

Abstract
Caliciviruses are single-stranded RNA viruses that cause a wide range of diseases in both humans and animals, but little is known about the regulation of cellular translation during infection. We used two distinct calicivirus strains, MD145-12 (genus Norovirus) and feline calicivirus (FCV) (genus Vesivirus), to investigate potential strategies used by the caliciviruses to inhibit cellular translation. Recombinant 3C-like proteinases (r3CLpro) from norovirus and FCV were found to cleave poly(A)-binding protein (PABP) in the absence of other viral proteins. The norovirus r3CLpro PABP cleavage products were indistinguishable from those generated by poliovirus (PV) 3Cpro cleavage, while the FCV r3CLpro products differed due to cleavage at an alternate cleavage site 24 amino acids downstream of one of the PV 3Cpro cleavage sites. All cleavages by calicivirus or PV proteases separated the C-terminal domain of PABP that binds translation factors eIF4B and eRF3 from the N-terminal RNA-binding domain of PABP. The effect of PABP cleavage by the norovirus r3CLpro was analyzed in HeLa cell translation extracts, and the presence of r3CLpro inhibited translation of both endogenous and exogenous mRNAs. Translation inhibition was poly(A) dependent, and replenishment of the extracts with PABP restored translation. Analysis of FCV-infected feline kidney cells showed that the levels of de novo cellular protein synthesis decreased over time as virus-specific proteins accumulated, and cleavage of PABP occurred in virus-infected cells. Our data indicate that the calicivirus 3CLpro, like PV 3Cpro, mediates the cleavage of PABP as part of its strategy to inhibit cellular translation. PABP cleavage may be a common mechanism among certain virus families to manipulate cellular translation.
The family Caliciviridae is comprised of four genera: Vesivirus (including feline calicivirus [FCV]), Lagovirus (including rabbit hemorrhagic disease virus [RHDV]), Norovirus (including Norwalk virus and MD145-12), and Sapovirus (including Sapporo virus and porcine enteric calicivirus [PEC]) (21). They are single-stranded, positive-sense RNA viruses that share evolutionary relatedness with the picornaviruses. The major caliciviruses associated with human disease belong to the genus Norovirus. The noroviruses (designated here as NoV) are responsible for the majority of food- and waterborne outbreaks of viral gastroenteritis worldwide (15, 26), but the development of control strategies for NoV disease has been complicated by the absence of tissue culture systems to study replication (23). Analysis of calicivirus proteins in comparative studies with the picornaviruses might yield insight into their functions during viral replication and facilitate the development of treatment strategies for calicivirus-associated diseases.
In this study, we investigated possible strategies that caliciviruses might use to inhibit cellular translation. For picornaviruses, previous studies had implicated the cleavage of eIF4G by certain 2A proteinases (2Apro) and the cleavage of poly(A)-binding protein (PABP) by the 3C cysteine proteinase (3Cpro) in the shutoff of cellular mRNA translation during viral replication (8, 20, 32, 34, 39, 40, 42, 46). Caliciviruses have only one known viral proteinase (45, 53, 58), designated as a 3C-like proteinase (3CLpro) because of its sequence similarity to the picornavirus 3Cpro in the active site region. The picornavirus 3Cpro and calicivirus 3CLpro enzymes belong to a family of viral proteinases that fold into a bilateral beta sheet domain similar to the cellular chymotrypsin-like serine proteinases. However, the nucleophilic residue of the viral enzymes is a cysteine instead of serine (4, 9). Atomic structures for some picornavirus 3Cpro enzymes have been solved (1, 7, 47). Amino acid residues His40, Glu71, and Cys147 constitute the catalytic triad in the poliovirus (PV) 3Cpro (49), and His40, Glu71, and Cys146 form the catalytic triad in the rhinovirus 3Cpro (47). The calicivirus 3CLpro structure has not yet been determined, but site-directed mutagenesis has been used to identify residues His27, Asp44, and Cys104 as part of the active site of the RHDV 3CLpro (9) and residues His30 and Cys139 as part of a catalytic dyad in the NoV 3CLpro (53). The presence of conserved histidine and cysteine residues in the active site is a feature that is conserved between the picornavirus and calicivirus proteinases in spite of considerable diversity in the primary amino acid sequence.
PABP and the poly(A) tail of eukaryotic mRNAs play an important role in stimulating translation initiation (31, 52). The PABP molecule is comprised of two functional domains, an N-terminal domain with four RNA recognition motifs (RRM) and a C-terminal domain (CTD) (19, 50). The N-terminal domain of PABP (RRM2) binds to eIF4G and to the poly(A) tail of mRNAs. eIF4G binds the cap-binding protein eIF4E. Combined, these protein-protein interactions facilitate the 5′-3′ end circularization of mRNA and also stimulate translation by 4- to 10-fold (30, 60, 63). The mechanism of this cap-poly(A) synergy is unclear, but PABP binding was proposed to induce cooperative conformational changes in eIF4E and eIF4G that enhance the stability of initiation complexes on capped mRNAs (62). There is also evidence that some ribosomes may recycle after reaching termination codons and that PABP or mRNA circularization may facilitate this process (3, 61). Though the PABP CTD seems to be dispensable for RNA circularization, it has been implicated in translation. The CTD contains a cleft that binds several proteins involved in translation regulation (38), including translation initiation factor eIF4B (a cofactor of RNA helicase eIF4A) (14), PABP-interacting proteins Paip-1 (16) and Paip-2 (35), and eukaryotic release factor 3 (eRF3) (29). A recent report has shown that a point mutation in the CTD could eliminate its binding to eRF3 and in turn inhibit cap-poly(A)-dependent translation (29, 61). It was recently demonstrated that PV 3Cpro cleaves PABP to release the CTD from the N-terminal domain, and it was proposed that the removal of CTD inhibits translation by a novel mechanism without affecting binding of the N-terminal domain of PABP to poly(A) RNA and eIF4G (40).
We report here that a strategy to inhibit cellular translation likely exists during calicivirus infection, and we provide evidence that the calicivirus 3CLpro, like that of the picornavirus 3Cpro, may play a role in this inhibition by the cleavage of PABP. Our data suggest that cleavage of PABP may be a common mechanism used by diverse virus families to manipulate host cell mRNA translation and further confirms the importance of the CTD from PABP in translation.
MATERIALS AND METHODS
Viruses and cells.
The caliciviruses analyzed in this study were the Urbana strain of FCV (GenBank accession no. L40021) (54) and the MD145-12 strain of NoV (GenBank accession no. AY032605) (22). FCV was grown in Crandell-Rees feline kidney (CRFK) cells (54), and FCV-infected total cell lysates were prepared as described previously (24).
Analysis of protein synthesis in FCV-infected cells.
To study the effect of calicivirus replication on host protein synthesis, CRFK monolayers (4 × 106 cells) were infected with FCV at a multiplicity of infection (MOI) of 10 (or mock infected) and incubated at 37°C. For radiolabeling of viral and host cell proteins, the cells were washed with methionine-free growth medium at different times postinfection and incubated in the same medium for 30 min. [35S]methionine (>1,000 Ci/mmol) (Amersham Biosciences Corp., Piscataway N.J.) was added at a concentration of 150 μCi/ml, and the cells were incubated for an additional hour. Cells at each time point were then collected, washed, and lysed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer with 2% mercaptoethanol prior to analysis in a 12% Tris-Gly polyacrylamide gel by SDS-PAGE and autoradiography. Signals were detected with a phosphorimager (Molecular Dynamics, Amersham), and the IP Lab Gel suite (Signal Analytics, Vienna, Va.) was used for the analysis.
Plasmids and recombinant proteins.
pGEM-4Z (Promega Corporation, Madison, Wis.) containing the firefly luciferase gene was a kind gift of P. Sarnow. This plasmid was linearized with HpaI or BamHI to produce runoff templates for synthesis of polyadenylated (30-nucleotide [nt]) and nonpolyadenylated luciferase RNAs, respectively.
