

Abstract
Intraperitoneal injection of the Gram-negative bacterial endotoxin lipopolysaccharide (LPS) elicits a rapid innate immune response. While this systemic inflammatory response can be destructive, tolerable low doses of LPS render the brain transiently resistant to subsequent injuries. However, the mechanism by which microglia respond to LPS stimulation and participate in subsequent neuroprotection has not been documented. In this study, we first established a novel LPS treatment paradigm where mice were injected intraperitoneally with 1.0 mg/kg LPS for four consecutive days to globally activate CNS microglia. By using a reciprocal bone marrow transplantation procedure between wild-type and Toll-like receptor 4 (TLR4) mutant mice, we demonstrated that the presence of LPS receptor (TLR4) is not required on hematogenous immune cells but is required on cells that are not replaced by bone marrow transplantation, such as vascular endothelia and microglia, to transduce microglial activation and neuroprotection. Furthermore, we showed that activated microglia physically ensheathe cortical projection neurons, which have reduced axosomatic inhibitory synapses from the neuronal perikarya. In line with previous reports that inhibitory synapse reduction protects neurons from degeneration and injury, we show here that neuronal cell death and lesion volumes are significantly reduced in LPS-treated animals following experimental brain injury. Together, our results suggest that activated microglia participate in neuroprotection and that this neuroprotection is likely achieved through reduction of inhibitory axosomatic synapses. The therapeutic significance of these findings rests not only in identifying neuroprotective functions of microglia, but also in establishing the CNS location of TLR4 activation.
Introduction
In neuroprotective preconditioning, a moderate primary stimulus can protect the CNS against more destructive secondary insults. A wide range of stimuli have been used in preconditioning paradigms, including lipopolysaccharide (LPS; bacterial endotoxin), global and focal ischemia, cortical spreading depression, seizures, anesthetic inhalants, 3-nitropropionic acid, and hypo-/hyperthermia (Baughman et al., 1988; Kitagawa et al., 1990; Kawahara et al., 1995; Kobayashi et al., 1995; Chen and Simon, 1997; Tasaki et al., 1997; Nishio et al., 1999; Ota et al., 2000; Kapinya et al., 2002). The prevailing theme among preconditioning treatments is the ability to afford protection at subthreshold doses (Kirino, 2002). While the protection/destruction doses of preconditioning agents have been studied in detail, causative molecular and cellular mechanisms require further examination.
LPS is perhaps the most intriguing neuroprotective stimulus because it requires no disruption of the CNS. A peripheral injection of 0.05–1.0 mg/kg LPS can induce transient neuroprotection against ischemia, stroke, and more destructive LPS treatments (Shpargel et al., 2008). Due to its ease of application, unraveling the molecular pathways in LPS neuroprotective signaling has tremendous potential for therapeutic applications. It is possible that injections of LPS initiate signaling through macrophages that are highly enriched in the peritoneal cavity; LPS may also diffuse into the bloodstream, where it stimulates circulating immune cells (Hirano, 1996). Toll-like receptor 4 (TLR4) is a pattern recognition receptor that binds to pathogen-associated molecular patterns in LPS and initiates a signaling cascade through nuclear factor κB (NF-κB)-dependent and NF-κB-independent pathways (Pålsson-McDermott and O'Neill, 2004). The peripheral LPS signal permeates the CNS and activates resident microglia within the brain (Rivest, 2003). However, it remains to be determined whether peripherally injected low doses of LPS directly stimulate microglia or whether they induce microglial activation through downstream surrogate messengers that are produced by immune cells or brain microvascular endothelial cells.
Microglia are resident myelocytic cells in the CNS that perform innate immune functions. Upon activation, microglia can become phagocytic and secrete a multitude of noxious cytokines, chemokines, and oxygen radicals. Activated microglia can also perform neuroprotective functions (Ransohoff and Perry, 2009). Activated microglia produce anti-inflammatory factors and can even ensheathe neurons to provide trophic support under damaging or regenerating conditions (Blinzinger and Kreutzberg, 1968; Streit, 2002). To date, the mode of action and the precise roles of microglia in LPS preconditioning are unknown (Rosenzweig et al., 2007), possibly because microglial activation is not usually observed in most current preconditioning models. In this study, we first established a new LPS-preconditioning paradigm where we injected mice with a low dose of LPS for four consecutive days to globally activate microglia. We sought to identify the functional locale of TLR4 receptors in LPS neuroprotective preconditioning by using a bone marrow chimeric approach with microglial activation as a defining feature. In our model, microglial activation in response to peripheral LPS largely depended on nonhematogenous TLR4 receptors. Activated microglia ensheathe neurons with reduced axosomatic synapses, which is probably part of a neuroprotective mechanism against experimental brain injury.
Materials and Methods
Animals.
C57BL/6J, C57BL/10ScNJ, C3H/HeJ, C3H/HeOuJ mice were purchased from Jackson Labs; B6-LY5.2/Cr mice were purchased from NCI-Frederick (Frederick, MD). The animals were housed under pathogen-free conditions in a temperature- and humidity-controlled environment and given access to food and water ad libitum. All mice used in this study are males and are between the ages of 8 and 12 weeks. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic.
LPS injection and immunohistochemistry.
Mice 8–12 weeks of age received 1, 2, or 4 daily intraperitoneal injections of saline or 1.0 mg/kg LPS (E. coli, serotype 055:B5, Sigma). Their body weights were monitored daily; in a subset of animals, core body temperature was also monitored (DC temperature controller, World Precision Instrument). Twenty-four hours after the final injection, mice were anesthetized with ketamine/xylazine (75 mg/kg and 10 mg/kg, respectively) and transcardially perfused with 4% paraformaldehyde/0.08 m phosphate buffer and the brains were removed and fixed for an additional 24 h. Brain samples were cryoprotected in 20% glycerol overnight and sectioned at a 30 μm thickness on a freezing sliding microtome. Free-floating sections were microwaved in 0.01 m citrate buffer, pH 6.0, followed by pretreatment in 3% hydrogen peroxide/5% Triton X/PBS. Sections were blocked in 3% normal goat serum/0.01% Triton X/PBS and incubated with primary antibody diluted in blocking reagent overnight at 4°C (rat-anti-F4/80, AbD Serotec, 1:200; mouse-anti-Iba-1, CCF Hybridoma Core, 1:500). Following incubation in secondary antibodies (biotinylated goat anti-rat or mouse; 1:500), sections were stained by the avidin-biotin complex (Vector Laboratories), diaminobenzidine (Sigma), enhanced with 0.04% osmium tetroxide (Electron Microscopy Sciences), and mounted on glass slides in glycerol. To quantify Iba-1-positive microglia across different treatments, four random fields in the cerebral cortex were imaged at 20× with a light microscope (Leica). Microglial cell bodies with clear morphology were counted.
