

Abstract
The re-emergence of interest in intermediary metabolism and the development of metabolomics in relation to cancer and other diseases provide a timely reason to revisit issues of tumour cell metabolism. In this review, we address the issue of the role of high aerobic glycolysis, which is commonly associated with the metabolism of many tumour cells. The concept presented emphasises the importance of the glycolysis-citrate-lipogenesis pathway in providing the synthetic and bioenergetic requirements that are essential for the growth and proliferation of tumour cells. We hope that our discussion will be informative and instructive, and will stimulate interest and research regarding the intermediary metabolism and its regulation in tumour cells. We express our appreciation to the many pioneering and contemporary researchers whose studies provide much of the basis for this presentation.
Keywords: tumour cell metabolism, glycolysis, lipogenesis, citrate, m-aconitase, prostate cell metabolism, mitochondrial citrate export
Introduction
With great respect, we borrow the question raised by the distinguished biochemist, Dr. Racker, and colleague in 1981 [1] in their statement, `Malignant tumours exhibit a high rate of glycolysis. Only few investigators studied this phenomenon and for 50 years no answer was forthcoming to the basic question: Why do tumour cells glycolyse?' They were referring to the 50 years since the hallmark studies of Otto Warburg reported in 1926 [2]. The focal observation was the shift in intermediary metabolism from the typical aerobic metabolism of the normal mammalian cells to the high level of `aerobic glycolysis' and low respiration that characterised many tumour cells. The question was raised by Racker and Spector in regard to the issue of the mechanism responsible for the acceleration of aerobic glycolysis in tumour cells. We employ this question to address a different issue; namely, what is the importance of or reason for the transition to a high aerobic metabolism by tumour cells. This too has been an intriguing question that over the past 70 years has been addressed in numerous and excellent reports (e.g. [1–13], and many others that are worthy of citation).
The current re-emergence of interest in intermediary metabolism and the development of metabolomics in relation to cancer and other diseases provide a timely reason to revisit this issue of tumour metabolism. Therefore, we will present our concept regarding `Why do tumour cells glycolyse?' In so doing, we must acknowledge that we are drawing upon existing information, and our assimilation of the data and views of others along with our experiences. The question, while important, is essentially rhetorical for which a definitive answer cannot be proven. Concurrence or disagreement with the viewpoint that will be presented becomes the decision of the reader. Moreover, we hope and welcome concurring and opposing responses; as well as expanded information that we might have omitted due to oversight and space limitations of this presentation.
Defining a tumour cell: A parasitic existence
The intermediary metabolism of any cell is determined by and geared to the activities of the cell. The cellular metabolism provides the bioenergetic and synthetic/degradation requirements in support of the cell's function, growth, and reproductive activities. An understanding of the `purpose' of a cell's existence at any point of time in its life provides information of the role of its intermediary metabolism. Therefore, we begin to address the question, `Why do tumour cells glycolyse?' by describing our view of a tumour cell's existence. By reverting to an early graduate training (LCC) in parasitology, it becomes evident that the tumour cell is a `parasitic cell'. As such, it has one function in life. Its purpose is to grow and proliferate. It lives to reproduce; to insure its generational propagation. It does so at the expense and destruction of its host. These are the criteria of a parasitic life-style. It is interesting to note the similar view that was expressed by Bagetto [5], `… cancer cells will kill neighbouring cells … since it is compatible with the existence of these parasites of the living organisms'.
It is an error, in our view, to consider or to describe tumour cells as `de-differentiated' or `un-differentiated' cells (with the notable exception of a clinical connotation). To do so place tumour cells in the same category as normal undifferentiated cells (e.g. stem cells, mesenchymal cells and others that we will refer to collectively as `stem' cells). Stem cells, like tumour ells, also exist to grow and proliferate; but they do so for the purpose of differentiating into specialised cells that perform specific functions. They proliferate to maintain a continual population for further differentiation. However, unlike the parasitic tumour cells, these cells grow/proliferate in harmony with the host tissue; i.e. they exhibit a symbiotic life-style. In this sense these are `sane' cells and tumour cells are `insane' cells.