To express the recombinant (r) FCV 3CL proteinase (which occurs in infected cells as part of a proteinase-polymerase precursor protein, ProPol) (58), the corresponding ProPol region of the virus genome was PCR amplified from the full-length FCV cDNA clone pQ14 (54) as template and cloned into bacterial expression vector pET-28b (Novagen, Madison, Wis.). The region was amplified using oligonucleotides 5′-ACATATAAGCTTTCTGGACCCGGCACC-3′ (corresponding to the beginning of the ProPol sequence at nt 3233 to 3247 with a HindIII site shown underlined) and 5′-ATTATACTCGAGTCAAACTTCGAACACATC-3′ (complementary to the 3′ end of the ORF1 at nt 5294 to 5311 with a termination codon [bold] and a XhoI site [underlined]). The purified PCR fragment was treated with HindIII and XhoI and ligated into HindIII-XhoI-linearized pET-28b. The resulting plasmid, designated pHPP, contained the FCV ProPol sequence fused to the vector sequence encoding an N-terminal His6 tag under control of the T7 RNA polymerase promoter and placed downstream from the bacterial ribosome binding site. The expression and purification of FCV rProPol in Escherichia coli BL21(DE3) cells were performed as previously described (5). The mature r3CL proteinase of the MD145-12 NoV strain (designated NoV r3CLpro), engineered with a His6 tail at its carboxyl terminus, was expressed in bacteria and purified as previously described (5). Protein quantity was estimated by the Bradford method (11), and the enzyme was stored in buffer containing 300 mM NaCl, 50 mM Tris, pH 8, 2 mM dithiothreitol, and 10% glycerol at −70°C. The NoV r3CLpro was inactivated by heating at 80°C for 25 min, and this preparation was designated 3CL inactivated (NoV r3CL-In).
The expression and purification of recombinant human His-PABP, coxsackie B3 virus (CVB3) 2Apro, and PV 3Cpro proteins have been previously described (32, 41). Plasmid pET28a-PABP, encoding human PABP, was constructed by excising the PABP open reading frame from plasmid pGEX-4T2-PABP with BamHI and NotI and inserting the DNA fragment into the unique BamHI/NotI sites of pET-28a. The predicted cleavage sites Q413/T414, Q437/G438, and Q537/G538 in the PABP protein recognized by PV 3C (41) were each altered by site-directed mutagenesis of the pET28a-PABP plasmid. The primers (Invitrogen, Carlsbad, Calif.) used for the mutagenesis are described in Table 1. The resulting plasmids were designated pET-Q413A/PABP (A413/T414), pET-Q437A/PABP (A437/G438), and pET-Q537A/PABP (A537/G538). All plasmid constructions were verified by sequence analysis. Another set of PABP plasmid derivatives was obtained by inserting a stop codon at each mapped cleavage site such that full-length translated PABP products were equivalent to the N-terminal cleavage fragments generated by PV 2Apro or 3Cpro (Table 1).
TABLE 1.
Primers used for site-directed mutagenesis of PABP
| Mutation | Primer sequencea |
|---|---|
| Q413Ab | GCAGCTATCCCAgcGACTCAGAACCG |
| Q437Ab | GCTGGACTGCTgcGGGTGCCAGACC |
| Q537Ab | CTGTTCATGTAgcAGGTCAGGAAC |
| T414Xc | CTTCATGGCAGCTATCCCACAGtgaCAGAACGCTGCTGCATACTAT |
| G448Xc | CACATCAACACAGACAATGtGaCCACGTCCTGCAGCTGCA |
| G538Xc | CAGCCTGCTGTTCATGTACAAtgaCAGGAACCTTTGACTGCTTCC |
Only the forward-sense primer sequence is shown. Nucleotide substitutions are shown in lowercase.
The Glu residue at the P1 position was replaced by an Ala in each of the previously established cleavage sites in PABP recognized by PV 3Cpro (41).
The codon for the amino acid at the P1′ position was replaced with a stopcodon (X).
HeLa cell fractionation.
For fractionation of HeLa cells, an S10 lysate was prepared as described previously (12) and centrifuged at 200,000 × g for 1 h using a 70 Ti rotor in a Beckman ultracentrifuge. The supernatant containing non-ribosome-associated PABP was retained as the non-ribosome-associated fraction (S200). The pellet was resuspended in one cell volume of lysis buffer containing 0.5 M KCl to dissociate ribosome-associated proteins and resedimented at 200,000 × g for 45 min. The supernatant was dialyzed and used as the crude translation initiation factor extract designated RSW. The pellet was resuspended in lysis buffer and used as the ribosome-enriched fraction (designated ribosome). All fractions contained PABP and were used in assays as substrates for viral proteinases.
In vitro cleavage assays.
In vitro cleavage assays of cellular PABP were performed using essentially the same conditions regardless of the source of the substrate, which included S10, S200, RSW, and ribosome-enriched fractions of HeLa cells. These substrates and various amounts of proteinases (final concentration, 3 to 30 μg/ml) were incubated at 37°C for 3.5 h in cleavage buffer (100 mM NaCl, 5 mM MgCl2, 10 mM HEPES-KOH, pH 7.4). All reactions were stopped by the addition of SDS-PAGE sample buffer and were analyzed in 10% (wt/vol) acrylamide gels by SDS-PAGE, followed by immunoblotting or autoradiography.
Two approaches were used to analyze the cleavage of recombinant PABP in vitro. The first method used as substrate radiolabeled human PABP protein derived by in vitro translation of purified RNA in reticulocyte lysates as described previously (32). The second method used radiolabeled human PABP protein synthesized in a coupled transcription-translation (TNT) reaction containing pET/T7-PABP plasmid (or its derivatives). Each 25-μl TNT reaction contained 1 μg of plasmid DNA and 15 μCi of 35S-labeled Met (1000 Ci/mmol) (Amersham) and was performed in the presence of T7 RNA polymerase at 30°C for 1.5 h as recommended by the manufacturer (Promega). An aliquot (4 μl) of the PABP TNT reaction was mixed with 6 μl of phosphate-buffered saline (PBS) (Quality Biological Inc., Gaithersburg, Md.), pH 7.4, containing 5 μg of FCV ProPol incubation at 30°C, overnight. The entire reaction was then resolved in a 12% Tris-Gly polyacrylamide gel by SDS-PAGE, prior to staining with Coomassie blue (Pierce, Rockford, Ill.). The gel was then dried and subjected to autoradiography. Alternatively, aliquots (1 μl) of the human PABP TNT reaction were mixed with cytoplasmic extracts from FCV-infected cells (10 μl) and incubated for 3.5 h at 30°C before analysis by SDS-PAGE and autoradiography.
Translation assays in HeLa cell extracts.
HeLa S3 cells were maintained in suspension cultures at a concentration of 7 × 105 to 9 × 105 cells/ml at 37°C and 5% CO2 in Joklik's medium, supplemented with 1% fetal calf and 9% calf serum. Cells were disrupted in a Dounce homogenizer with five to seven strokes until 80% lysis occurred, and then extracts were prepared as previously described (6, 40). In vitro translation assays were typically performed in 40-μl reaction mixtures containing a mix of cell lysate-translation cocktail (l:1) {1.8 mM HEPES, pH 7.6, 25 μM amino acids minus methionine, 50 μg of creatine phosphokinase/ml, 3 mM 2-aminopurine, 25 mM creatine phosphate, 1 mM ATP, 0.5 mM GTP, 2 mM dithiothreitol, 90 mM KCl, 1.8 mM Mg(OAc)2, and 30μCi [35S]methionine-cysteine [trans label]} (1,200 Ci/mmol; ICN). HeLa extracts were preincubated with CVB3 2Apro, PV 3Cpro, NoV 3CL, FCV 3CL, or buffer for 5 min at 30°C as indicated in the figure legends. The translation cocktail was added to the extracts pretreated with proteinases at the same time as exogenous RNA (100 to 300 ng) and incubated at 37°C. Incorporation of 35S-trans label into translation products was analyzed by densitometry of SDS-PAGE autoradiograms with NIH Image J software or by precipitation of proteins with trichloroacetic acid and scintillation counting. For PABP restoration experiments, PABP (200 μM) was preincubated with capped, polyadenylated, or nonpolyadenylated luciferase RNA at room temperature for 10 min. RNA/PABP complexes were added to the non-nuclease-treated HeLa extracts pretreated with buffer or the proteinase of interest for 5 min at 30°C.
Luciferase assays.
Luciferase activity was determined by luciferase assay system reagents (Promega) according to the manufacturer's instructions with photon emission measured using a Sirius luminometer (Berthold, Oak Ridge, Tenn.).
Antibody production and immunoblots.
