

Abstract
A major aim of proteomics is to comprehensively identify and quantify all protein species variants from a given biological source. However, in spite of its tremendous utility, bottom-up proteomic strategies can do little to provide true quantification of distinct whole protein species variants given its reliance on proteolysis. This is particularly true when molecular size information is lost as in gel-free proteomics. Crk and CrkL comprise a family of adaptor proteins that couple upstream phosphotyrosine signals to downstream effectors by virtue of their SH2 and SH3 domains respectively. Here we compare the identification and quantification of CrkL-SH3 binding partners between embryonic murine brain and liver. We also uncover and quantify tissue-specific variants in CrkL-SH3 binding proteins.
Keywords: Quantitative Proteomics, CrkL, Crk, SH3, C3G, Splice Variants, Protein Species Variants
Graphical abstract
1. Introduction
The proto-oncogene c-Crk (CT10 regulator of kinase) and its relative CrkL (Crk-like) are cellular homologs of the viral oncogene v-Crk carried by the avian sarcoma virus CT10 [1, 2]. Although devoid of enzymatic activity, the Crk and CrkL proteins facilitate signal transduction by linking proteins containing phosphorylated tyrosine residues to downstream effectors. Crk and CrkL perform this role by virtue of their simple dual domain structure consisting of an amino-terminal Src Homology 2 (SH2) domain and either one or two carboxyl-terminal SH3 domains. Their SH2 domains bind to proteins with phosphorylated tyrosine in YxxP motifs and their SH3 domain binds to proteins harboring PxxPxK motifs [2, 3]. The substrates of many tyrosine kinases recruit Crk and CrkL and thereby regulate an array of signaling pathways [2–4]. Crk and CrkL play overlapping roles and are essential for proper development in the mouse, most evident by the early lethal phenotype that results in compound c-Crk and Crkl mutants [5–7]. However, genetic disruption of only one family member can still have important effects [5–7]. Previously we found Crk and CrkL were recruited to tyrosine phosphorylated Disabled-1 (Dab1), a critical scaffold regulating Reelin signaling in mammalian brain development [8]. The essential nature of Crk and CrkL in Reelin signaling was given genetic support using Cre-Lox-mediated compound disruption of their encoded genes conditionally in the developing nervous system [7]. The recruitment of Crk and CrkL to phoshpho-tyrosyl Dab1 co-translocates their SH3-binding proteins including the Rap-GEF C3G (CrkL SH3-binding Guanine Nucleotide-Releasing Protein) [8]. Furthermore, we recently identified several additional CrkL-SH3 binding partners from embryonic murine brain [3]. Given Reelin’s ability to cluster several receptors [9, 10] this leads to the potential of multiple complex intracellular signaling assemblages proximal to Reelin receptors.
In order to determine if the CrkL-SH3 binding proteins we identified in embryonic brain were distinct from CrkL-SH3 binding partners in other tissue types, we used quantitative mass spectrometry to compare CrkL-SH3 binding proteins from embryonic murine brain and liver lysates. CrkL-SH3 binding proteins were eluted and subjected to SDS-PAGE. Protein regions from the entire gel were subjected to in-gel tryptic digestion. Extracted peptides were subjected to labeling by reductive amination using reagents with differential masses based on stable isotopes. Following liquid chromatography tandem mass spectrometry (LC-MS/MS), a total of 40 CrkL-SH3 binding proteins common to two biological replicates were quantified. 30 were enriched in the brain pulldowns while three were enriched in the liver pulldowns. Three proteins showed no enrichment in the pulldowns while four of the proteins showed the striking behavior of variant-specific enrichment, at least when considering differences in molecular weight as a proxy for protein species variants. This concept is further discussed with particular consideration paid to signatures of protein species variants in quantitative bottom-up proteomic workflows.
2. Material and methods
2.1. Mice, plasmids and antibodies
Timed pregnant CD-1 mice were obtained from Charles River Laboratories, Canada (Saint-Constant, Québec, Canada) and treated according to an institutionally-approved IACUC protocol (#10-068). Mice were euthanized after a brief isoflurane administration when embryos were at embryonic day 16.5 (E16.5). The embryonic brains and livers were carefully dissected and tissue was lysed as described below. The bacterial expression plasmid encoding GST (pGEX-4T-1) was from Stratagene/Agilent Technologies (Santa Clara, CA, USA) and the plasmid encoding GST-CrkL-SH3 was a gift of Akira Imamoto (University of Chicago, USA) [5]. The following antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA): α-C3G (H-300), α-DDEF2/ASAP2 (H-300), α-N-WASP (H-100), and α-PEX13 (H-300).