The primary purpose of the intermediary metabolism of the tumour cell must be to facilitate the major purpose of its existence; which is its dedication to a parasitic existence of reproduction, i.e. growth and proliferation. There is no other purpose or function of tumour cells! Essentially all the malignant activities of tumour cells (e.g. host tissue digestion and invasion; metastasis) exist to accommodate their parasitic existence. The intermediary metabolism must provide the bioenergetic and synthetic requirements that are most important to support growth and proliferation of the parasitic tumour cell. Therefore, one can ask, `What synthetic role does glucose metabolism play in proliferating cells?' (Note: obviously glucose metabolism has multiple involvements; but for this discussion we highlight a singular overriding role.) We propose the answer to be its role in de novo lipogenesis/cholesterogenesis (we will simply refer to as lipogenesis); which is the key metabolic activity (along with protein synthesis) required for growth and proliferation. The increase in total cell mass of the proliferating cancer cells necessitates a net increase in lipid biosynthesis, especially for increased membranogenesis. In other words, proliferating tumour cells are prolific lipogenic cells. Indeed, the importance of increased lipogenesis to cancer cell existence is well exemplified in the recent thoughtful paper of Menendez et al. [6].
The pathways of glucose utilisation: `Aerobic glycolysis' re-defined
To address this question, we propose that new metabolism definitions are essential. The term, `glycolyse', was used by Racker and Spector [1] in reference to the tumour cell pathway of `aerobic glycolysis'. `Aerobic glycolysis' implies the conversion of glucose to lactic acid (i.e. anaerobic glycolysis) under conditions of oxygen availability. However, under aerobic conditions glycolysis is defined as the conversion of glucose to pyruvate, and occurs in essentially all mammalian cells. The difference is whether pyruvate is reduced to lactic acid as the end-product; or if pyruvate is further metabolised. McKeehan [13] addressed this issue by correctly defining the former as the `glycolytic' pathway and the latter as the Emden-Meyerhoff pathway. However, contemporarily, the two pathways are commonly used synonymously. As such, the term `aerobic glycolysis' becomes confusing and perhaps misleading, particularly in the context of the intermediary metabolism of cells in situ.
For the following discussion, we will re-define the terminology of the pathways of glucose utilisation in cells in relation to their tissue environment. When glucose is converted to lactate only, we propose the condition to be referred to as `anoxic glycolysis' rather than `aerobic glycolysis'. We employ the term `anoxia' as a physiological condition in which the availability of oxygen to the cell is insufficient to support cellular oxidative metabolism. This, in our view, is more appropriate than the term `anaerobic' that implies an environment that is devoid of oxygen. We propose the term `normoxic glycolysis' for the pathway in which glucose is converted to pyruvate, which is subsequently utilised. Based on these relationships, the following pathways and bioenergetics of glucose utilisation are relevant to the question at hand.
-
(1)Anoxic glycolytic pathway
-
(2)Normoxic glycolytic pathway
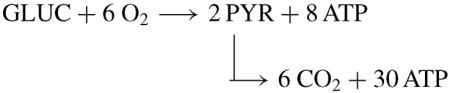
-
(2A)The overall reaction is
-
(2A)
The production of eight ATP/ glucose presumes the shuttle of NADH to mitochondrial oxidative phosphorylation so that 2 NADH → 2 NAD + 6 ATP. However, Argiles and Lopez-Soriano [7] point out that the NADH shuttle systems might be defective in tumour cells. In contrast, the extensive review by Pedersen [8] leads to the conclusion that highly glycolytic tumour cells do retain shuttle capacity for oxidation of glycolytic generated NADH.
However, neither pathway (1) nor (2) is applicable to the metabolism of proliferating tumour cells. These cells require a pathway in which citrate production is an end-product of glucose utilisation. Consequently, a third pathway must be defined.
-
(3)Hypoxic/normoxic glycolytic-citrate pathway
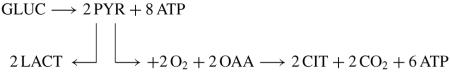
-
(3A)The overall reaction is
-
(3A)
We elect the term `Hypoxic' because it is evident that tumour cells spend much of their existence in a tissue environment that is neither anoxic nor normoxic, but is hypoxic. Therefore, this pathway exists in these cells in a hypoxic environment. We cannot imply that limited cellular oxygen availability is the direct cause of the failure of citrate oxidation. It is possible that hypoxic induced signalling pathways are involved. It is also possible for this pathway to exist under normoxic conditions, as it does in normal citrate-producing prostate epithelial cells [14–18]. In those cells, m-aconitase activity is inhibited by mitochondrial zinc, which prevents citrate oxidation.