Rabbit anti-human PABP antibody was raised against a synthetic peptide sequence (GIDDERLRKEFSPFGTC) in the RRM4 of PABP. Immunoblots (with the exception of that shown in Fig. 1) were incubated with a 1:1,000 dilution of the PABP peptide-specific antibody in BLOTTO (5% dry milk, 20 mM Tris, pH 7.4) overnight at 4°C. After washing three times with PBST (PBS containing 0.05% Tween 20), blots were incubated with a 1:1,000 dilution of horseradish peroxidase-conjugated goat anti-rabbit antibody (Calbiochem, San Diego, Calif.) in BLOTTO at room temperature for an hour. Blots were washed three times with PBST and developed by enhanced chemiluminescence (Pierce). Signals were detected with a Kodak chemiluminescent image analyzer (Eastman Kodak, Rochester, N.Y.). The immunoblot shown in Fig. 1 was analyzed with antibodies raised against recombinant PABP as described previously (41). Immunoblot analysis of eIF4GI was carried out with rabbit polyclonal antisera raised versus a peptide sequence (RPDDRSQGAIIADRPGLPGPEH) located in the N-terminal domain of human eIF4GI (amino acid 231 to 252). Immunoblots were incubated with a 1:1,000 dilution of primary antibody in BLOTTO overnight at 4°C, followed by secondary antibodies as described above.
FIG. 1.

Comparison of the cleavage activity of NoV r3CLpro with that of PV 3Cpro and 2Apro on the cellular protein substrates, PABP and eIF4G. (A) The structural organization of human PABP and the previously mapped cleavage sites recognized by the PV 3C and 2A proteinases (32, 41). (B and C) NoV r3CLpro cleaves PABP. HeLa S3 cells were fractionated into cytoplasmic (S10) (B) and ribosome-associated fractions (C). In vitro cleavage reactions were assembled in these fractions and proteins were analyzed by SDS-10% PAGE followed by immunoblotting with PABP-specific antibodies. Numbers on the right show the migration of molecular weight markers (in thousands). HeLa S10 lysates (B) were incubated for 3.5 h at 37°C with the following: lane 1, mock incubation control (indicated by C); lane 2, heat-inactivated NoV proteinase, 3CL-In; lane 3, NoV r3CLpro, 80 ng/μl; lane 4, PV r3Cpro, 40 ng/μl; and lane 5, PV r3Cpro, 80 ng/μl. The HeLa ribosomal fraction (C) was incubated for 3.5 h at 37°C with the following: lane 1, mock control (indicated by C); lane 2, PV r3Cpro, 40 ng/μl; lane 3, PV r3Cpro, 80 ng/μl; and lane 4, NoV r3CLpro, 80 ng/μl. (D) NoV 3CLpro does not cleave eIF4G. Prior to immunoblot analysis with eIF4G antibody, HeLa S10 lysates were incubated for 3.5 h at 37°C with the following: lane 1, mock control (indicated by C); lane 2, PV r3Cpro, 40 ng/μl; lane 3, PV r3Cpro, 80 ng/μl; lane 4, NoV r3CLpro, 40 ng/μl; lane 5, NoV r3CLpro, 80 ng/μl; lane 6, CVB3 r2Apro, 40 ng/μl; and lane 7, CVB3 r2Apro, 80 ng/μl. The 115-kDa N-terminal cleavage fragment of eIF4G is indicated as cpN.
RESULTS
Cleavage of PABP by NoV 3CLpro and comparison with PV 3Cpro.
A comparison of the viral polyprotein dipeptide cleavage sites recognized by the 3Cpro of PV with those of the 3CLpro of two caliciviruses, NoV strain MD145 and vesivirus strain FCV, shows certain similarities and differences (Table 2). Substrate recognition by PV has been shown to depend on the Q/G dipeptide sequence flanking the scissile bond, hydrophobic residues in the P4 position, and conformational constraints (44). In regard to the amino acid residue in the P1 position of the viral polyprotein cleavage site, the PV 3Cpro has a high requirement for Q (37, 44, 51). FCV 3CLpro recognizes E (55), and the NoV 3CLpro recognizes either Q or E (5, 27). We recently showed that PV 3Cpro cleaves PABP at specific Q/G or Q/T amino acid pairs (41) to generate three possible N-terminal cleavage products (cp) designated 3C (61 kDa), 3Calt′ (49 kDa), and 3Calt (45 kDa) (illustrated in Fig. 1A). The cleavage efficiency of PV 3Cpro at the various sites was found to vary depending on the form of the PABP substrate presented to the proteinases. The similarities in dipeptide recognition sites between PV 3Cpro and the NoV 3CLpro prompted us to investigate whether the NoV 3CLpro might also recognize these or similar sites on human PABP.
TABLE 2.
Comparison of viral polyprotein cleavage sites recognized by the cysteine proteinases of two caliciviruses (MD145 and FCV) and a picornavirus (PV) with PABP cleavage sitesa
| Strain (genus) or site | Position in polyprotein of dipeptide cleavage site | Sequence of dipeptide cleavage site and surrounding regionb
|
||||||
|---|---|---|---|---|---|---|---|---|
| P4 | P3 | P2 | P1 | P1′ | P2′ | P3′ | ||
| MD145 (Norovirus) | 330/331 | Y | E | L | Q | G | P | E |
| 696/697 | Y | E | L | Q | G | P | T | |
| 875/876 | I | K | T | E | G | K | K | |
| 1008/1009 | L | S | F | E | A | P | P | |
| 1189/1190 | A | T | L | E | G | G | D | |
| FCV (Vesivirus) | 46/47 | I | R | A | E | A | C | P |
| 331/332 | F | R | S | E | D | V | A | |
| 685/686 | F | E | A | E | N | G | H | |
| 960/961 | P | K | S | E | A | K | G | |
| 1071/1072 | F | A | E | E | S | G | P | |
| PV (Poliovirus) | 341/342 | P | R | L | Q | G | L | P |
| 579/580 | A | L | A | Q | G | L | G | |
| 1030/1031 | A | M | E | Q | G | I | T | |
| 1127/1128 | V | I | K | Q | G | D | S | |
| 1456/1457 | A | L | F | Q | G | P | L | |
| 1543/1544 | A | G | H | Q | G | A | Y | |
| 1565/1566 | A | K | V | Q | G | P | G | |
| 1748/1749 | T | Q | S | Q | G | E | I | |
| PABP 3C site | 534/539 | V | H | V | Q | G | Q | N |
| PABP 3Calt′ site | 434/439 | W | T | A | Q | G | A | R |
| PABP 3Calt site | 410/415 | A | I | P | Q | T | Q | E |
In vitro cleavage reactions were performed in which PABP present in either HeLa S10 cytoplasmic extracts or in isolated ribosome fractions was incubated with similar concentrations of PV 3Cpro, NoV r3CLpro, or PV 2Apro (Fig. 1B and C). When PV 3Cpro was incubated with the HeLa S10 lysate, PABP was cleaved (at about 70% efficiency) in a dose-dependent manner and the 3C cp (61 kDa) was detected along with a minor amount of 3Calt cp (46 kDa) (Fig. 1B, lanes 4 and 5). Consistent with a previous study (41), a proportion of PABP in the S10 fraction remained resistant to cleavage by PV 3Cpro. When NoV r3CLpro was incubated with these fractions, the PABP was cleaved to release a similar cleavage product of about 61 kDa, but at a lower cleavage efficiency than with PV 3Cpro (Fig. 1B, lane 3). The control for this reaction was heat-inactivated NoV proteinase (r3CL-In), which did not cleave PABP (Fig. 1B, lane 2). The activity of the NoV r3Clpro was confirmed by efficient in vitro trans cleavage of a 76-kDa MD145-12 viral Pro−Pol substrate containing an intact cleavage site but an inactivated viral proteinase to prevent autocleavage (see Fig. 5C) (5). It had previously been shown that the pool of PABP associated with the polyribosomes was cleaved more efficiently by PV 3Cpro than in the cytoplasmic S10 extract, and the predominant cleavage product was 3Calt cp (46 kDa) (41). We examined whether the NoV r3CLpro could also cleave ribosome-associated PABP (Fig. 1C). Consistent with the previous study (41), ribosome-associated PABP was cleaved in the PV 3Cpro control experiment in a dose-dependent manner (Fig. 1C, lanes 2 and 3) to yield both the 3C cp and a larger amount of 3Calt cp. Similarly, NoV 3CLpro cleaved PABP more efficiently in the ribosome fractions to yield both 61- and 46-kDa proteins that comigrated with the cleavage products produced by PV 3Cpro (compare Fig. 1B, lane 3 with C, lane 4). At the highest concentration tested (80 ng/μl), both proteinases cleaved the full-length PABP completely (Fig. 1C, lanes 3 and 4). These results suggested that both proteinases specifically target the PABP associated with the translation machinery of the ribosomes more efficiently than the various cytoplasmic forms of PABP and that the NoV 3CLpro likely recognizes the same cleavage sites as PV 3Cpro on PABP. Further, both proteases display a switch in cleavage site preference to the 3Calt site when presented with polysome-associated PABP.
