakkara A, Bevans S, Li J, Smith PL. Polotsky VY. Intermittent hypoxia has organ-specific effects on oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1274–R1281. doi: 10.1152/ajpregu.90346.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai W. Schwarzkopf K. Hypoxemia during one-lung ventilation: prediction, prevention, and treatment. Anesthesiology. 2009;110:1402–1411. doi: 10.1097/ALN.0b013e31819fb15d. [DOI] [PubMed] [Google Scholar]
- Kim I, Yang D, Carroll JL. Donnelly DF. Perinatal hyperoxia exposure impairs hypoxia-induced depolarization in rat carotid body glomus cells. Respir Physiol Neurobiol. 2013;188:9–14. doi: 10.1016/j.resp.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. Systemic effects resulting from carotid body stimulation. Invited article. Adv Exp Med Biol. 2009;648:223–233. doi: 10.1007/978-90-481-2259-2_26. [DOI] [PubMed] [Google Scholar]
- Lim J, Yang C, Hong SJ. Kim KS. Regulation of tyrosine hydroxylase gene transcription by the cAMP-signaling pathway: involvement of multiple transcription factors. Mol Cell Biochem. 2000;212:51–60. [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH. Mitchell GS. Attenuation of the hypoxic ventilatory response in adult rats following one month of perinatal hyperoxia. J Physiol. 1996;495:561–571. doi: 10.1113/jphysiol.1996.sp021616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH. Mitchell GS. Developmental plasticity of the hypoxic ventilatory response. Respir Physiol. 1997;110:261–268. doi: 10.1016/s0034-5687(97)00091-1. [DOI] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH. Mitchell GS. Slow recovery of impaired phrenic responses to hypoxia following perinatal hyperoxia in rats. J Physiol. 1998;511:599–603. doi: 10.1111/j.1469-7793.1998.599bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SF, Crawley DE, Barnes PJ. Evans TW. Endothelium-derived relaxing factor inhibits hypoxic pulmonary vasoconstriction in rats. Am Rev Respir Dis. 1991;143:32–37. doi: 10.1164/ajrccm/143.1.32. [DOI] [PubMed] [Google Scholar]
- Lu W, Wang J, Peng G, Shimoda LA. Sylvester JT. Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2009;297:L17–L25. doi: 10.1152/ajplung.00063.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BE, Marshall C, Frasch F. Hanson CW. a Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. 1. Physiologic concepts. Intensive Care Med. 1994;20:291–297. doi: 10.1007/BF01708968. [DOI] [PubMed] [Google Scholar]
- Marshall BE, Hanson CW, Frasch F. Marshall C. b Role of hypoxic pulmonary vasoconstriction in pulmonary gas exchange and blood flow distribution. 2. Pathophysiology. Intensive Care Med. 1994;20:379–389. doi: 10.1007/BF01720916. [DOI] [PubMed] [Google Scholar]
- Mir AK, Al-Neamy K, Pallot DJ. Nahorski SR. Catecholamines in the carotid body of several mammalian species: effects of surgical and chemical sympathectomy. Brain Res. 1982;252:335–342. doi: 10.1016/0006-8993(82)90401-2. [DOI] [PubMed] [Google Scholar]
- Mukundan H1, Kanagy NL. Resta TC. 17-beta estradiol attenuates hypoxic induction of HIF-1alpha and erythropoietin in Hep3B cells. J Cardiovasc Pharmacol. 2004;44:93–100. doi: 10.1097/00005344-200407000-00013. [DOI] [PubMed] [Google Scholar]
- Murray TR, Chen L, Marshall BE. Macarak EJ. Hypoxic contraction of cultured pulmonary vascular smooth muscle cells. Am J Respir Cell Mol Biol. 1990;3:457–465. doi: 10.1165/ajrcmb/3.5.457. [DOI] [PubMed] [Google Scholar]
- Nagendran J, Stewart K, Hoskinson M. Archer SL. An anesthesiologist's guide to hypoxic pulmonary vasoconstriction: implications for managing single-lung anesthesia and atelectasis. Curr Opin Anaesthesiol. 2006;19:34–43. doi: 10.1097/01.aco.0000192777.09527.9e. [DOI] [PubMed] [Google Scholar]
- Ng LC, Ramduny D, Airey JA, Singer CA, Keller PS, Shen XM, Tian H, Valencik M. Hume JR. Orai1 interacts with STIM1 and mediates capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2010;299:C1079–C1090. doi: 10.1152/ajpcell.00548.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes P. Demaurex N. Redox regulation of store-operated Ca2+ entry. Antioxid Redox Signal. 2014;21:915–932. doi: 10.1089/ars.2013.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa A, Firth AL, Smith KA, Maliakal MV. Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2012;302:C405–C411. doi: 10.1152/ajpcell.00337.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olschewski A, Hong Z, Nelson DP. Weir EK. Graded response of K+ current, membrane potential, and [Ca2+]i to hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1143–L1150. doi: 10.1152/ajplung.00104.2002. [DOI] [PubMed] [Google Scholar]
- Patterson AJ, Xiao D, Xiong F, Dixon B. Zhang L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCε gene in foetal rat hearts. Cardiovasc Res. 2012;93:302–310. doi: 10.1093/cvr/cvr322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia MT, Almaraz L. Gonzalez C. Effects of different types of stimulation on cyclic AMP content in the rabbit carotid body: functional significance. J Neurochem. 1990;55:1287–1293. doi: 10.1111/j.1471-4159.1990.tb03137.x. [DOI] [PubMed] [Google Scholar]