A replaceable source of OAA is required in this metabolic pathway. In prostate epithelial cells, mitochondrial aspartate transamination with glutamate provides the source of OAA [14, 15, 17]. However, normal prostate cells have the unique function of production and secretion of enormously high citrate levels, which we refer to as `net citrate production'. In those cells, citrate is the major end-product of intermediary metabolism and involves the loss of six carbons from the metabolic pool. In contrast, the incorporation of citrate for lipogenesis results in the utilisation of two carbons (AcCoA); so that a four-carbon source of OAA can be regenerated for additional citrate synthesis. Therefore, the critical pathway in tumour cells becomes
-
(4)Glycolytic-lipogenic pathway
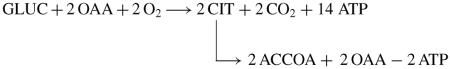
-
(4A)Overall reaction
-
(4A)
The overall reaction (4A) presumes that citrate export occurs via a mechanism such as the citrate/malate shuttle, and that the OAA produced by the ATP-citrate lyase reaction is recycled into mitochondria for synthesis of citrate. If not, a new source of OAA must be provided. This could arise from glutamate oxidation to a-ketoglutarate entry into the Krebs cycle and direct conversion to OAA as is described for tumour cell glutaminolysis.
An alternative pathway involves the transamination of glutamate with aspartate as a source of OAA as shown below. This is the pathway of net citrate production (Fig. 1) in prostate cells that we call the `glutamate-aspartate-citrate pathway' [14–19]. The overall reaction is
We relate tumour cell metabolism to normal prostate cell metabolism as both being citrate-producing cells. The essential difference is that the fate of citrate in the prostate cells is its accumulation for secretion rather than for utilisation in lipogenesis. Therefore, we offer the `glutamate-aspartatecitrate' pathway as a possibility in tumour cell metabolism. The operation of this pathway depends upon the utilisation and depletion of OAA, and a continual available source of aspartate. Otherwise the direction of the mAAT reaction will be reversed. Prostate cells contain a high-affinity aspartate transporter that provides for cellular import of aspartate to maintain its high cellular availability for citrate production. Advantages of the `glutamate-aspartate-citrate' pathway include the regeneration of glutamate and the possible coupling of the GDH reaction generation of NADH to terminal oxidation, which could yield additional ATP production. For this pathway to occur in tumour cells would require that any operation of glutaminolysis does not involve the glutamate-OAA transaminase reaction.
Fig. 1.

The pathway of net citrate production in normal prostate epithelial cells. Zinc inhibits m-aconitase activity. Aspartate transamination provides the source of OAA and glucose provides acetyl CoA for continued citrate synthesis.
`Why do tumour cells glycolyse?' … and the answer is `from glycolysis through citrate to lipogenesis'
The description of a `high aerobic glycolysis' (that we now describe as `anoxic glycolysis') that tumour cells employ results in lactic acid production to generate 2 ATP/glucose, presumably to replace the lost ATP production due to defective oxidation via Krebs cycle/terminal oxidation/coupled phosphorylation. If bioenergetics were the important metabolic concern of the tumour cells as is often proposed, the `anoxic glycolytic' pathway would be a costly adaptation. The tumour cell glucose utilisation would have to increase 19-fold to achieve the same bioenergetic results of the normoxic glycolytic pathway. However, the glycolytic production of ATP occurs at a rate that is 100-times faster than the mitochondrial oxidation to CO2 + ATP. This is the nature of a successful parasitic existence. The tumour cells efficiently extract glucose from and at the expense of the host, and then employ effective adaptive metabolic activities suited to their circumstances.