Morphometric analysis.
Quantification of area occupied by microglia was performed as previously described (Trapp et al., 2007). Briefly, Images of Iba-1 immunoreactivity from cerebral cortical regions were digitally photographed using a Leica DMR microscope fitted with an Optronics Magna Fire CCD color video camera and image acquisition system. Digital images from three control and three experimental animals were captured at 40× magnification. Using ImageJ software (available at http://rsbweb.nih.gov/ij/), cortical area occupied by Iba-1 immunoreactivity was determined by measuring number of pixels above a set threshold value and expressed as a percentage of total pixels within the microscope field. The percentage of neurons associating with microglia was measured in the cerebral cortex of saline- and LPS-injected animals using confocal microscopy. Images of brain sections stained for neurons and microglia were captured at 40× magnification. Four images per cerebral cortex (layers III-V) were analyzed. Neurons either directly associated with microglial cell bodies or with at least 25% of their circumference occupied by microglial processes were counted. The number of neurons associated with microglia over the total number of neurons in a micrograph field was calculated and expressed as a percentage.
Flow cytometry analysis of CNS-derived cells.
One day following the desired intraperitoneal injection time course, mice were transcardially perfused with cold PBS. Brains were dissected and minced to small pieces < 1 mm3. Single cell suspension was made by incubating the brain tissues with 0.25% trypsin and 20 U DNase (Worthington Biochemical) at 37°C for 1 h with occasional agitation. Cell suspension was washed and centrifuged at 800 × g over a 30%/70% Percoll (GE Healthcare Life Sciences) gradient. Purified cells were analyzed on a FACSCalibur flow cytometer (Becton Dickinson) using CD45-APC (BD Biosciences) and CD11b-PerCP antibodies (BD Biosciences). Resulting data were analyzed with Flowjo 7.1 software (TreeStar).
Microarray analysis for characterization of microglial activation.
Mice were injected with LPS or saline as described above (n = 6/group). Brains were removed and cortices were dissected and snap frozen at −80°C. Total RNA were isolated using TRIzol reagent (Invitrogen), and biotinylated cRNA probes were generated and hybridized to 430A mouse arrays (Affymetrix). Following hybridization with biotinylated probes, arrays were washed, stained, and scanned using the Gene Array scanner (Affymetrix). Scanned images were inspected and quality control procedures standardized in the laboratory and recommended by the manufacturer were used to eliminate outliers. Initial data analysis was performed using Microarray Analysis Suite software 5.0 (MAS 5.0, Affymetrix). Resultant data files were used to compile the gene profiles of LPS-treated microglia by grouping into either M1 (classically activated) or M2 (alternatively activated) phenotypes.
Generation of bone marrow chimera.
Bone marrow cells were collected from hindlimb femurs and tibias of donor mice. Recipient mice at the age of 4–5 weeks were irradiated with 950 rads (Shepherd Mark 137Cs irradiator) and subsequently injected intravenously with 2 × 107 bone marrow cells through the retro-orbital sinus. Recipient mice received 1.2 mg/ml gentamicin through drinking water for 10 d. After 6 weeks, 50 μl of blood was collected through an incision in the tail vein and placed in 10 U of heparin. The erythrocytes were lysed (0.83% NH4Cl, 0.1% KHCO3, 3 mm EDTA, pH = 7.4) and the leukocytes were washed in PBS and analyzed by flow cytometry for CD45.1 (FITC) and CD45.2 (PE, BD Biosciences) to determine the extent of chimeric marrow contribution. C3H/HeJ and C57BL/10ScNJ mice express the CD45.2 allele before bone marrow transplantation; C3H/HeOuJ and B6-LY5.2/Cr mice express CD45.1.
Immunofluorescence and confocal microscopy.
For double or triple staining, brain sections (30 μm free-floating) were incubated with primary antibodies specific for Iba-1 (1:500) and rabbit-anti-synaptophysin (1:200, Millipore) for 2 d at 4°C. The sections were washed and then stained with species-specific secondary antibodies conjugated to Alexa Fluor 488 and 647 (Invitrogen) for 2 h at room temperature. During the last 20 min of incubation, neurons were stained using Neuro Trace Nissl stain 530/615 (Invitrogen) at 1:200. Sections were rinsed, mounted with Vectashield (Vector Labs) and examined on a Leica TCS confocal microscope (Leica Microsystems).
Pre-embedding immunohistochemistry and electron microscopy.
Free-floating tissue sections of motor cortex from saline- and LPS-injected mice were pretreated with 0.1% Triton X-100 and immunostained with Iba-1 antibody and DAB (3,3′-diaminobenzidine tetrahydrochloride) as described above, postfixed in 2.5% glutaraldehyde, placed in 2% osmium tetroxide for 2 h and embedded in Epon. Ultrathin (100 nm) sections were cut, mounted on Formvar-coated grids, and photographed in a Philips CM100 electron microscope.
Cellular proliferation examined by bromodeoxyuridine incorporation assay.
Wild-type (WT) mice were injected with either LPS or saline as described above, followed by a daily dose of bromodeoxyuridine (BrdU; 30 mg/kg, i.p.; Invitrogen) 1 h later. Alternatively, on the last day of drug administration, all mice received three BrdU injections (30 mg/kg, i.p., once every 2 h) before they were killed 2 h after the last BrdU pulse. The brains were then dissected as described above. Brain sections (30 μm) were treated with 2N HCl for 20 min, 4% Na2B4O7 for 10 min, blocked in 3% normal goat serum for 60 min, and incubated in Alexa Fluor 555-conjugated mouse-anti-BrdU monoclonal antibody (1:50, Invitrogen) at 4°C for 2 d. During the last 10 min of incubation, the sections were stained using TO-PRO-3 Iodide (Invitrogen) at a 1:2000 dilution. Sections were rinsed, mounted with Vectashield (Vector Labs) and examined on a Leica TCS confocal microscope (Leica Microsystems).
Experimental brain injury and lesion quantification.