- Perez-Garcia MT, Almaraz L. Gonzalez C. Cyclic AMP modulates differentially the release of dopamine induced by hypoxia and other stimuli and increases dopamine synthesis in the rabbit carotid body. J Neurochem. 1991;57:1992–2000. doi: 10.1111/j.1471-4159.1991.tb06414.x. [DOI] [PubMed] [Google Scholar]
- Platoshyn O, Brevnova EE, Burg ED, Yu Y, Remillard CV. Yuan JX. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol. 2006;290:C907–C916. doi: 10.1152/ajpcell.00028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto-Lloret J, Caceres AI, Obeso A, Rocher A, Rigual R, Agapito MT, Bustamante R, Castañeda J, Perez-Garcia MT, Lopez-Lopez JR. Gonzalez C. Ventilatory responses and carotid body function in adult rats perinatally exposed to hyperoxia. J Physiol. 2004;554:126–144. doi: 10.1113/jphysiol.2003.049445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero M, Gonzalez-Martin MC, Vega-Agapito V, Gonzalez C, Obeso A, Farre R, Agapito T. Yubero S. The effects of intermittent hypoxia on redox status, NF-κB activation, and plasma lipid levels are dependent on the lowest oxygen saturation. Free Radic Biol Med. 2013;65:1143–1154. doi: 10.1016/j.freeradbiomed.2013.08.180. [DOI] [PubMed] [Google Scholar]
- Ramirez M, Gallego-Martin T, Olea E, Rocher A, Obeso A. Gonzalez C. Serotonin dynamics and actions in the rat carotid body: preliminary findings. Adv Exp Med Biol. 2012;758:255–263. doi: 10.1007/978-94-007-4584-1_35. [DOI] [PubMed] [Google Scholar]
- Saugstad OD, Aune D, Aguar M, Kapadia V, Finer N. Vento M. Systematic review and meta-analysis of optimal initial fraction of oxygen levels in the delivery room at ≤32 weeks. Acta Paediatr. 2014;103:744–751. doi: 10.1111/apa.12656. [DOI] [PubMed] [Google Scholar]
- Say L, Gülmezoglu AM. Hofmeyr GJ. Maternal oxygen administration for suspected impaired fetal growth. Cochrane Database Syst Rev. 2003;1:CD000137. doi: 10.1002/14651858.CD000137. [DOI] [PubMed] [Google Scholar]
- Schafer FQ. Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Singh SN, Vats P, Kumria MM, Ranganathan S, Shyam R. Arora MP. Effect of high altitude (7620 m) exposure on glutathione and related metabolism in rats. Eur J Appl Physiol. 2001;84(3):233–237. doi: 10.1007/s004210170010. [DOI] [PubMed] [Google Scholar]
- Song MY, Makino A. Yuan JX. STIM2 contributes to enhanced store-operated Ca entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ. 2011;1:84–94. doi: 10.4103/2045-8932.78106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson ER. Hypoxic pulmonary vasoconstriction. High Alt Med Biol. 2013;14:101–110. doi: 10.1089/ham.2013.1010. [DOI] [PubMed] [Google Scholar]
- Sylvester JT, Shimoda LA, Aaronson PI. Ward JP. Hy-poxic pulmonary vasoconstriction. Physiol Rev. 2012;92:367–520. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo L, Lopez-Lopez JR, Castañeda J. Gonzalez C. Carbon monoxide inhibits hypoxic pulmonary vasoconstriction in rats by a cGMP-independent mechanism. Pflugers Arch. 1997;434:698–704. doi: 10.1007/s004240050454. [DOI] [PubMed] [Google Scholar]
- Tracy M, Downe L. Holberton J. How safe is intermittent positive pressure ventilation in preterm babies ventilated from delivery to newborn intensive care unit? Arch Dis Child Fetal Neonatal Ed. 2004;89:F84–F87. doi: 10.1136/fn.89.1.F84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario I, Rigual R, Obeso A. Gonzalez C. a Characterization of the synthesis and release of catecholamine in the rat carotid body in vitro. Am J Physiol Cell Physiol. 2000;278:C490–C499. doi: 10.1152/ajpcell.2000.278.3.C490. [DOI] [PubMed] [Google Scholar]
- Vicario I, Obeso A, Rocher A, Lopez-Lopez JR. Gonzalez C. b Intracellular Ca(2+) stores in chemoreceptor cells of the rabbit carotid body: significance for chemoreception. Am J Physiol Cell Physiol. 2000;279:C51–C61. doi: 10.1152/ajpcell.2000.279.1.C51. [DOI] [PubMed] [Google Scholar]
- Wang J, Shimoda LA, Weigand L, Wang W, Sun D. Sylvester JT. Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1059–L1069. doi: 10.1152/ajplung.00448.2004. [DOI] [PubMed] [Google Scholar]
- Ward MP, Milledge JS. West JB. High Altitude Medicine and Physiology. 2nd edn. London: Chapman & Hall Medical; 1995. pp. 366–87. [Google Scholar]
- Wenninger JM, Olson EB, Wang Z, Keith IM, Mitchell GS. Bisgard GE. Carotid sinus nerve responses and ventilatory acclimatization to hypoxia in adult rats following 2 weeks of postnatal hyperoxia. Respir Physiol Neurobiol. 2006;150:155–164. doi: 10.1016/j.resp.2005.05.014. [DOI] [PubMed] [Google Scholar]
- Yeh DY, Kao SJ, Feng NH, Chen HI, Wang D. Increased nitric oxide production accompanies blunted hypoxic pulmonary vasoconstriction in hyperoxic rat lung. Chin J Physiol. 2006;49:305–12. [PubMed] [Google Scholar]