The incorporation of an increased `anoxic' glycolysis could be important when the tumour cells are exposed to a physiological anoxic environment. This could provide the bioenergetic requirements for the interim survival of the tumour cells in an arrested state while awaiting a forthcoming oxygenating environment. However, the `anoxic' glycolytic pathway is incompatible with the lipogenic requirements for tumour cell growth/proliferation. We propose that the accelerated aerobic glycolysis exists primarily for the purpose of providing a readily available source of AcCoA for mitochondrial synthesis of citrate. The citrate then provides the source of cytosolic AcCoA for the lipogenic demands of the tumour cells for their avid purpose of growth and proliferation. In addition, this pathway provides an increased production of ATP (12 ATP/glucose) compared to anoxic glycolysis (2 ATP/glucose) to meet the bioenergetic requirements of the malignant/proliferation activities of the tumour cells. This is also consistent with the conclusion of Pedersen [9] that 60–85% of the ATP production by tumour cells is mitochondrial-generated.
A number of relationships provide support for this concept (Fig. 2) regarding the reason why tumour cells `glycolyse'.
Fig. 2.
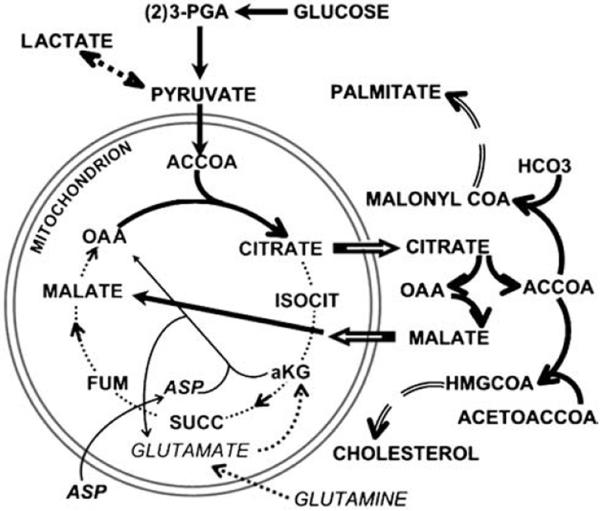
The glucose-citrate-lipogenesis pathway. The thick solid arrows represent the pathway in which the lipogenic carbon source is totally derived from glucose. The thin arrows represent the aspartate-glutamate transamination pathway as an alternate source of OAA (as occurs in normal prostate cells, Fig. 1). The dotted arrows represent a pathway of glutaminolysis as a source of OAA.
Lipogenesis (lipogenesis/cholesterogenesis) is the origin of membrane biosynthesis, which in turn is an essential requirement (along with associated anabolic pathways) for cell growth/proliferation. The metabolic shift to lipogenesis is also evident from the up-regulation of key lipogenic enzymes in tumour cells.
Glucose (with some possible exception described below) provides the carbon source for de novo fatty acid synthesis. The synthesis of 1 mol of typical fatty acid requires the utilisation of 10 mol of glucose. Therefore, it takes a prolific amount of glucose conversion to fulfil the lipogenic requirements of growing/proliferating tumour cells. Neither anoxic glycolysis that produces only lactic acid nor normoxic glycolysis that completely oxidises glucose, regardless of the bioenergetic consequences of each, is compatible with the lipogenic requirements of tumour cells.
The essential building-block intermediate for fatty acid synthesis is cytosolic AcCoA. Cytosolic AcCoA is derived from mitochondrial production of citrate.
The critical metabolic crossroad is citrate. Mitochondrial synthesis is the cellular source of citrate. It is synthesised from pyruvate oxidation that, in the absence of an alternative pyruvate source, is derived from glycolysis. Therefore, the first requirement is that some portion of utilised glucose must be converted to pyruvate, which then must be oxidised to AcCoA for condensation with OAA to produce citrate.
CITRATE – The metabolic switch
Once synthesised, the fate of mitochondrial citrate becomes critical. There are two alternatives for mitochondrial citrate utilisation: (a) oxidation, or (b) export.