Saline- and LPS-treated animals (n = 11/group) were anesthetized with intraperitoneal injections of ketamine and xylazine as described above. The surgeon was blinded to the treatment given to the mice. A 10 mm midline sagittal incision was made in the skin over the skull and the sagittal cranial suture and right parietal region were visualized. A unilateral aseptic cryogenic injury of the right parietal cortex was induced by applying a 3.5 mm diameter metallic probe, which was precooled in liquid nitrogen, on the skull for 15 s, resulting in blanching of the skull surface. Thereafter, the skin incision was closed by stainless steel wound clips using a clip applier (Reflex, CellPoint Scientific) and mice were allowed to recover under a heating lamp for 2 h. Seventy-two hours after injury, mice were deeply anesthetized and transcardially perfused with 4% paraformaldehyde and brains were collected for assessment of brain injury. Serial coronal brain sections (30 μm) were cut on a cryostat (Leica) and collected every 300 μm throughout the entire brain and stained with Giemsa stain (Sigma). Microscope slides were then scanned on a slide scanner (Microtek) and analyzed with Adobe Photoshop for morphometric measurements. Two measurements were made for each slide and a mean value generated. Lesion volumes were calculated by multiplying the total lesion area by the section interval thickness and expressed in cubic millimeters.
Quantification of apoptotic cells by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay.
Apoptotic cells characterized by DNA fragmentation were detected using a Klenow-FragEL DNA fragmentation detection kit (Calbiochem). Methyl green counterstaining was used for the morphological evaluation and characterization of normal and apoptotic cells. Two slides from each brain collected from the center of the lesion 4 h postinjury were examined. Cell counting was performed in the penumbra of the lesion defined by a 150 μm thick area at the deepest border of the lesion as depicted in Figure 5D (see below). To this end, cells with nuclear staining characteristic of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay-positive (TUNEL+) cells were counted in at least 5 mice per group.
Figure 5.

Synapses cannot be seen where microglia appose neuronal cell bodies. A, In PBS-injected animals, microglia (green, Iba-1) are small and round with fine processes. Synaptic terminals (red, synaptophysin) are evenly distributed on the circumference of the neuronal perikarya (blue, Nissl). B, In LPS-injected animals, microglia have larger cell bodies, thicker processes, and can be observed partially ensheathing neuronal cell bodies. No synaptic staining is visible where microglia appose neuronal perikarya (arrowhead). Scale bar, 10 μm. C, Immunoelectron micrograph shows an Iba-1-positive microglia in direct contact with the plasma membrane of a neuronal cell body (arrows). Scale bar, 1 μm. D, The percentage of neurons in the cerebral cortex that are contacted by microglia is significantly increased following LPS injections compared with controls (n = 3/group). A–C represent at least three different experiments. *p < 0.05.
Statistical analysis.
The difference between two groups was analyzed with Student's t test. For multigroup comparisons, one-way ANOVA was used. Statistical significance was established to be p < 0.05. Analyses were performed either with Microsoft Excel or GraphPad Prism 5.0. Unless otherwise indicated, data are presented as mean ± SD.
Results
Four daily LPS injections promote widespread microglial activation in the CNS
Our laboratory has previously established a model for microglial activation through intracerebral injection of Bacillus Calmette–Guerin (BCG) bacteria followed by peripheral subcutaneous BCG challenge 10 d later (Trapp et al., 2007). The BCG injections activated microglia at the primary cortical injection site with no obvious hallmarks of neuronal pathology. As an alternative and less invasive model, we peripherally injected 1.0 mg/kg LPS into the mouse peritoneal cavity. We performed temporal experiments to identify the injection regimen required to globally activate brain microglia. Microglial activation was assessed morphologically by immunohistochemistry with the classic antibody specific for ionized calcium binding adaptor molecule 1 (Iba-1) and subsequently confirmed by enhanced expression of the activation marker F4/80, expressed by activated but not resting microglia.
Microglial activation was not detectable 24 h after the first LPS injection (Fig. 1B) and microglial morphology was similar to that of the PBS-injected animals (Fig. 1A). Twenty-four hours after the second LPS injection, microglia began to retract their processes and enlarge their cell bodies. Interestingly, numerous spiny protrusions were seen at this interval, giving them a “bushy” appearance, indicating transition to activation (Fig. 1C). One day after the fourth and final LPS treatment, microglia demonstrated asymmetrical process distributions, dense cell bodies and thickened proximal dendrites (Fig. 1D). Their cell bodies often change from spherical to curved contour (Fig. 1D, arrows), resembling a “cup shape” as previously described (Stence et al., 2001). With the increasing numbers of daily LPS injections, the microglial activation profile expanded throughout the cortical and subcortical gray matter (Fig. 1E). The density of the microglial cells was quantified by Iba-1 immunostaining (Fig. 1F). In vehicle-treated animals, resting microglia occupied 6.0 ± 1.56% of the total cortical area examined, whereas in LPS-treated mice the activated microglia occupied 8.8 ± 1.60% of the area. The extent of this increase (48.0%, p < 0.05) suggests that the multiple LPS injection paradigm induced a pronounced hypertrophy of microglial cells. This hypertrophic reaction is not location-dependent, as microglial activation is seen homogeneously distributed among various cortical and subcortical regions (Fig. 1E). Cortical microglial activation was further confirmed by F4/80 immunoreactivity (Fig. 1G). F4/80 staining is not discernable in PBS-treated mice, but becomes apparent in LPS-injected animals. To establish that TLR4 underlies this process, LPS was injected into the peritoneum of TLR4-null mice and brain sections were analyzed for microglial responses. No microglial activation was observed in TLR4-deficient mice (Fig. 1G).
Figure 1.

Microglia in the brain parenchyma are globally activated by four daily intraperitoneal injections of 1.0 mg/kg LPS. A–E, Microglial morphology was visualized by DAB immunohistostaining with mouse-anti-Iba-1 antibody. The morphology of microglia was similar in LPS-injected animals 24 h after the first injection (B) to microglia in PBS-injected animals (A), both having small rounded cell bodies and long, fine processes. Twenty-four hours after 2 LPS injections, processes were retracted and thickened and cell bodies were expanded (C). Microglial activation was apparent 24 h after 4 LPS injections (D), as evidenced by thick cell bodies and asymmetric cell processes. Blue arrows denote cup-shaped microglia. Scale bar, 60 μm. E, The observed microglial activation was global throughout the brain parenchyma. Scale bar, 120 μm. F, Percentage of area occupied by microglia over the total area of interest was significantly higher in 4 daily LPS-treated mice than in PBS-treated mice (representative of three replicates). G, Microglial activation is further confirmed by F4/80 antibody, which stains activated but not resting microglia. The specificity of TLR4 in this process was confirmed by injecting LPS into TLR4-deficient mice. No microglial activation was detected in TLR4−/− mice, as indicted by a lack of F4/80 positivity. Scale bar, 60 μm. H, LPS treatment does not induce microglial proliferation. BrdU+ cells cannot be observed in the cerebral cortex. However, BrdU+ cells (red) are easily identifiable in the SVZ and dentate gyrus regions (images represent three replicates of two different approaches). Nuclei are counterstained in blue. Scale bar, 120 μm. ctx, Cortex; SVZ, subventricular zone; LV, lateral ventricle.