FIG. 5.

Analysis of PABP cleavage in FCV-infected cells and in vitro with the FCV 3CL proteinase (rProPol). (A) Comparison of PABP cleavage between PV-infected HeLa cells and FCV-infected CRFK cells. CRFK cells were either mock or FCV infected and harvested after 6 hpi. HeLa cells were mock or PV infected (MOI of 3) and harvested 6 hpi. An aliquot of FCV-infected lysates (lane 4) was compared to a PV-infected HeLa cell lysate (lane 2) by SDS-10% PAGE followed by immunoblotting with human PABP-specific antibodies. Numbers on the right show the migration of molecular weight markers (in thousands). Previously identified PABP cleavage fragments are indicated (3C, 61-kDa N-terminal cleavage fragment of PABP; 3Calt, 46-kDa N-terminal cleavage fragment of PABP; 2A, 57-kDa N-terminal cleavage fragment of PABP generated by 2Apro; FCV 3CLpro cp, FCV-induced cleavage fragment of PABP). (B) Comparison of PV 3Cpro and FCV 3CLpro in an in vitro cleavage assay of rPABP. RNA transcripts encoding PABP were translated in a reaction mixture containing 35S-labeled methionine. Translation mixtures were then incubated with 10 ng of PV 3Cpro/μl (lane 2) or FCV rProPol (lane 3) at 30°C. The translation mixture incubated without proteinase was used as negative control (indicated by C, lane 1). Marker proteins corresponding to three N-terminal fragments of PABP were produced by translation of three PABP RNAs bearing individual stop codons at positions 537 (PABP537), 487 (PABP487), or 413 (PABP413) are included as a control (lane 4). These correspond to N-terminal PABP cleavage fragments released by cleavage at the 3C, 2A, or 3Calt sites, respectively. (C) Mapping of the FCV 3CLpro cleavage site on PABP by site-directed mutagenesis of the Q413/T414, Q437/T438, and Q537/G538 dipeptides. For each construct indicated above the gel (PABP, Q413A, Q437A, and Q537A), 4 μl of a 25-μl reaction mixture was either incubated with 6 μl of PBS, pH 7.4, (odd-numbered lanes and minus signs) or 6 μl of PBS containing 5 μg of FCV 3CL (rProPol) (even-numbered lanes and plus signs). Reaction mixtures were incubated overnight at 30°C prior to separation in a 12% Tris-Gly polyacrylamide gel and autoradiography. To assess calicivirus protease activity, a MD145-12 NoV Pro−Pol substrate (76 kDa) was translated and incubated alone or with proteases similarly (lanes 9 to 11). Molecular weight markers (in thousands) are indicated on the left. (D) FCV 3CL inhibits cellular mRNA translation in vitro. HeLa lysates were treated with buffer (indicated by C) or increasing amounts of FCV 3CL (10, 20, and 40 ng/μl) as described in Materials and Methods. An autoradiogram of SDS-PAGE is shown and percent translation quantified by densitometry is shown below the lanes. Migration of molecular mass standards (in kDa) is shown on the right.
eIF4G is not cleaved by NoV 3CLpro.
Certain picornavirus proteinases cleave translation initiation factor eIF4GI, and this cleavage is involved also in the mechanism responsible for the shutoff of host cell translation in virus-infected cells (18). During PV, coxsackievirus, or rhinovirus infection, the cleavage of eIF4GI is mediated by 2Apro and cellular proteases (10, 43, 64), and for foot-and-mouth disease virus, it is mediated by the leader proteinase Lpro (36). Because the caliciviruses apparently do not encode a 2A-like proteinase, we examined the ability of the NoV r3CLpro to cleave eIF4G (Fig. 1D). The HeLa S10 lysates were incubated with the PV 3C, CVB3 2A, or NoV 3CL proteinases and then analyzed in an immunoblot developed with anti-eIF4GI antibodies. A control was included without proteinase to illustrate the presence of intact eIF4GI in HeLa cell lysates. As expected, eIF4GI was cleaved completely by CVB3 2Apro, and the typical 110- to 130-kDa N-terminal cleavage fragments were generated (Fig. 1D, lanes 6 and 7). In contrast, neither PV 3Cpro or NoV 3CLpro (at the same concentrations used in Fig. 1B and C) cleaved eIF4GI (Fig. 1D, lanes 2 to 5). FCV rProPol also did not cleave HeLa eIF4GI when incubated under similar conditions (data not shown). These results indicate that the NoV r3CLpro does not cleave human eIF4GI in vitro, despite the existence of several potential dipeptide cleavage site sequences in eIF4GI (data not shown).
NoV 3CLpro inhibits endogenous mRNA translation in HeLa translation extracts.
We examined whether the observed cleavage of PABP by NoV r3CLpro might affect protein synthesis in a HeLa lysate-based translation assay. We used non-nuclease-treated HeLa translation extracts in order to preserve competition for ribosomes from endogenous mRNAs and retain cap-poly(A) synergy. As previously reported, poly(A) tails on mRNA could stimulate translation 5- to 10-fold compared to nonpolyadenylated counterparts in this system, effectively mimicking in vivo cap-poly(A) synergy (6). Viral proteinases were preincubated with translation extracts before testing their effects on translation of endogenous HeLa mRNA (Fig. 2). Preincubation with rNoV 3CL resulted in a dose-dependent inhibition of translation as determined by the analysis of newly synthesized radiolabeled proteins (Fig. 2, lanes 2 to 4), whereas inactivated rNoV 3CL only slightly decreased translation (lane 5). Preincubation of the lysate with a high concentration of NoV r3CLpro that cleaved over half the ribosome-associated PABP (80 ng/μl) reduced translation about 60% compared to the buffer-treated control (Fig. 2). The level of translation inhibition induced by NoV 3CL was similar to the inhibition resulting from incubation with PV 3Cpro (lane 7). These data indicate that NoV r3CLpro is associated with inhibition of translation in this in vitro assay and that it can mediate this effect in the absence of other viral proteins.
FIG. 2.
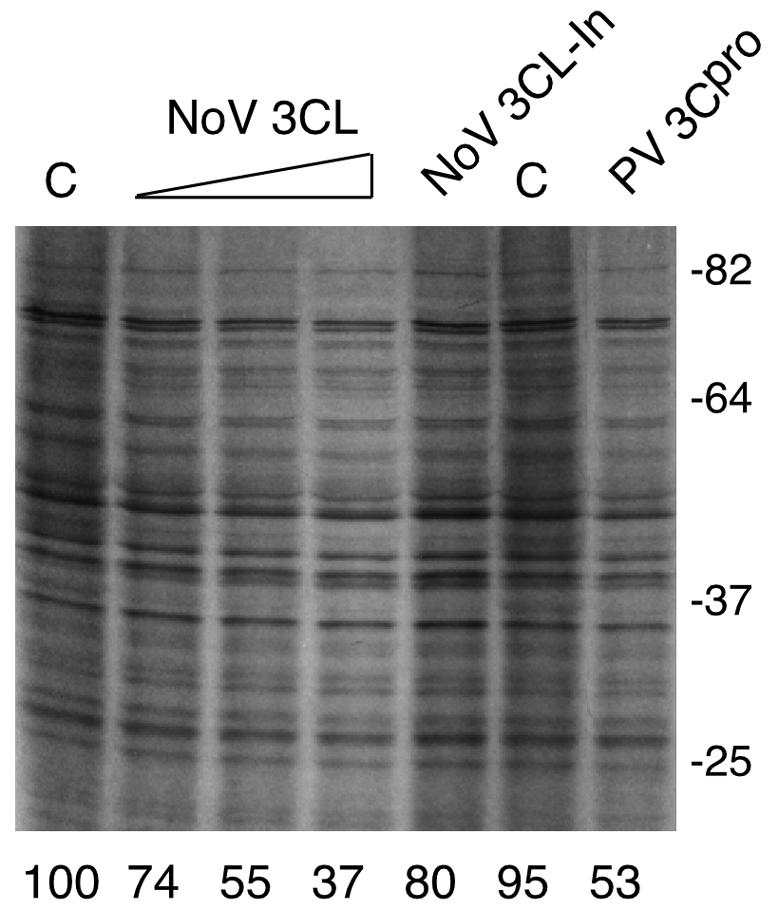
Inhibition of cellular mRNA translation by NoV r3CLpro. Autoradiogram of a 10% (wt/vol) polyacrylamide SDS-PAGE gel showing [35S]methionine-cysteine incorporation into newly synthesized proteins in HeLa translation extracts that were treated with buffer only (lanes marked C), increasing concentrations of NoV r3L Cpro at 20, 40, or 80 ng/μl (lanes 2 to 4, respectively), inactivated NoV r3CL (lane 5), or PV r3CLpro at 60 ng/μl (lane 7). Migration of molecular mass standards (in kilodaltons) is shown on the right. Relative translation intensity compared to control was determined by densitometry (percentage of control) and is shown below each lane.