2.2 Affinity chromatography and GST fusion protein pulldown assays
E16.5 murine whole brain and liver extracts were generated by dounce homogenization in ice-cold brain complex lysis buffer (BCLB: 25 mM Tris pH 7.2, 137 mM NaCl, 10% glycerol, 1% Igepal, 25 mM NaF, 10 mM Na4P2O7, 1 mM Na3VO4, 1 mM PMSF, 10 μg/ml leupeptin, and 10 μg/ml pepstatin A). The supernatants were collected after lysates were centrifuged at 4° C for 20 minutes at 16,000 × g. Three ml of each supernatant, corresponding to five mg of protein was pre-cleared by rocking with 100 μl of a 50% slurry of BCLB-washed glutathione agarose resin (G-Biosciences, Maryland Heights, MO, USA) for one hour at 4° C. After centrifugation for ten minutes at 16,000 × g the supernatants were collected. Each supernatant was then similarly pre-cleared by rocking for three hours at 4°C with 70 μl of a 50% slurry (in BCLB) of glutathione agarose bound to 35 μg of GST. The resins were collected by brief centrifugation and the supernatants were retained. The GST resin was then washed three times with BCLB, drained and proteins were eluted from the resin with protein sample buffer (125 mM Tris pH 6.8, 2% SDS, 5% β-mercaptoethanol, 7.5% glycerol) at 95° C for five minutes. The eluates were stored at −20° C for later use. The GST pre-cleared supernatants were rocked overnight at 4° C, each with 70 μl of a 50% glutathione agarose slurry (in BCLB) bound to 35 μg GST-CrkL-SH3. Resins and their associated proteins were collected by centrifugation and washed three times with BCLB and drained. Protein sample buffer was added and samples, including the GST pre-cleared resins, were heated to 95° C for five minutes. Eluted proteins were separated using a 7.5% (37.5:1 acrylamide:bis-acrylamide) SDS-PAGE gel and then stained with coomassie blue.
2.3 In-gel reduction, alkylation and tryptic digestion
The lanes of the coomassie-stained gel from the brain and liver pulldowns were each cut into 15 equal regions above the CrkL-SH3 band. The gel pieces were diced into one mm cubes, washed with one ml HPLC-grade water and incubated in one ml destain solution (50 mM ammonium bicarbonate and 50% acetonitrile (MeCN)) for 30 minutes at 37° C. For full removal of the stain, destaining was repeated once again and then gel slices were subjected to dehydration by adding 100% MeCN for 10 minutes. The MeCN was then removed and the gel slices were dried. The proteins in the gel pieces were then reduced in 10 mM DTT in 50 mM ammonium bicarbonate and by incubation at 56° C for 20 minutes. The samples were allowed to cool down to room temperature and the overlay solution was drained. The gel pieces were then dehydrated with 100% MeCN, rehydrated with water and then dehydrated again with MeCN. Proteins were next alkylated in 50 mM iodoacetamide in 50 mM ammonium bicarbonate while incubating in the dark at room temperature for one hour. After removing the samples from the dark, the alkylation solution was discarded and the gel pieces were washed with one ml of HPLC-grade water and then with 500 μl destain solution. The gel pieces were then dehydrated using 100% MeCN, allowing five minutes for each step. The washing was repeated twice and the gel pieces were then dehydrated using 100% MeCN and then dried in a speed vacuum for 15 minutes. Proteins were cut into peptides using sequencing grade modified trypsin (Promega, Madison, WI, USA) at a concentration of 6 ng/μl in 50 mm ammonium bicarbonate at 37° C overnight. The in-gel digests were centrifuged for five minutes at 13,000 × g and the supernatant was transferred to a 0.6 ml tube. Extraction solution (50% MeCN, 2.5% formic acid (FA)) was then added to the gel pieces and they were centrifuged at 13,000 × g for 15 minutes. Extractions were combined with appropriate digests and dried in a speed vacuum.