To examine whether LPS treatment also induces microglial proliferation, we performed a BrdU incorporation assay following LPS or PBS treatment (Fig. 1H). Two injection paradigms were used. In the first set of experiments, BrdU was injected daily along with LPS or PBS. No BrdU immunopositivity can be discerned in the cerebral cortex where activated microglia are abundant. However, BrdU-positive cells can be easily identified in the subventricular zone or subgranular zone of the dentate gyrus in the hippocampus, the regions which are considered adult neuronal stem cell niches (Temple, 2001). To rule out the possibility that extensive microglial proliferation occurs later in the process of LPS treatment when activation is at its peak levels, we performed BrdU pulses on the final day of LPS treatment in a second set of experiments. The results were consistent with the first set, with representative images shown in Figure 1H. Further, we did not notice any differences between LPS- or PBS-treated mice, indicating that this proliferation—possibly neurogenesis—is independent of LPS injections. Additionally, observation of microglial cells by light microscopy between controls and LPS-injected animals (Fig. 1A–D) did not reveal any statistically significant differences in numbers, suggesting no microglial proliferation occurs following LPS treatment (p = 0.93 by ANOVA, n = 4 fields/animal, representative of triplicate).
Within the brain areas examined as indicated in Figure 1E, we did not observe end-stage phagocytic amoeboid or “gitter” cells. While the activation of microglia is pronounced, this dose of LPS is below the threshold for neuronal injury, as degenerating neurons were not detected in brains of LPS-treated mice with TUNEL staining (data not shown). There was no seizing behavior or fatalities observed after LPS treatment, albeit changes in body weight and body temperature were observed. The biggest drop in bodyweight occurred after the first LPS injection, followed by a plateau and a rebound in body weight. The body temperature was moderately elevated (1.7 ± 0.74°C, n = 3) during the entire course of the paradigm and returned to slightly above normal at the time of injury (37.4 ± 0.78°C, n = 3), which is 24 h after the final injection.
LPS-primed microglia are skewed toward an M2 phenotype
To further characterize the activated microglia displaying either M1 (classically activated) or M2 (alternatively activated) phenotypes, we performed gene profiling by mining through our microarray database (Fig. 2). Microglia isolated from mouse treated with LPS show remarkably elevated expression of M2-type genes, including Ym1 (also known as Chi3l3), Socs3, Il4ra, Ptprc, Cd163, Il1ra, Mrc1, and Arg1, compared with control mice. Conversely, several M1-related genes, such as Ccr2, Nos2 (also known as iNos), Tnf, Ly6c, IL12a, IL12b, and Icam1 either remain unchanged or were reduced. These results suggest that LPS-treatment primes microglia toward differentiating into an M2 phenotype, which could potentially contribute to neuroprotection by establishing an anti-inflammatory, tissue-repairing microenvironment.
Figure 2.

Multiple low-dose LPS treatment skews activated microglia toward an M2-like phenotype. Microarray analysis (n = 6/group) of cortical tissues shows that M2-related genes are significantly increased following LPS treatment, but that M1-related genes remain either unchanged or reduced when compared with PBS-treated controls.
Infiltration of peripheral immune cells after LPS treatment is marginal
The innate immune system is a major target of LPS, so we analyzed the extent of peripheral leukocyte infiltration into the brain. Following LPS or vehicle injections, we collected mouse brain cells over Percoll gradients and performed flow cytometry to distinguish peripheral monocytes/neutrophils (CD45hiCD11b+) from resident brain microglia (CD45lowCD11b+; Ford et al., 1995). CD45hiCD11b− cells, infiltrating lymphocytes, were also analyzed and compared. A series of four daily 1.0 mg/kg LPS intraperitoneal injections only slightly raised the average peripheral immune cell infiltration into the brain (Fig. 3A). The percentages of infiltrating leukocytes over total CD45+ cells were not significantly different between the LPS-treated and saline control groups (6.1 ± 2.0% vs 4.1 ± 2.3% for monocytes/neutrophils, p = 0.29; and 3.2 ± 0.5% vs 1.8 ± 0.8% for lymphocytes, p = 0.53). The percentage of resident microglia over total CD45+ cells was also not significantly different between the two groups (90.7 ± 2.5% following LPS-treatment vs 94.1 ± 3.0% in controls, p = 0.31; Fig. 3B). Furthermore, the infiltrating peripheral monocytes and lymphocytes in the brains of LPS-treated mice were minimal compared with the abundance of resident microglial cells. This suggests that microglial activation was unlikely to be mediated by infiltrating immune cells through local paracrine actions. However the possibility remains that hematogenous immune cells contribute to CNS microglial activation through the production of soluble mediators (e.g., cytokines) released into the systemic circulation. In an effort to examine this prospect, we generated TLR4 mutant bone marrow chimeric mice.
Figure 3.

Intraperitoneal LPS injections did not lead to infiltration of peripheral leukocytes into the CNS. A, Brain cells analyzed by flow cytometry displayed similar patterns of CD11b and CD45 expression between LPS- and saline-treated mice. B, The percentages of lymphocytes, monocytes/neutrophils, and microglia over total CD45+ cells were quantified and were not significantly different (n = 3/group).