PABP cleavage by 3CLpro inhibits poly(A)-dependent translation.
PABP/poly(A)-dependent translation stimulation is demonstrated most effectively using non-nuclease-treated yeast or HeLa translation extracts, and depletion of PABP in these extracts affects only the translation of polyadenylated RNAs (6, 59). Our next experiments examined whether the inhibition of cellular translation by NoV 3CLpro was associated with a poly(A)/PABP-dependent mechanism. RNA transcripts (with or without polyadenylation) encoding firefly luciferase were generated in vitro for use as a reporter for translation in HeLa lysates. Figure 3A (left panel) shows that preincubation of lysates with NoV r3CLpro inhibited translation of polyadenylated RNAs in a dose-dependent manner. When nonpolyadenylated luciferase RNA was examined, the overall translation rates were approximately fourfold lower than polyadenylated RNA, showing poly(A) stimulation to be functional in these extracts (Fig. 3A, right panel). Addition of NoV r3CLpro did not significantly affect translation levels of nonpolyadenylated RNAs (Fig. 3A, right panel).
FIG. 3.

NoV 3CLpro specifically inhibits poly(A)-dependent translation, and translation is restored by the addition of PABP. (A) Effect of NoV 3CLpro on translation efficiency of polyadenylated mRNA. Graphic representation of luciferase activity generated (relative light units [RLU] set to 100% of control reactions) using capped and polyadenylated luciferase RNA (left panel) or nonpolyadenylated luciferase RNA (right panel) in HeLa translation extracts incubated with increasing concentrations of NoV r3CLpro. Data represent the means ± standard deviations of four individual experiments. (B) Addition of exogenous PABP restores translation efficiency of polyadenylated mRNA in the presence of NoV 3CLpro. Comparison of polyadenylated (left panel) or nonpolyadenylated (right panel) luciferase RNA translation in HeLa translation extracts pretreated with buffer or 10 ng of NoV r3CLpro/μl and supplemented with His-PABP. Black bars represent the percent translation (light units relative to buffer control) in 3CLpro-treated lysates. Gray bars indicate luciferase translation levels after addition of His-PABP. Data represent the means ± standard deviations of three individual experiments.
To test whether 3CLpro-mediated translation inhibition was reversible and linked to PABP cleavage, 3CLpro-treated extracts were supplemented with purified recombinant His-PABP. Increasing concentrations of His-PABP were incubated together with a constant amount of either polyadenylated or nonpolyadenylated RNA. This RNA/protein complex was used to supplement extracts pretreated with 3CLpro. As before, 10 ng of 3CLpro/μl significantly inhibited reporter RNA translation (Fig. 3B, left panel). Addition of increasing concentrations of His-PABP restored translation in extracts pretreated with 3CLpro in a dose-dependent manner (Fig. 3B, left panel). 3CLpro did not affect translation of nonpolyadenylated luciferase RNA (Fig. 3B, right panel), and addition of His-PABP did not stimulate translation of nonpolyadenylated luciferase RNA after NoV 3CLpro treatment. Collectively, the results show that PABP supplementation restores poly(A)-dependent translation after inhibition by 3CLpro. This restoration is specific for polyadenylated mRNAs because PABP supplementation did not stimulate translation of non-poly(A) mRNAs. Furthermore, the data support the hypothesis that NoV r3CLpro-mediated translation inhibition is PABP dependent.
Evidence for shutoff of host cell protein synthesis during FCV infection.
Although the in vitro data showed evidence for cleavage of human PABP by the NoV r3CLpro, the absence of a cell culture system precluded testing of PABP cleavage during NoV infection. Because FCV grows well in cell culture, we examined whether there was indeed evidence for a decrease in cellular protein synthesis during calicivirus infection. CRFK cells were infected with FCV (or mock infected) and pulse-labeled at hourly time points beginning at 3 h postinfection (hpi) (Fig. 4). Analysis of the FCV-infected cell lysates by SDS-PAGE and autoradiography showed a visible increase in radiolabeled virus-specific proteins over time and a decrease in the accumulation of cellular proteins when compared to the mock-infected control cell lysates (Fig. 4A). Measurement and comparison of the signals generated by representative cellular proteins present in mock- or FCV-infected cells showed that FCV infection was associated with a decrease in cellular translation beginning at 5 hpi (Fig. 4B). In comparison, the FCV capsid protein showed a marked increase at 5 hpi and peaked in intensity at 6 hpi (Fig. 4B). This result was consistent with the presence of a mechanism for the preferential translation of viral proteins at the expense of host cell translation during calicivirus replication.
FIG. 4.

Comparison of proteins produced during FCV infection with those produced in mock-infected cells over time. (A) Analysis of proteins by SDS-PAGE and autoradiography. Feline kidney cells (4 × 106 cells) were either infected with FCV at an MOI of 10 or mock infected. After 3 h and then at subsequent hourly time points, medium was removed and the cells were pulse-radiolabeled for 1.5 h. The cells were then collected, washed, and boiled in SDS sample buffer prior to analysis by SDS-PAGE in a 12% polyacrylamide gel and autoradiography. Viral proteins ProPol, capsid, p39, and p32 are indicated. The asterisks illustrate two cell-associated proteins that decrease in intensity over time during FCV infection. (B) Phosphorimager quantitation of signals associated with selected cellular and viral proteins. Bands 1 and 2 correspond to the cellular proteins indicated with an asterisk in panel A, with band 1 corresponding to the slower migrating protein. The signals generated by the cellular proteins were compared to that of the 60-kDa mature FCV capsid protein. RLU, relative light units.
PABP is cleaved in FCV-infected cells.
We then used the FCV cell culture system to examine whether cleavage of PABP occurred during infection. Although the NoV r3CLpro clearly cleaved PABP and caused an inhibition of translation in in vitro assays, the absence of PABP cleavage in an authentic calicivirus infection would argue against its biological relevance. It should be noted that although PABP is highly conserved across species, the feline PABP has not yet been characterized. Mock- or virus-infected feline CRFK cell lysates were analyzed by immunoblotting with anti-human PABP antibodies (Fig. 5A). The anti-human PABP antibodies detected proteins consistent with PABP in the mock-infected feline cell lysates (Fig. 5A, lane 3), which then allowed us to examine PABP cleavage in FCV-infected cells. A mock- or PV-infected HeLa cell lysate were included in the immunoblot as controls (Fig. 5A, lanes 1 and 2, respectively). The PV-infected cell lysate control contained the two predominant PABP N-terminal cleavage products (the 61-kDa 3C cp and 46-kDa 3Calt cp) derived by 3Cpro cleavage and the previously described N-terminal cleavage product (55 kDa) of PABP generated by 2Apro cleavage. In contrast, a protein of approximately 50 kDa that corresponded to a potential PABP cleavage product was detected in the FCV-infected cell lysate (Fig. 5A, lane 4). Cleavage of feline eIF4GI could not be determined because of a lack of reactivity with anti-human eIF4GI antiserum (data not shown).