-
(a)
In typical normal mammalian cell metabolism, citrate is converted to isocitrate (m-aconitase reaction), which provides the entry step for the oxidative stage of the Krebs cycle; and is critical for the complete oxidation of glucose and fatty acids that is coupled to ATP production. Obviously, the oxidation of citrate eliminates its availability for lipogenesis. Typically, m-aconitase is in excess and is not a rate-limiting enzyme. Several studies [8, 9, 19–21] report that downstream reactions from isocitrate to OAA are operational in tumour cells, and that citrate oxidation (i.e. m-aconitase) is not impaired in tumour cells. In contrast, Hernanz and de la Fuente [22] provide evidence for a decreased m-aconitase activity in tumour cells. In most studies, the oxidation of citrate and m-aconitase activity was determined in isolated mitochondrial preparations and under conditions to maximise the activities. It is important to recognise that these conditions might not reflect the activities in situ. It is becoming apparent that, within cells, m-aconitase activity is susceptible to a variety of reversible inhibitory influences that would not be represented in disrupted cellular preparations, including isolated mitochondria. An important and informative measurement that has not been reported is the steady-state cellular citrate/isocitrate ratio; which is established by m-aconitase. Due to the equilibrium established by m-aconitase, mammalian cells typically exhibit a citrate/isocitrate ~10/1, which is independent of the cellular concentration of citrate. If m-aconitase activity is functionally limiting, the citrate/isocitrate ratio will increase significantly, provided that citrate synthesis occurs. This is precisely the condition that occurs with citrate-producing prostate epithelial cells [23, 24]. The inhibition of m-aconitase activity by zinc results in a cellular citrate/isocitrate ratio ~30/1. Elimination of the inhibition so that citrate oxidation occurs reduces the citrate/isocitrate ratio to ~10/1. Based on this relationship, measurements of citrate and isocitrate in tumour cells or tumour tissue would be extremely informative.
-
(b)
Regardless of the issue of citrate oxidation, tumour cells exhibit an increase in the export of citrate from mitochondria [8, 19]. This is the essential metabolic adaptation for lipogenic cells. Although the identification of the mitochondrial citrate exporter and the export mechanism requires further study, the up-regulation of the process in tumour cells is well established. This export of citrate is the initiating step in lipogenesis. In the cytosol, citrate is cleaved by ATP-citrate lyase to AcCoA + OAA; and the AcCoA is carboxylated for incorporation into fatty acid/cholesterol synthesis. The significance of this utilisation of citrate for tumour cell existence is further revealed by the bioenergetic consequence. The tumour cells preferentially elect to sacrifice 12 ATP/citrate exported (24 ATP/glucose utilised) rather than being oxidised to obtain the additional ATP production.
This leads to another consideration. If m-aconitase activity is not limiting in tumour cells, the intramitochondrial citrate is subjected to both oxidation and export. Parlo and Coleman [8, 19] suggest that the up-regulation and rapidity of the citrate exporter depletes the availability of citrate for oxidation via the Krebs cycle. However, if m-aconitase activity is not limiting, the intramitochondrial citrate will exist in equilibrium with isocitrate, which is rapidly oxidised by IDH. Under these conditions, the extent of citrate availability for export will be dependent upon the rapidity of the export process. Therefore, both citrate oxidation for energy production and citrate export for lipogenesis will co-exist. The combination of both events would become dependent upon the rate of citrate synthesis. The apparent `truncation' of the Krebs cycle at the citrate step in tumour cells, as has been described [8, 19], would be due to a declining availability of citrate. To maintain a continual citrate availability to sustain its oxidation and export will require an increased production of pyruvate and OAA and a non-limiting citrate synthase activity. Under these conditions, the citrate/isocitrate ratio would be maintained at ~10/1. If, on the other hand, m-aconitase activity is inhibited and limiting, citrate export will occur, but the citrate/isocitrate ratio would be markedly increased >10/1.
The consideration of these relationships leads to some dilemma. The stoichiometery of glucose utilised and products produced needs to be addressed. This is based on the acceptance of the concept that the anoxic glycolytic pathway might be involved in the survival of arrested/dormant tumour cells; but is incompatible with requirements of growing tumours. Then one would expect that the major end-products of glucose utilisation would be some combination of lactic acid, citrate, and CO2. As best that we could ascertain from a literature search, no such information has been reported. There are many reports of product-formation from glucose, but (apparently) none that provides this stoichiometry. Nevertheless, it would be important in any such study to account for citrate production that would be `swept' into fatty acid synthesis. A simple measurement of the existing level of cit-rate even with CO2 production would not be equivalent to the level of citrate production.