Bone marrow chimera mice reveal that hematogenous TLR4 is not required for microglial activation
To maintain MHC haplotype compatibility, we used C3H/HeJ mice carrying a spontaneous point mutation of Tlr4. C3H/HeOuJ mice were used as a WT strain and can be distinguished based on flow cytometric analysis of CD45 isoforms: C3H/HeJ mice carry the CD45.2 allele before bone marrow transplantation and C3H/HeOuJ mice carry CD45.1. Based on CD45 expression, we have achieved an average replacement of 90% TLR4 mutant bone marrow in WT recipient mice (TLR4−/−→WT) and 95% WT marrow in TLR4 mutant recipient (WT→TLR4−/−; Fig. 4A). Intraperitoneal LPS injections into TLR4 mutant mice carrying WT marrow resulted in defective microglial activation (Fig. 4B). In stark contrast, intraperitoneal LPS injections into WT mice carrying TLR4 mutant bone marrow resulted in excessive microglial activation, even stronger F4/80 expression and thicker processes than in the WT→WT controls (Fig. 4B). Collectively, in the peripheral intraperitoneal LPS injection model, microglial activation requires neither peripheral immune cells themselves nor any soluble factors they produce or release. Instead, LPS-induced microglial activation highly likely depends on the integrity of TLR4 receptors within the CNS, either in the brain parenchyma or on CNS vascular endothelia. This observation is supported by the excessive activation of microglia in TLR4−/−→WT mice, possibly because with a TLR4 mutant periphery, more LPS will be available in the bloodstream to signal through the CNS. In addition, we also repeated this bone marrow chimerism experiment in another pair of congenic mouse strains: B6-Ly5.2/Cr mice were used as WT and C57BL/10ScNJ mice, which carry genomic deletion of Tlr4, were used as TLR4−/−. The results were similar to that of the chimeras between the C3H mice (data not shown). To rule out the possibility that total body irradiation can induce microglial activation via a breached blood–brain barrier (BBB) (Mildner et al., 2007), we examined microglial morphology at 1, 2 and 3 weeks after bone marrow transplant by immunohistochemistry as described above. We did not observe microglial activation compared with control wild-type mice injected with PBS (data not shown).
Figure 4.
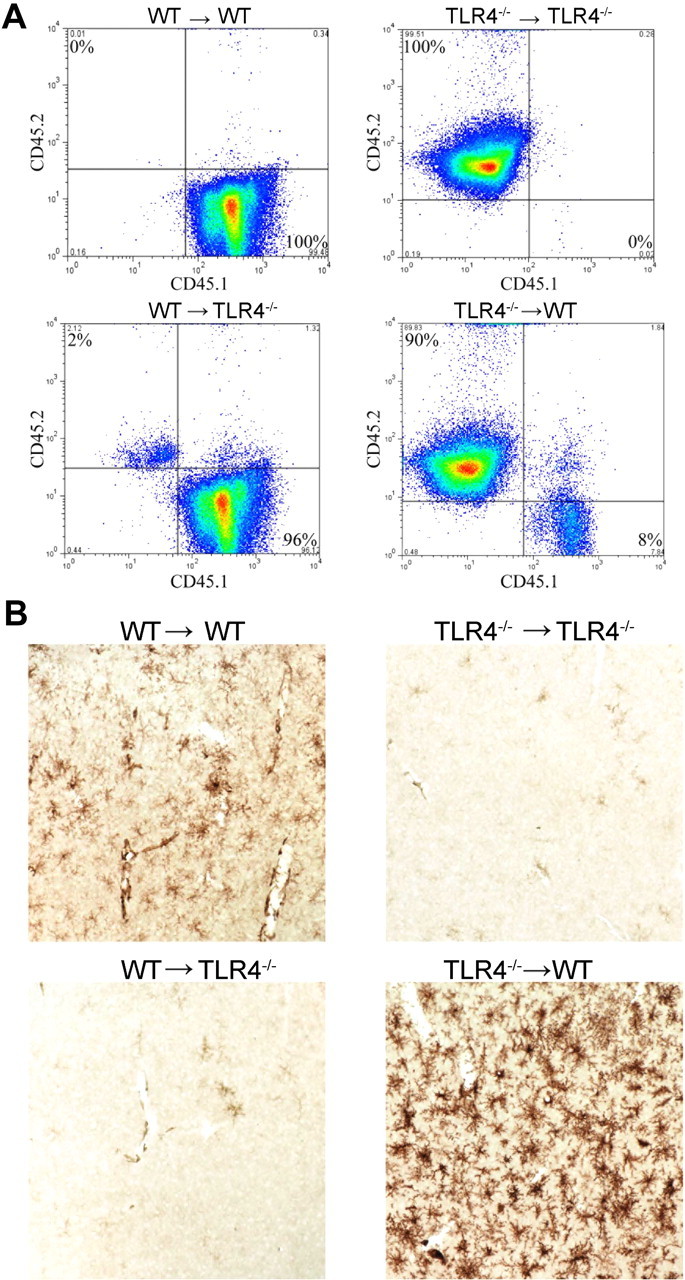
Bone marrow chimera mice show that peripheral hematogenous TLR4 is not required for LPS-induced microglial activation. A, Blood cells taken from mice that underwent bone marrow transplant surgery were analyzed using flow cytometry and showed that most of cells in the WT→TLR4−/− chimera displayed the WT isoform of CD45 and vice versa. B, No microglial activation was seen in the TLR4−/−→TLR4−/− or the WT→TLR4−/− chimeras, as evidenced by the lack of DAB immunohistostaining using the anti-F4/80 antibody. Microglial activation was observed in the WT→WT chimera and was more robust in the TLR4−/−→WT chimera. Images represent at least 12 different chimerae.
Activated microglia remove perikaryal/proximal synapses
Having established that microglia activation is not dependent on the presence of peripheral hematogenous TLR4, we next set out to examine the impact of microglial morphological change on neuronal synaptic networks, based on reports of activated microglia stripping afferent synapses from neuronal perikarya as part of a neuroprotective response (López-Redondo et al., 2000; Streit, 2002). Brain sections from either PBS- or LPS-treated WT mice were triple-labeled with fluorescent markers to identify synapses (synaptophysin), microglia (Iba-1), or neuronal somata (Nissl). In PBS-treated mice microglia are quiescent. The axosomatic synapses can be easily identified evenly distributed around the neuronal surface (Fig. 5A). In contrast, following LPS treatment, activated microglia often appear to be perineuronally located and the curvature of their somata seem to tightly ensheathe neuronal perikarya. Where the microglia appose neuronal cell bodies, there is no longer discernible synaptic staining (Fig. 5B). To confirm the physical loss of synapses at the ultrastructural level we performed immunoelectron microscopy on LPS-treated, DAB-stained mouse brain sections. The electron micrograph substantiates that Iba-1-positive microglia directly appose the surface of cortical neurons (Fig. 5C, arrows). The percentage of neurons that are associated with microglia was quantified and compared between vehicle- and LPS-injected animals. In control mice, ∼5.7% of cortical neurons examined had brief contact with microglia; whereas following LPS treatment, this percentage increased more than two-folds to 12.5% (p < 0.01, Fig. 5D). These data suggest that activated microglia displace axosomatic synapses from neurons. This synaptic displacement was also observed in TLR4−/−→WT bone marrow chimeric mice to a similar extent following LPS treatment, suggesting that microglial activation is an integral component of this synaptic modification.