It was possible that the difference in size of the major PABP cleavage product detected in FCV-infected cells was due to amino acid sequence diversity between PABPs from different species. We examined whether FCV 3CLpro (in the form of rProPol) could cleave human PABP (Fig. 5B). In vitro cleavage reactions were assembled by incubating FCV rProPol or rPV 3Cpro with radiolabeled human PABP and the resulting products were compared directly. The rPV 3Cpro released the expected 61-kDa 3C cp (Fig. 5B, lane 2) that migrated similarly to the 61-kDa N-terminal PABP fragment produced by translation of a PABP construct engineered with a stop codon at the PABP 537 cleavage site (Fig. 5B, lane 4). The FCV 3CLpro cleaved human PABP to generate a 50-kDa cleavage product (Fig. 5B, lane 3) that was similar in size to the feline PABP cleavage product detected in feline kidney cells. This result suggested marked conservation of amino acid sequence between the human and feline PABP, consistent with the observed cross-reactivity of human PABP antibodies with feline PABP noted above. However, this 50-kDa cleavage product produced by FCV rProPol did not comigrate with PABP fragments truncated at the 3Calt, 2A, or 3C sites (lane 4). It should be noted that the size of the observed FCV 3CLpro PABP cleavage product was consistent with that of the previously described 49-kDa 3Calt′, a minor cleavage product that has been observed only occasionally in studies of PV 3Cpro (41). In addition, the FCV 3CLpro characteristically recognizes dipeptide cleavage sites with an E in the P1 position when processing viral proteins, although mutational analysis previously indicated that the FCV 3CLpro was not restricted to this sequence (57). To further map the cleavage site, wild-type and mutant forms of human PABP were incubated with FCV rProPol in an in vitro cleavage assay (Fig. 5C). The FCV rProPol cleaved wild-type PABP efficiently to yield two proteins of approximately 50 and 28 kDa (Fig. 5C, lane 2), with the 50-kDa protein consistent in size with the cleavage product observed in FCV-infected cells. Mutation of the PABP 3Calt′ cleavage site at position 437 (Q to A) abolished cleavage by FCV rProPol (Fig. 5C, lane 6), suggesting that the FCV 3CLpro recognized this site preferentially over those utilized predominantly by PV 3Cpro, and likely, NoV 3CLpro. When FCV rProPol (3CL) was incubated with HeLa translation lysates, a dose-dependent decrease in mRNA translation was observed, although the levels of inhibition were slightly less than that observed with NoV 3CL (Fig. 5D). This suggests that cleavage of PABP at the alternate 3Calt′ site also results in a similar inhibition of translation.
Finally, we investigated the kinetics of PABP cleavage in FCV-infected cells. Immunoblot analysis of infected CRFK cells at time points during FCV infection revealed that PABP cleavage was detected initially at 5 hpi and was maximal at 7 hpi (Fig. 6A). We also mixed infected cell extracts with radiolabeled human PABP generated in in vitro TNT reaction extracts and detected PABP cleavage activity at 5 hpi that peaked at 7 hpi, closely correlating with PABP cleavage and levels of ProPol expression in infected cells (Fig. 4). Thus, the kinetics of PABP cleavage paralleled translation inhibition, both beginning at 5 hpi and becoming maximal at 7 hpi. Further, the inhibition of translation in FCV-infected cells correlated with partial PABP cleavage, similar to what has been reported previously in PV-infected cells (32, 41).
FIG. 6.

Kinetic analysis of PABP cleavage in FCV-infected cells. CRFK cells were infected with FCV as described previously, and cells were isolated at hourly time points (indicated above lanes) postinfection for analysis. (A) Immunoblot analysis of feline PABP over time in FCV-infected cells detected with antibodies raised against human PABP. An analysis of mock-infected cells harvested after 8 h incubation is shown (M). (B) Analysis of FCV-infected cell lysates in the in vitro assay for human PABP cleavage. Aliquots of infected cell extracts from time points 1 to 8 hpi were mixed with radiolabeled human PABP and incubated for 3.5 h at 30°C. Controls include radiolabeled PABP incubated with recombinant FCV ProPol enzyme, mock-infected cell extract (M), and buffer alone (C). Autoradiogram of SDS-polyacrylamide gel is shown. Arrowheads indicate PABP cleavage products. Migration of molecular weight markers is indicated on the right. The human PABP-specific antibody used in the immunoblot (A) is peptide-specific (sequence located in RRM4) and recognizes only the amino-terminal cleavage product of PABP.
DISCUSSION
Viruses require the use of the host cell translation machinery to produce viral proteins in an infected cell. Thus, most viruses have evolved with strategies to effectively compete with cellular mRNAs for ribosomes and a limited number of translation initiation factors. The mechanisms by which caliciviruses manipulate cellular translation had not been established. Here, we report evidence that host cell translation is inhibited during calicivirus replication and demonstrate that PABP, a key protein involved in the translation of polyadenylated cellular mRNAs, can be cleaved and apparently inactivated by the calicivirus 3CLpro. The NoV 3CLpro cleaved PABP similarly to PV 3Cpro, including a recognition of PABP associated with ribosomes that is more efficient than for PABP associated with a less-purified cytoplasmic fraction. This observation indicates that both enzymes selectively target PABP associated with the translation apparatus, possibly to inhibit PABP function in translation. We recently reported that PV 3Cpro selectively inhibits poly(A)-dependent translation by removal of the CTD of PABP (40). This cleavage inhibits translation by a unique mechanism without affecting PABP interactions with eIF4GI or the poly(A) tail, since N-terminal cleavage fragments of PABP retain intact RRMs that bind RNA (40). It was proposed that cleavage of PABP likely inhibits translation by disrupting interactions with eIF4B and eRF3 and possibly affects late events in translation such as ribosome recycling (40). The striking similarity in the cleavage specificities between the PV and NoV proteinases suggests that study of both virus systems could be useful in further defining common themes used by these viruses to manipulate cellular translation.
We examined a cultivatable calicivirus, FCV, to verify the occurrence of cellular PABP cleavage during replication. Antibodies generated against human PABP cross-reacted with feline PABP, enabling the identification of a PABP cleavage fragment in FCV-infected cell lysates. Of interest, the FCV 3CLpro (rProPol) cleaved both feline and human PABP similarly, and the cleavage site was provisionally mapped to a site on human PABP rarely utilized by PV 3Cpro. This alternate cleavage site (3Calt′) is only 24 amino acids away from the 3Calt site, thus a similar effect on PABP function would likely occur with cleavage by either viral proteinase. Examination of cleavage site sequences does not reveal an obvious reason for the differential selection of PABP cleavage sites by the various proteinases. PV 3Cpro cleavage depends on both conformational constraints and primary sequence which includes a preference for A/V-X-X-Q/G in the P4 through P1′ positions (44). The sequences surrounding the three PV 3Cpro cleavage sites on PABP are different; however, all contain Q/G or Q/T dipeptides (Table 2) (41). NoV 3CL recognizes amino acid pairs with E or Q in the P1 position (45) and, therefore, might share specificity for Q/G pairs in PABP with PV 3Cpro. The cleavage pair recognized in human PABP by FCV 3CL is likely Q437/G, although FCV 3CL apparently prefers cleavage sites with E (Table 2). However, its ability to recognize cleavage sites with Q in the P1 position has been also demonstrated (57). In addition, comparison of the sequences adjacent to known cleavage sites of FCV showed a preference for a large, hydrophobic amino acid residue in the P4 position (55), and the presence of a tryptophan residue in the P4 position of the PABP Q437/G cleavage site may provide further explanation for efficient recognition of this particular site by FCV 3CL (Table 2). Another factor in this recognition could be the folding of the PABP that provides the FCV 3CL proteinase access to this particular cleavage site. It is likely that the cleavage site selection by these proteases requires conformational constraints and perhaps even unknown cofactors. Despite the differences in PABP cleavage site selection, the ability of FCV to cleave PABP strongly suggests that this translation inhibition mechanism occurs during an authentic calicivirus infection. It may prove useful to develop homologous reagents for further study of the effect of FCV replication on cellular proteins in permissive feline cells.
Similar to PV 3Cpro, the NoV r3CLpro did not cleave eIF4G in an in vitro assay. It remains possible that eIF4GI may be cleaved during NoV infection by a viral or cellular proteinase, but our in vitro studies suggest that its cleavage is not essential for the translation inhibition associated with the PABP cleavage. We could not rule out the absence of cleavage of eIF4GI during FCV infection because the anti-human and -mouse eIF4G antibodies we tested did not recognize feline eIF4G in CRFK cells (data not shown). Furthermore, it should be noted that FCV (2, 56) and other caliciviruses such as RHDV (33) induce apoptosis in infected cells, and this process could also affect the cellular translation machinery.