Although we propose that the critical role of accelerated glycolysis is to provide acetyl CoA for lipogenesis (the glycolytic-citrate-lipogenic pathway), the fact that a considerable portion of the glucose utilised under normoxic/hypoxic conditions ends up as lactate cannot be ignored. This reflects the major metabolic adaptation and priority of tumour cells to optimise their capability to import and glycolytically utilise glucose; i.e. they are prolific consumers of host glucose. The accelerated rate of conversion of glucose to pyruvate exceeds the rate of pyruvate oxidation. As concluded by Pedersen [9], this accumulating pyruvate is subjected to reduction of lactate. The oxidation of pyruvate might be further impaired by the export of citrate. The truncated Krebs cycle eliminates the re-cycling of OAA, which is essential for condensation of the acetyl CoA produced by pyruvate oxidation. The AcCoA imposes a product inhibition on pyruvate dehydrogenase activity. Therefore, the further oxidation of pyruvate must await the production of OAA from an alternative source. Until then, the excess pyruvate is reduced to lactic acid. This insures that a sufficient pool of pyruvate is available for lipogenesis. Again, it is the host, not the parasitic tumour cells, that suffers the consequences of excessive (wasteful) utilisation of glucose. Moreover, some excess lactate can re-enter the host circulation for utilisation by host tissues. If the tumour cells are capable of completely oxidising pyruvate via the Krebs cycle as discussed above, one must expect that synthesised citrate that is not rapidly exported to the cytosol can be oxidised to CO2 + ATP. An appropriate stoichiometric analysis of overall glucose utilisation by the combined pathways should be
where citrate = (citrate conc) + (`lipogenic/cholesterogenic' AcCoA).
These considerations provide our answer to the question, `Why do tumour cells glycolyse?' They do so to meet the de novo lipogenic/cholesterogenic requirements that are essential for their parasitic existence of growth and proliferation. The critical pathway is glycolysis-citrate-lipogeneis, which along the way is also a fairly efficient bioenergetic pathway.
The alternative paths to lipogenesis
It is evident that tumour cells, of necessity for their parasitic existence, are essentially lipogenic cells. That glucose provides the acetyl CoA for de novo lipogenesis is well established. However, not all tumour cells exhibit a `high aerobic glycolysis'. If lipogenesis is essential for the growth/proliferation of all tumour cells, those that do not `glycolyse' must posses an alternative pathway. Moreadith and Lehninger [25] observed, `In fact, many malignant cell lines, as well as some normal cells, do not have an absolute requirement for glucose per se … ' They proposed and provided evidence for the possible involvement of a glutamate pathway to lipogenesis in tumour cells. They stated `The two major products of the [glutamate oxidation] pathway described here, citrate and alanine, have important roles in tumour metabolism. Citrate is required as the major source of cytosolic acetyl-coA for fatty acid and cholesterol biosynthesis'. As they proposed, the glutamate pathway could provide both OAA and AcCoA for citrate → lipogenesis, in the absence of glucose contribution. McKeehan [13] proposed the utilisation of glutaminolysis as a source of pyruvate in tumour cells. The following is a modified representation of the pathway of glutaminolysis through citrate to lipogenesis:
Another alternative is the possible cytosolic direct production of AcCoA. This could be achieved by the action of AcCoA synthetase. Loikkanen et al. [26] reported that expression of cytosolic AcCoA synthetase in adult mouse tissues that is regulated during embryogenesis; thereby suggesting its possible role in providing cytosolic AcCoA for lipogenesis and cholesterogenesis. Sone et al. [27] reported that up-regulation of AcCoA synthetase in specific circumstances in lipogenic tissues could provide the supply of acetylCoA directly from cellular acetate for lipogenesis. Oikawa et al. [28] reported that AcCoA synthetase could provide the acyl-CoA utilised for the synthesis of cellular lipids in proliferating preadipocytes. To our knowledge no information exists regarding this enzyme and lipogenic pathway in tumour cells. Nevertheless, the need for de novo lipogenesis in growing/proliferating tumour cell seems evident regardless of the metabolic pathway employed for its achievement. Those cells that exhibit a high hypoxic/normoxic glycolysis do so to accommodate their synthetic/bioenergetic requirements for lipogenesis.