Peripheral LPS injections protects neurons from experimental brain injury
To determine whether 4 daily intraperitoneal injections of LPS is neuroprotective, adult WT mice were divided randomly into two groups, which received LPS (1.0 mg/kg) or saline injections followed by cryogenic injury to the surface of the skull over the motor cortex 24 h later. Animals were perfused 72 h following the cryogenic injury and processed for immunohistochemical analysis. The lesion site was grossly evident as a hemorrhagic and necrotic area on the cortical surface as depicted by Giemsa staining (Fig. 6A,B). Quantification of serial brain sections showed that LPS-treated mice displayed a smaller lesion volume than saline-injected control mice (Fig. 6C). A similar reduction (30%) in lesion volume was also observed in experimentally injured TLR4−/−→WT chimeric mice when compared with WT→TLR4−/− chimeras (data not shown). The decreased lesion volume in LPS-treated brain suggests a reduction in the number of apoptotic cells immediately following cryogenic injury. To test this possibility, we quantified TUNEL-positive cells in the cortex of saline- and LPS-treated mice 4 h after cryogenic injury. TUNEL-positive cells were quantified in the penumbra of the cortical lesions as depicted in Figure 6D. There was a significant reduction (p < 0.05) in apoptotic cells in this area, establishing reduced neuronal apoptosis in LPS-treated mice following traumatic injury. Collectively, these results demonstrate a neuroprotective capacity of our LPS preconditioning paradigm against experimental brain injury.
Figure 6.

LPS-treated mice had a smaller lesion volume and reduced apoptotic cells following cryogenic injury. A–C, Mice treated with 4 daily injections of PBS or LPS underwent cryogenic injury and were killed 72 h postinjury. Serial sections were visualized with Giemsa stain (discoloration in A and B demarcates the lesions). LPS-treated animals (B) had a significantly smaller lesion volume than those treated daily with PBS (A) (n = 11/group). The lesion size was quantified in C. D, Significantly fewer TUNEL-positive cells (brown) were present in the penumbra region (illustrated) in LPS-treated animals compared with controls (n = 3/group). Data are expressed as mean ± SEM. *p < 0.05.
Discussion
The goal of this study was to determine the underlying cellular and molecular mechanisms and mode of action of peripheral LPS treatment in CNS microglial activation and prophylactic neuroprotection. Through bone marrow chimera construction between WT and TLR4-deficient mice, we have identified activation of nonhematogenous TLR4 as the essential initiating locale responsible for microglial activation upon peripheral multiple low dose LPS treatment. Activated microglia target and ensheathe cortical neurons and disrupt synaptic connections from the neuronal perikarya, a phenomenon termed “synaptic stripping” (Blinzinger and Kreutzberg, 1968). We demonstrate here that as part of LPS-triggered CNS innate immune responses, activated microglia displace inhibitory axosomatic synapses from cortical projection neurons, which may serve as a protective mechanism that protects these neurons from experimental brain trauma. These results are consistent with previous studies that have established that reduced axosomatic GABAergic innervation protects neurons against noxious insult (Hardingham et al., 2002; Hardingham and Bading, 2003).
An initial interest of our research was to understand whether the actions of activated microglia are neuroprotective or neurotoxic. Microglial activation has been observed in many neurodegenerative diseases such as Alzheimer's (Haga et al., 1989), HIV dementia (Yoshioka et al., 1992), and multiple sclerosis (Peterson et al., 2001). It is highly debated whether these activated microglia are helpful or harmful to the CNS, as evidence supports both perspectives. To address this challenging question and recapitulate the activated microglia phenotype in animal models, we developed an experimental paradigm where global microglial activation throughout the CNS was induced by peripherally injecting 1.0 mg/kg LPS for 4 consecutive days (Fig. 1). Our results support the notion that this microglial activation paradigm offers prophylactic protection to neurons, particularly in the context of traumatic brain injury (Fig. 6). Our unique paradigm is different from other LPS preconditioning regimens which typically consist of a single injection of LPS before experimentally induced damage (Rosenzweig et al., 2004). Interestingly, the neuroprotection induced by 4 daily LPS injections is insult-specific (B. D. Trapp, unpublished observation). A single intraperitoneal injection of LPS transiently protects against ischemic infarcts but does not activate microglia nor protect against cryogenic-induced cortical injury; on the other hand, four intraperitoneal injections of LPS confer protection against traumatic brain injury but not ischemic stroke. Thus it appears that two LPS-induced neuroprotective pathways can be induced: one protecting against ischemic brain injury and one against traumatic brain injury. The former is likely to be mediated at the level of CNS vasculature and to include interferon signaling (Stevens et al., 2011), while the latter is likely a parenchymal response that involves microglial activation, reduced GABAergic innervation of cortical neurons and increased NMDA signaling (discussed below).
A rather unexpected but interesting observation is that the activated microglia are not actually proliferating (Fig. 1H). It remains an enduring and contentious debate as to whether microglial activation and proliferation are equally involved in synaptic remodeling. Studies using proliferation inhibitors preventing microglial division after hypoglossal axotomy (Svensson and Aldskogius, 1993) or CSF-1 deficient mice which have impaired microglial proliferation (Kalla et al., 2001) have shown that microglial proliferation is not an essential process in synaptic remodeling. These observations prompted further argument that microglia are not actively involved in synaptic withdrawal (Perry and O'Connor, 2010). However, here we present evidence that microglial activation and proliferation are indeed dissociated and that synaptic remodeling does not depend on microglial proliferation, but rather on activation.
TLR4 bone marrow chimera experiments identified CNS TLR4 as, at least partially, the origin of LPS signaling resulting in microglial activation and neuroprotection, and the absence of TLR4 from peripheral leukocytes had no effect on the ability of intraperitoneal LPS injections to activate microglia (Fig. 4). These results are in agreement with a previous study in which a similar reciprocal bone marrow transplantation between TLR4 mutant and WT mice was performed (Chakravarty and Herkenham, 2005). In that study, by monitoring cytokine production in either the CNS or peripheral compartment after challenging the animals with LPS, the authors were able to demonstrate that a long-lasting inflammatory response in the CNS depends on parenchymal TLR4, but not on TLR4 expressed by hematogenous cells. Together, these observations suggest that peripheral leukocytes, in particular blood-borne monocytes, are not a major contributor to LPS-induced neuroprotection in the CNS, which is further underscored by our findings that infiltration of peripheral white blood cells following LPS treatment is marginal (Fig. 3). This conclusion is also in line with recent studies demonstrating that anti-inflammatory tissue-repairing macrophages are locally renewed and do not rely on recruitment of circulating monocytes (Jenkins et al., 2011; Randolph, 2011).