The mechanism of viral mRNA translation initiation during calicivirus infection remains unclear. The calicivirus genome is capable of serving as an mRNA but lacks either a 5′-end cap structure or any identified internal ribosomal entry site. The genome contains a VPg protein covalently linked to the 5′ ends of both the genome and the subgenomic RNAs (13, 48), which may play a role in translation initiation by recruiting the 40S ribosomal subunit through interactions with translation initiation factor eIF3 (17, 28). In addition, the calicivirus VPg itself contains sequence homology with certain translation initiation factors, indicating that it might directly compete with cellular host factors for binding to the ribosome (57). Both genomic and subgenomic calicivirus RNAs possess a poly(A) tail at the 3′ end, and a recent study has shown that the 3′-end nontranslated region of the Norwalk virus genome with a poly(A) tail of 24 nt binds directly to PABP (25). However, it is not clear whether PABP binding of the calicivirus poly(A) tail stimulates translation of viral mRNAs since there is no 5′ cap structure. PABP may interact with VPg at the 5′ end of calicivirus RNA in an analogous fashion; however, this has not been tested. Since the poly(A) tail itself stimulates translation of capped and internal ribosomal entry site-containing mRNAs, it will be important to determine whether poly(A) tails stimulate VPg-dependent translation of calicivirus mRNAs. However, if the poly(A) tail is indeed important for viral mRNA translation, it might be predicted that 3CLpro-mediated cleavage of PABP could also inhibit viral protein synthesis. A mechanism would be needed to allow discrimination between polyadenylated cellular and viral RNAs in infected cells so that host cell mRNAs are selectively inhibited during early phases of infection. It is possible that cleavage of PABP may also be a requirement for the switch between translation and RNA replication, converting the viral genome from a translation-competent to replication-competent status. The elucidation of these mechanisms for both caliciviruses and picornaviruses will be relevant to the understanding of how viral messages are translated at such high efficiency in infected cells and yet allow RNA replication to begin.
In summary, we show that calicivirus 3CLpro cleaves PABP in vitro and during infection. We propose that the calicivirus 3CLpro specifically targets a pool of PABP involved in cellular protein translation. Furthermore, this cleavage likely inhibits cellular mRNA translation by removal of CTD from PABP independently of eIF4G cleavage. Cleavage of PABP may be a strategy used by caliciviruses to down-regulate cellular mRNA translation in infected cells to more effectively synthesize viral proteins. The cleavage of PABP by viral proteinases may be a common mechanism used by different viruses to gain control of host cell mRNA translation, and this may be crucial for the successful life cycle of these RNA viruses.
Acknowledgments
This work was supported by NIH grants AI50237 and GM59803 to R.E.L.
We thank P. Younan and C. Rivera for technical assistance.
REFERENCES
- 1.Allaire, M., M. M. Chernaia, B. A. Malcolm, and M. N. G. James. 1994. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature 369:72-76. [DOI] [PubMed] [Google Scholar]
- 2.Al-Molawi, N., V. A. Beardmore, M. J. Carter, G. E. Kass, and L. O. Roberts. 2003. Caspase-mediated cleavage of the feline calicivirus capsid protein. J. Gen. Virol. 84:1237-1244. [DOI] [PubMed] [Google Scholar]
- 3.Asselbergs, F., W. Peters, W. van Venrooij, and H. Bloemendal. 1978. Diminished sensitivity of re-initiation of translation to inhibition by cap analogues in reticulocyte lysates. Eur. J. Biochem. 88:483-488. [DOI] [PubMed] [Google Scholar]
- 4.Bazan, J. F., and R. J. Fletterick. 1988. Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc. Natl. Acad. Sci. USA 85:7872-7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belliot, G., S. V. Sosnovtsev, T. Mitra, C. Hammer, M. Garfield, and K. Y. Green. 2003. In vitro proteolytic processing of the MD145 norovirus ORF1 nonstructural polyprotein yields stable precursors and products similar to those detected in calicivirus-infected cells. J. Virol. 77:10957-10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergamini, G., T. Preiss, and M. W. Hentze. 2000. Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA 6:1781-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bergmann, E. M., S. C. Mosimann, M. M. Chernaia, B. A. Malcolm, and M. N. G. James. 1997. The refined crystal structure of the 3C gene product from hepatitis A virus: specific proteinase activity and RNA recognition. J. Virol. 71:2436-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein, H. D., N. Sonenberg, and D. Baltimore. 1985. Poliovirus mutant that does not selectively inhibit host cell protein synthesis. Mol. Cell. Biol. 5:2913-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boniotti, B., C. Wirblich, M. Sibilia, G. Meyers, H. J. Thiel, and C. Rossi. 1994. Identification and characterization of a 3C-like protease from rabbit hemorrhagic disease virus, a calicivirus. J. Virol. 68:6487-6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bovee, M. L., W. E. Marissen, M. Zamora, and R. E. Lloyd. 1998. The predominant eIF4G-specific cleavage activity in poliovirus-infected HeLa cells is distinct from 2A protease. Virology 245:229-240. [DOI] [PubMed] [Google Scholar]
- 11.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 12.Brown, B. A., and E. Ehrenfeld. 1979. Translation of poliovirus RNA in vitro: changes in cleavage pattern and initiation sites by ribosomal salt wash. Virology 97:396-405. [DOI] [PubMed] [Google Scholar]
- 13.Burroughs, J. N., and F. Brown. 1978. Presence of a covalently linked protein on calicivirus RNA. J. Gen. Virol. 41:443-446. [DOI] [PubMed] [Google Scholar]
- 14.Bushell, M., L. McKendrick, R. U. Janicke, M. J. Clemens, and S. J. Morley. 1999. Caspase-3 is necessary and sufficient for cleavage of protein synthesis eukaryotic initiation factor 4G during apoptosis. FEBS Lett. 451:332-336. [DOI] [PubMed] [Google Scholar]
- 15.Chris, A. 2003. Norwalk-like viruses: when the runs can slow you down. Can. Med. Assoc. J. 168:64-67. [PMC free article] [PubMed] [Google Scholar]
- 16.Craig, A. W. B., A. Haghighat, A. T. K. Yu, and N. Sonenberg. 1998. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 392:520-523. [DOI] [PubMed] [Google Scholar]
- 17.Daughenbaugh, K. F., C. S. Fraser, J. W. Hershey, and M. E. Hardy. 2003. The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 22:2852-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etchison, D., S. C. Milburn, I. Edery, N. Sonenberg, and J. W. B. Hershey. 1982. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eukaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 257:14806-14810. [PubMed] [Google Scholar]
- 19.Gorlach, M., C. G. Burd, and G. Dreyfuss. 1994. The mRNA poly(A)-binding protein: localization, abundance, and RNA-binding specificity. Exp. Cell Res. 211:400-407. [DOI] [PubMed] [Google Scholar]
- 20.Gradi, A., Y. V. Svitkin, H. Imataka, and N. Sonenberg. 1998. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. USA 95:11089-11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green, K. Y., M. S. Ando, M. S. Balayan, T. Berke, I. N. Clarke, M. K. Estes, D. O. Matson, S. Nakata, J. D. Neill, M. J. Studdert, and H. J. Thiel. 2000. Taxonomy of the caliciviruses. J. Infect. Dis. 181:S322-S330. [DOI] [PubMed] [Google Scholar]
- 22.Green, K. Y., G. Belliot, J. L. Taylor, J. Valdesuso, J. F. Lew, A. Z. Kapikian, and F.-Y. Lin. 2002. A predominant role for Norwalk-like viruses as agents of epidemic gastroenteritis in Maryland nursing homes for the elderly. J. Infect. Dis. 185:133-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green, K. Y., R. M. Chanock, and A. Z. Kapikian. 2001. Human caliciviruses, p. 841-874. In D. Knipe and P. Howley (ed.), Fields virology. Lippincott, Williams, and Wilkins, Philadelphia, Pa.