Concluding remarks
`Why do tumour cells glycolyse?' We have offered our concept and answer to the question. There will be no right or wrong answer to such a question. Other alternatives that we have not described also exist. However, merely thinking about the question and offering a response caused us to re-educate ourselves to the integrated aspects of intermediary metabolism and cell function. We hope that we might have raised issues for the reader that are worthy of further consideration and indicative of needed areas of research in tumour metabolism. The most satisfying aspect of our preparation of this paper was the re-visitation and review of the early papers (circa 1950–1980) of some of the `pioneers and giants' of the golden era of intermediary metabolism. We would urge all contemporary researchers in intermediary metabolism/tumour metabolism to read and to ponder the exquisite thoughtfulness and capability of that earlier generation of pioneering biochemists.
Our final point is one of caution. It is a mistake to expect that all tumour cells must exhibit a universal adaptive intermediary metabolism. The use of an elected variety of tumour cells to establish a commonality for all tumour cells leads to inductive arguments. Another danger is in regard to the extrapolations that can be made from the experimental systems that are employed. Pathways and reactions of intermediary metabolism that are defined in isolated mitochondrial preparations do not necessarily establish their operation in the intact cellular environment. Pathways and reactions established in studies with tumour cells in vitro do not necessarily reflect the pathways that are operating in the tumour cells in situ in their host tissue environment. As long as these limitations are recognised and the interpretation of the studies is tempered, it is productive to pursue the intriguing issues of tumour cell metabolism.
Acknowledgments
The studies of LCC and RBF described in this review were supported in part by NIH grants CA71207, CA21097, CA79903 and CA93443.
References
- 1.Racker E, Spector M. Warburg effect revisited: merger of biochemistry and molecular biology. Science. 1981;213:303–307. doi: 10.1126/science.6264596. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F, Negelein E. Uber den Stoffwechsel von Tumouren im Korper. Klin Woch. 1926;5:829–832. [Google Scholar]
- 3.Chance B, Hess B. Spectroscopic evidence of metabolic control. Science. 1959;129:700–708. doi: 10.1126/science.129.3350.700. [DOI] [PubMed] [Google Scholar]
- 4.Dang CV, Samenza GL. Oncogenic alterations of metabolism. Trends Bio Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 5.Baggetto LG. Deviant energetic metabolism of glycolytic cancer cells. Biochimie. 1992;74:959–974. doi: 10.1016/0300-9084(92)90016-8. [DOI] [PubMed] [Google Scholar]
- 6.Menendez JA, Colomer R, Lupu R. Why does tumour-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids? Med Hypoth. 2005;64:342–3499. doi: 10.1016/j.mehy.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 7.Argiles JM, Lopez-Soriano FJ. Way do cells have such a high glycolytic rate. Med Hypoth. 1990;32:151–155. doi: 10.1016/0306-9877(90)90039-h. [DOI] [PubMed] [Google Scholar]
- 8.Parlo RA, Coleman PS. Enhanced rate of citrate export from cholesterol-rich hepatoma mitochondria. J Biol Chem. 1984;259:997–10003. [PubMed] [Google Scholar]
- 9.Pedersen PL. Tumour mitochondria and the bioenergetics of cancer cells. Prog Exp Tumour Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- 10.Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion. 2004;4:755–762. doi: 10.1016/j.mito.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 11.Matsuno T. Bioenergetics of tumour cells: glutamine metabolism in tumour cell mitochondria. Int J Biochem. 1987;19(4):303–307. doi: 10.1016/0020-711x(87)90002-4. [DOI] [PubMed] [Google Scholar]