How do peripheral injections of LPS activate microglia in the CNS? It has been shown that TLR4 expression in the brain is largely restricted to endothelial cells, epithelial cells and microglia (Chakravarty and Herkenham, 2005). LPS contained within the peritoneal cavity can diffuse into the bloodstream (Hirano, 1996), where it can activate microglia, though this is unlikely to occur by penetration of the BBB, which is known to be impermeable to LPS (Singh and Jiang, 2004). However, it is possible that LPS enters the CNS by circumventing this barrier at locations with a weaker BBB, such as the circumventricular organs (CVOs) (Rivest, 2003). Although likely, this “leaky” model cannot completely explain our observation that microglial activation following 4 LPS injections is global throughout the CNS, because this model predicts a spatial distribution of differentially activated microglia between CVOs and the brain parenchyma. A more plausible explanation is that circulating LPS signals through endothelial cells, initiating a cascade of cytokines to induce microglial activation. TNFα (tumor necrosis factor α), for example, has been shown to be necessary for LPS-mediated preconditioning (Rosenzweig et al., 2007) and injections of TNFα-neutralizing antibody into the cerebral ventricles prevented microglial cytokine production (Nadeau and Rivest, 2000). Moreover, intraventricular cytokine delivery protected the CNS from future insults, whereas intravenous injection of the same cytokines failed to reproduce LPS preconditioning when BBB integrity was preserved (Nawashiro et al., 1997; Maier et al., 2006). Taking these results together, it is plausible to hypothesize that the activation of TLR4 on the luminal surface of CNS endothelial cells results in local cytokine secretion to activate resident microglia. This hypothesis could best be examined by constructing a mouse model that exclusively expresses TLR4 receptors on CNS endothelial cells (Meng et al., 2007).
Our data establish that microglia actively participate in an LPS-induced neuroprotective response through close and intimate association with neuronal cell bodies and proximal dendrites. This study is the first of its kind to associate neuroprotective preconditioning with microglial activation. Several previous studies using single-LPS-injection preconditioning paradigms have largely focused on cytokine profiles. It has been shown in an ischemic stroke model that LPS preconditioning increases type I interferon expression in the CNS in addition to altering the transcription of the cytokine-responsive genes that are activated by the transcription factors IRFs (interferon regulator factors) (Stevens et al., 2011). T helper 2 (Th2)-associated anti-inflammatory cytokines were also increased following LPS pretreatment, which tips the balance of immune response toward anti-inflammatory and tissue-repairing functions (Yu et al., 2010). Here we show that in our LPS pretreatment paradigm, activated microglia display an M2-like phenotype (Fig. 2), which is important in fostering a neuroprotective environment. While containing inflammation and restoring homeostasis are important in a diseased brain, an amiable environment alone cannot account for the increased resistance of neurons to noxious stimuli. We hypothesize that LPS treatment intrinsically enhances neuronal survival following injury or damage, and further that synaptic displacement is essential for this neuroprotection. It has recently been shown with in vivo live imaging that even at resting state, microglia are essentially “restless”—their processes constantly extend and retract, making frequent but transient contact with synapses (Nimmerjahn et al., 2005). This surveillance is important for synaptic formation and maturation during postnatal development because microglia actively participate in pruning of the newly created synapses by engulfing redundant synaptic components (Paolicelli et al., 2011). Here we offer evidence that microglia retain the ability to make intimate contact with neurons and replace synaptic terminals into adulthood, both at a basal level and to a much more heightened extent when activated (Fig. 5D), and that this synaptic modification could serve as a protective mechanism in the adult brain. However, it remains to be determined whether the neuro-microglial contact duration is much more prolonged under distressed situation than under physiological conditions.
A direct relationship between synaptic remodeling and neuroprotection is derived from investigations of NMDA receptor function (Hardingham et al., 2002). Synaptic activity controls nuclear signaling and transcription. Stimulation of NMDA receptors by synaptic activity (glutamate) robustly activates cAMP response element-binding protein (CREB), which increases BDNF (brain-derived neurotrophic factor) gene expression and facilitates neuronal survival. The majority of synapses that terminate on cortical neuronal perikarya use GABA as their neurotransmitter (Mallat and Chamak, 1994). GABAergic axosomatic synapses are inhibitory synapses, which function to oppose the excitatory effects of NMDA receptors. Transient elimination of axosomatic synapses by activated microglia (Fig. 5) preferentially reduces inhibitory innervation and therefore diminishes the inhibition exerted on NMDA receptors. It has been shown that pharmacological antagonism of GABAergic receptors can result in increased firing of synaptic NMDA receptors, which stimulates anti-apoptotic and/or anti-necrotic signaling pathways through CREB, and thereby promotes neuronal survival (Hardingham and Bading, 2003). Synaptic reduction by activated microglia may induce neuroprotection by a similar mechanism. Conformational studies are currently ongoing to examine the activation of CREB and its related pro-survival proteins following LPS preconditioning.
The peripheral LPS injection paradigm may prove to be a valuable asset in understanding neuroprotection. This paradigm induces transient CNS inflammation and neuroprotection without any apparent detrimental effects. Establishing a detailed mechanistic pathway for LPS neuroprotective preconditioning could ultimately result in broad therapeutic applications that benefit patients who suffer from neurodegenerative diseases or brain trauma.
Footnotes
This investigation is supported by the Bakken Heart and Brain Institute and the Nancy Davis Foundation. Z.C. is supported by a fellowship from the Multiple Sclerosis Society of Canada and W.J. by a postdoctoral fellowship from the National Multiple Sclerosis Society (FG1711-A-1). We thank Dr. Christopher L. Nelson for scientific discussion and editing support and Dr. Robert Fairchild for advice and technical expertise.
The authors declare no competing financial interests.