- 24.Green, K. Y., A. Mory, M. H. Fogg, A. Weisberg, G. Belliot, M. Wagner, T. Mitra, E. Ehrenfeld, C. E. Cameron, and S. V. Sosnovtsev. 2002. Isolation of enzymatically active replication complexes from feline calicivirus-infected cells. J. Virol. 76:8582-8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutierrez-Escolano, A. L., M. Vazquez-Ochoa, J. Escobar-Herrera, and J. Hernandez-Acosta. 2003. La, PTB, and PAB proteins bind to the 3′ untranslated region of Norwalk virus genomic RNA. Biochem. Biophys. Res. Commun. 311:759-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardy, M. E. 1999. Norwalk and “Norwalk-like viruses” in epidemic gastroenteritis. Clin. Lab. Med. 19:675-690. [PubMed] [Google Scholar]
- 27.Hardy, M. E., T. J. Crone, J. E. Brower, and K. Ettayebi. 2002. Substrate specificity of the Norwalk virus 3C-like proteinase. Virus Res. 89:29-39. [DOI] [PubMed] [Google Scholar]
- 28.Herbert, T. P., I. Brierley, and T. D. Brown. 1997. Identification of a protein linked to the genomic and subgenomic mRNAs of feline calicivirus and its role in translation. J. Gen. Virol. 78:1033-1040. [DOI] [PubMed] [Google Scholar]
- 29.Hoshino, S., M. Imai, T. Kobayashi, N. Uchida, and T. Katada. 1999. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J. Biol. Chem. 274:16677-16680. [DOI] [PubMed] [Google Scholar]
- 30.Imataka, H., A. Gradi, and N. Sonenberg. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 17:7480-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson, A. 1996. Poly(A) metabolism and translation: the closed-loop model, p. 451-479. In J. W. B. Hershey, M. B. Mathews, and N. Sonenberg (ed.), Translational control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 32.Joachims, M., P. C. van Breugel, and R. E. Lloyd. 1999. Cleavage of poly(A)-binding protein by enterovirus proteases concurrent with inhibition of translation in vitro. J. Virol. 73:718-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung, J., B. Lee, J. Tai, J. Park, and Y. Lee. 2000. Apoptosis in rabbit haemorrhagic disease. J. Comp. Pathol. 123:135-140. [DOI] [PubMed] [Google Scholar]
- 34.Kerekatte, V., B. D. Keiper, C. Bradorff, A. Cai, K. U. Knowlton, and R. E. Rhoads. 1999. Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J. Virol. 73:709-717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khaleghpour, K., Y. V. Svitkin, A. W. Craig, C. T. DeMaria, R. C. Deo, S. K. Burley, and N. Sonenberg. 2001. Translational repression by a novel partner of human poly(A) binding protein, Paip2. Mol. Cell 7:205-216. [DOI] [PubMed] [Google Scholar]
- 36.Kirchweger, R., E. Ziegler, B. J. Lamphear, D. Waters, H. D. Liebig, W. Sommergruber, F. Sobrino, C. Hohenadl, D. Blaas, R. E. Rhoads, and T. Skern. 1994. Foot-and-mouth disease virus leader proteinase: purification of the lb form and determination of its cleavage site on eIF-4 gamma. J. Virol. 68:5677-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitamura, N., B. L. Semler, P. G. Rothberg, G. R. Larsen, C. J. Adler, A. J. Dorner, E. A. Emini, R. Hanecak, J. J. Lee, S. van der Werf, C. W. Anderson, and E. Wimmer. 1981. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature 291:547-553. [DOI] [PubMed] [Google Scholar]
- 38.Kozlov, G., J.-F. Trempe, K. Khaleghpour, A. Kahvejian, I. Ekiel, and K. Gehring. 2001. Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc. Natl. Acad. Sci. USA 98:4409-4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krausslich, H. G., M. J. H. Nicklin, H. Toyoda, D. Etchison, and E. Wimmer. 1987. Poliovirus proteinase 2A induces cleavage of eukaryotic initiation factor 4F polypeptide p220. J. Virol. 61:2711-2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuyumcu-Martinez, N. M., M. E. Van Eden, P. Younan, and R. E. Lloyd. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuyumcu-Martinez, N. M., M. Joachims, and R. E. Lloyd. 2002. Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J. Virol. 76:2062-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamphear, B. J., R. Kirchweger, T. Skern, and R. E. Rhoads. 1995. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases—implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem. 270:21975-21983. [DOI] [PubMed] [Google Scholar]
- 43.Lamphear, B. J., R. Q. Yan, F. Yang, D. Waters, H. D. Liebig, H. Klump, E. Kuechler, T. Skern, and R. E. Rhoads. 1993. Mapping the cleavage site in protein synthesis initiation factor-eIF-4γ of the 2A proteases from human coxsackievirus and rhinovirus. J. Biol. Chem. 268:19200-19203. [PubMed] [Google Scholar]
- 44.Leong, L. E.-C., C. T. Cornell, and B. L. Semler. 2002. Processing determinants and functions of cleavage products of picornavirus polyproteins, p. 187-197. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, D.C.
- 45.Liu, B. L., I. N. Clarke, and P. R. Lambden. 1996. Polyprotein processing in Southampton virus: identification of 3C-like protease cleavage sites by in vitro mutagenesis. J. Virol. 70:2605-2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lloyd, R. E., M. J. Grubman, and E. Ehrenfeld. 1988. Relationship of p220 cleavage during picornavirus infection to 2A proteinase sequences. J. Virol. 62:4216-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthews, D. A., W. W. Smith, R. A. Ferre, B. Condon, G. Budahazi, W. Sisson, J. E. Villafranca, C. A. Janson, H. E. Mcelroy, C. L. Gribskov, and S. Worland. 1994. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77:761-771. [DOI] [PubMed] [Google Scholar]
- 48.Meyers, G., C. Wirblich, and H. J. Thiel. 1991. Genomic and subgenomic RNAs of rabbit hemorrhagic disease virus are both protein-linked and packaged into particles. Virology 184:677-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosimann, S. C., M. M. Cherney, S. Sia, S. Plotch, and M. James. 1997. Refined x-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 273:1032-1047. [DOI] [PubMed] [Google Scholar]
- 50.Nietfeld, W., H. Mentzel, and T. Pieler. 1990. The Xenopus laevis poly(A) binding protein is composed of multiple functionally independent RNA binding domains. EMBO J. 9:3699-3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pallansch, M. A., O. M. Kew, B. L. Semler, D. R. Omilianowski, C. W. Anderson, E. Wimmer, and R. R. Ruekert. 1984. Protein processing map of poliovirus. J. Virol. 49:873-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sachs, A. B., and G. Varani. 2000. Eukaryotic translation initiation: there are (at least) two sides to every story. Nat. Struct. Biol. 7:356-361. [DOI] [PubMed] [Google Scholar]
- 53.Someya, Y., N. Takeda, and T. Miyamura. 2002. Identification of active-site amino acid residues in the Chiba virus 3C-like protease. J. Virol. 76:5949-5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sosnovtsev, S., and K. Y. Green. 1995. RNA transcripts derived from a cloned full-length copy of the feline calicivirus genome do not require VPg for infectivity. Virology 210:383-390. [DOI] [PubMed] [Google Scholar]
- 55.Sosnovtsev, S. V., M. Garfield, and K. Y. Green. 2002. Processing map and essential cleavage sites of the nonstructural polyprotein encoded by ORF1 of the feline calicivirus genome. J. Virol. 76:7060-7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sosnovtsev, S. V., E. A. Prikhod'ko, G. Belliot, J. I. Cohen, and K. Y. Green. 2003. Feline calicivirus replication induces apoptosis in cultured cells. Virus Res. 94:1-10. [DOI] [PubMed] [Google Scholar]
- 57.Sosnovtsev, S. V., S. A. Sosnovtseva, and K. Y. Green. 1998. Cleavage of the feline calicivirus capsid precursor is mediated by a virus-encoded proteinase. J. Virol. 72:3051-3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sosnovtseva, S. A., S. V. Sosnovtsev, and K. Y. Green. 1999. Mapping of the feline calicivirus proteinase responsible for autocatalytic processing of the nonstructural polyprotein and identification of a stable proteinase-polymerase precursor protein. J. Virol. 73:6626-6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tarun, S. Z., Jr., S. E. Wells, J. A. Deardorff, and A. B. Sachs. 1997. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proc. Natl. Acad. Sci. USA 94:9046-9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tarun, S. Z., and A. B. Sachs. 1996. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J. 15:7168-7177. [PMC free article] [PubMed] [Google Scholar]
- 61.Uchida, N., S.-I. Hoshino, H. Imataka, N. Sonenberg, and T. Katada. 2002. A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in cap/poly(A)-dependent translation. J. Biol. Chem. 277:50286-50292. [DOI] [PubMed] [Google Scholar]
- 62.Wei, C. C., M. L. Balasta, J. H. Ren, and D. J. Goss. 1998. Wheat germ poly(a) binding protein enhances the binding affinity of eukaryotic initiation factor 4f and (iso)4f for cap analogues. Biochemistry 37:1910-1916. [DOI] [PubMed] [Google Scholar]
- 63.Wells, S. E., P. E. Hillner, R. D. Vale, and A. B. Sachs. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 2:135-140. [DOI] [PubMed] [Google Scholar]
- 64.Zamora, M., W. E. Marissen, and R. E. Lloyd. 2002. Multiple eIF4GI-specific protease activities present in uninfected and poliovirus-infected cells. J. Virol. 76:165-177. [DOI] [PMC free article] [PubMed] [Google Scholar]