- 12.Sauer LA, Stayman JW, 3rd, Dauchy RT. Amino acid, glucose, and lactic acid utilization in vivo by rat tumours. Cancer Res. 1982;42:4090–4097. [PubMed] [Google Scholar]
- 13.McKeechan WL. Glycolysis, glutaminolysis and cell proliferation. Cell Biol Int Rep. 1982;6:635–650. doi: 10.1016/0309-1651(82)90125-4. [DOI] [PubMed] [Google Scholar]
- 14.Franklin RB, Costello LC. Glutamate dehydrogenase in rat ventral prostate and a proposed aspartate-glutamate pathway of citrate synthesis. J Urol. 1984;132:1239–1243. doi: 10.1016/s0022-5347(17)50113-5. [DOI] [PubMed] [Google Scholar]
- 15.Costello LC, Franklin RB. Prostate epithelial cells utilize glucose and aspartate as the carbon sources for net citrate production. Prostate. 1989;15:335–342. doi: 10.1002/pros.2990150406. [DOI] [PubMed] [Google Scholar]
- 16.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. J Biol Chem. 1997;272:28875–28881. doi: 10.1074/jbc.272.46.28875. [DOI] [PubMed] [Google Scholar]
- 17.Costello LC, Franklin RB. The intermediary metabolism of the prostate: a key to understanding the pathogenesis and progression of prostate malignancy. Oncology. 2001;59:269–282. doi: 10.1159/000012183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costello LC, Franklin RB. The novel role of zinc in the regulation of prostate citrate metabolism and its implications in prostate cancer. Prostate. 1998;35:285–296. doi: 10.1002/(sici)1097-0045(19980601)35:4<285::aid-pros8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 19.Parlo RA, Coleman PS. Continuous pyruvate carbon flux to newly synthesized cholesterol and the suppressed evolution of pyruvate-generated CO2 in tumours: further evidence for a persistent truncated Krebs cycle in hepatomas. Biochim Biophys Acta. 1986;886:169–176. doi: 10.1016/0167-4889(86)90134-5. [DOI] [PubMed] [Google Scholar]
- 20.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254:2669–2775. [PubMed] [Google Scholar]
- 21.Dietzen DJ, Davis EJ. Oxidation of pyruvate, malate, citrate, and cytosolic reducing equivalents by AS-30D hepatoma mitochondria. Arch Biochem Biophys. 1993;305:91–102. doi: 10.1006/abbi.1993.1397. [DOI] [PubMed] [Google Scholar]
- 22.Hernanz A, de la Fuente M. Characterization of aconitate hydratase from mitochondria and cytoplasm of ascites tumour cells. Biochem Cell Biol. 1988;66:792–795. doi: 10.1139/o88-090. [DOI] [PubMed] [Google Scholar]
- 23.Costello LC, Franklin RB. Concepts of citrate production and secretion by prostate. 1. Metabolic relationships. Prostate. 1991;18:25–46. doi: 10.1002/pros.2990180104. [DOI] [PubMed] [Google Scholar]
- 24.Franklin RB, Costello LC, Littleton G. Citrate uptake and oxidation by fragments of rat ventral prostate. Enzyme. 1977;22:45–51. doi: 10.1159/000458507. [DOI] [PubMed] [Google Scholar]
- 25.Moreadith RW, Lehninger AL. The pathways of glutamate and glutamine oxidation by tumour cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J Biol Chem. 1984;259:6215–6221. [PubMed] [Google Scholar]
- 26.Loikkanen I, Haghighi S, Vainio S, Pajunen A. Expression of cytosolic acetyl-CoA synthetase gene is developmentally regulated. Mech Dev. 2002;115:139–141. doi: 10.1016/s0925-4773(02)00097-7. [DOI] [PubMed] [Google Scholar]
- 27.Sone H, Shimano H, Sakakura Y, Inoue N, Amemiya-Kudo M, Yahagi N, Osawa M, Suzuki H, Yokoo T, Takahashi A, Iida K, Toyoshima H, Iwama A, Yamada N. Acetyl-coenzyme A synthetase is a lipogenic enzyme controlled by SREBP-1 and energy status. Am J Physiol Endocrinol Metab. 2002;282:E222–230. doi: 10.1152/ajpendo.00189.2001. [DOI] [PubMed] [Google Scholar]
- 28.Oikawa E, Iijima H, Suzuki T, Sasano H, Sato H, Kamataki A, Nagura H, Kang MJ, Fujino T, Suzuki H, Yamamoto TT. A novel acyl-CoA synthetase, ACS5, expressed in intestinal epithelial cells and proliferating preadipocytes. J Biochem (Tokyo) 1998;124:679–685. doi: 10.1093/oxfordjournals.jbchem.a022165. [DOI] [PubMed] [Google Scholar]