References
- Baughman VL, Hoffman WE, Miletich DJ, Albrecht RF, Thomas C. Neurologic outcome in rats following incomplete cerebral ischemia during halothane, isoflurane, or N2O. Anesthesiology. 1988;69:192–198. doi: 10.1097/00000542-198808000-00007. [DOI] [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motor neurons by microglial cells. Z Zellforsch Mikrosk Anat. 1968;85:145–157. doi: 10.1007/BF00325030. [DOI] [PubMed] [Google Scholar]
- Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci. 2005;25:1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Simon R. Ischemic tolerance in the brain. Neurology. 1997;48:306–311. doi: 10.1212/wnl.48.2.306. [DOI] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Haga S, Akai K, Ishii T. Demonstration of microglial cells in and around senile (neuritic) plaques in the Alzheimer brain. An immunohistochemical study using a novel monoclonal antibody. Acta Neuropathol. 1989;77:569–575. doi: 10.1007/BF00687883. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hirano S. Migratory responses of PMN after intraperitoneal and intratracheal administration of lipopolysaccharide. Am J Physiol. 1996;270:L836–L845. doi: 10.1152/ajplung.1996.270.5.L836. [DOI] [PubMed] [Google Scholar]
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalla R, Liu Z, Xu S, Koppius A, Imai Y, Kloss CU, Kohsaka S, Gschwendtner A, Möller JC, Werner A, Raivich G. Microglia and the early phase of immune surveillance in the axotomized facial motor nucleus: impaired microglial activation and lymphocyte recruitment but no effect on neuronal survival or axonal regeneration in macrophage-colony stimulating factor-deficient mice. J Comp Neurol. 2001;436:182–201. [PubMed] [Google Scholar]
- Kapinya KJ, Löwl D, Fütterer C, Maurer M, Waschke KF, Isaev NK, Dirnagl U. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke. 2002;33:1889–1898. doi: 10.1161/01.str.0000020092.41820.58. [DOI] [PubMed] [Google Scholar]
- Kawahara N, Ruetzler CA, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res. 1995;17:9–16. doi: 10.1080/01616412.1995.11740281. [DOI] [PubMed] [Google Scholar]
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab. 1995;15:721–727. doi: 10.1038/jcbfm.1995.92. [DOI] [PubMed] [Google Scholar]
- López-Redondo F, Nakajima K, Honda S, Kohsaka S. Glutamate transporter GLT-1 is highly expressed in activated microglia following facial nerve axotomy. Brain Res Mol Brain Res. 2000;76:429–435. doi: 10.1016/s0169-328x(00)00022-x. [DOI] [PubMed] [Google Scholar]
- Maier CM, Yu F, Nishi T, Lathrop SJ, Chan PH. Interferon-beta fails to protect in a model of transient focal stroke. Stroke. 2006;37:1116–1119. doi: 10.1161/01.STR.0000208214.46093.d5. [DOI] [PubMed] [Google Scholar]
- Mallat M, Chamak B. Brain macrophages: neurotoxic or neurotrophic effector cells? J Leukoc Biol. 1994;56:416–422. doi: 10.1002/jlb.56.3.416. [DOI] [PubMed] [Google Scholar]
- Meng F, Shi L, Cheng X, Hou N, Wang Y, Teng Y, Meng A, Yang X. Surfactant protein A promoter directs the expression of Cre recombinase in brain microvascular endothelial cells of transgenic mice. Matrix Biol. 2007;26:54–57. doi: 10.1016/j.matbio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor kappa B activity in the brain during endotoxemia. J Neurosci. 2000;20:3456–3468. doi: 10.1523/JNEUROSCI.20-09-03456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nishio S, Chen ZF, Yunoki M, Toyoda T, Anzivino M, Lee KS. Hypothermia-induced ischemic tolerance. Ann N Y Acad Sci. 1999;890:26–41. doi: 10.1111/j.1749-6632.1999.tb07978.x. [DOI] [PubMed] [Google Scholar]
- Ota A, Ikeda T, Xia XY, Xia YX, Ikenoue T. Hypoxic-ischemic tolerance induced by hyperthermic pretreatment in newborn rats. J Soc Gynecol Investig. 2000;7:102–105. [PubMed] [Google Scholar]
- Pålsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Perry VH, O'Connor V. The role of microglia in synaptic stripping and synaptic degeneration: a revised perspective. ASN Neuro. 2010;2:e00047. doi: 10.1042/AN20100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons and reduced inflammation in cortical MS lesions. Ann Neurol. 2001;50:389–400. doi: 10.1002/ana.1123. [DOI] [PubMed] [Google Scholar]
- Randolph GJ. Immunology. No need to coax monocytes. Science. 2011;332:1268–1269. doi: 10.1126/science.1208480. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- Rosenzweig HL, Minami M, Lessov NS, Coste SC, Stevens SL, Henshall DC, Meller R, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab. 2007;27:1663–1674. doi: 10.1038/sj.jcbfm.9600464. [DOI] [PubMed] [Google Scholar]
- Shpargel KB, Jalabi W, Jin Y, Dadabayev A, Penn MS, Trapp BD. Preconditioning paradigms and pathways in the brain. Cleve Clin J Med. 2008;75(Suppl 2):S77–S82. doi: 10.3949/ccjm.75.suppl_2.s77. [DOI] [PubMed] [Google Scholar]
- Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology. 2004;201:197–207. doi: 10.1016/j.tox.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci. 2011;31:8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Svensson M, Aldskogius H. Synaptic density of axotomized hypoglossal motorneurons following pharmacological blockade of the microglial cell proliferation. Exp Neurol. 1993;120:123–131. doi: 10.1006/exnr.1993.1046. [DOI] [PubMed] [Google Scholar]
- Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- Temple S. The development of neural stem cells. Nature. 2001;414:112–117. doi: 10.1038/35102174. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Wujek JR, Criste GA, Jalabi W, Yin X, Kidd GJ, Stohlman S, Ransohoff R. Evidence for synaptic stripping by cortical microglia. Glia. 2007;55:360–368. doi: 10.1002/glia.20462. [DOI] [PubMed] [Google Scholar]
- Yoshioka M, Shapshak P, Sun NCJ, Nelson SJ, Svenningsson A, Tate LG, Pardo V, Resnick L. Simultaneous detection of ferritin and HIV-1 in reactive microglia. Acta Neuropathol. 1992;84:297–306. doi: 10.1007/BF00227823. [DOI] [PubMed] [Google Scholar]
- Yu JT, Lee CH, Yoo KY, Choi JH, Li H, Park OK, Yan B, Hwang IK, Kwon YG, Kim YM, Won MH. Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal CA1 region preconditioned with lipopolysaccharide. J Neurol Sci. 2010;296:69–78. doi: 10.1016/j.jns.2010.06.004. [DOI] [PubMed] [Google Scholar]