2.3 Peptide labeling, liquid chromatography tandem mass spectrometry (LC-MS/MS), and data analysis
The labeling of tryptic peptides with mass tags was done using reductive amination (dimethylation) chemistry [11] in which peptides are modified using light (1H) or heavy (2H) formaldehyde and sodium cyanoborohydride. Heavy reagents were purchased from Cambridge Isotopes Laboratories, Tewksbury, MA, USA and light reagents were from Sigma (St. Louis, MO, USA). Embryonic brain and liver peptides derived from bound proteins in the CrkL-SH3 pulldowns were labeled using light and heavy reagents respectively. Peptides collected from each gel region were resuspended in 40 μl of 1M HEPES, pH 7.5. 4 μl of light labeling reagents (4% formaldehyde, 600 mM sodium cyanoborohydride) were added to each tube containing brain peptides and 4 μl of heavy labeling reagents (4% 2H2-formaldehyde, 600mM sodium cyanoborodeuteride) were added to each tube containing liver peptides. The reactions proceeded for 10 minutes and then the same volumes of labeling reagents were added again to push the reaction to completion. The reaction was allowed to continue for 10 additional minutes and then was quenched by adding 50 μl of 10% trifluoroacetic acid (TFA). Both light and heavy reactions were allowed to sit at room temperature for 1 hour and then mixed together carefully so that reactions representing each gel region from the brain samples were mixed with reactions representing the corresponding regions of the liver samples. Mixed peptides were then subjected to desalting using C18 spin columns (Thermo Electron, San Jose, CA, USA). Finally, the peptides eluted from the spin columns were dried in a speedvac and resuspended in 8 μl of 2.5% FA, 2.5% MeCN. 4 μl of the sample was loaded using a Micro AS autosampler (Thermo Electron) onto a microcapillary column of 100 μm inner diameter packed with 12 cm of reversed-phase Magic C18 packing material (5 μm, 200 Å; Michrom Bioresources, Inc., Auburn, CA, USA). After a 14.5 minute isocratic loading in 2.5% MeCN, 0.15% FA (Solvent A) peptides were eluted using a 5%−35% gradient of Solvent B (98.85% MeCN, 0.15% FA) over 30 min and electrosprayed into a linear ion trap-orbitrap (LTQ-Orbitrap) mass spectrometer (Thermo Electron). The precursor scan was followed by ten collision-induced dissociation (CID) tandem mass spectra for the top 10 most abundant ions. Dynamic exclusion was enabled with a repeat count of three and a repeat cycle of 180 seconds. Lock mass was enabled and set to calibrate on the mass of a polydimethylcyclosiloxane ion ([(Si(CH3)2O)5 + H+]+, m/z=371.10120). Tandem mass spectra were searched against a concatenated forward and reverse [12] mouse NCI protein database using SEQUEST (version 27 revision 12) requiring: fully tryptic peptides, a mass addition of 57.02146 Da for carbamidomethyl adduction on cysteines, a mass addition of 28.03130 Da on amino termini and lysines; and allowing for: a precursor mass tolerance of 40 PPM, a differential mass addition of 15.99491 Da on methionine, and a differential mass addition of 6.03766 Da on amino termini and lysines. SEQUEST matches in the first position were then filtered by XCorr scores of 1.0, 1.5, 1.8 and 2.0 for the charge states of plus one, two, three and four respectively. A ΔCn2 score of 0.2 was also required for each peptide. Protein matches from the GST-CrkL-SH3 pulldowns that were not identified in the GST pulldown, and were made with two or more peptides, were further considered. When such filters were applied to the data searched against the composite forward and reverse mouse NCI protein databases, no reverse peptide hits remained giving a false discovery rate at the peptide level of less than 0.01%. For protein quantification, peptides identified in the mass spectrometry analysis were quantified (heavy/light rations) by Vista [13] using an ≥85% peptide confidence score cutoff. In supplementary material of a previous publication we provide a brief review of SEQUEST and other parameters used here [14]. When manual quantification was done for presentation in figures, the raw data were analyzed in Xcalibur by generating extracted ion chromatograms and averaging MS1 scans for the co-eluting heavy and light peaks. When re-analyzing the data to identify as many C3G peptides as possible, including possible splice junctions, we created a database that contained fasta entries of each two exon sequence that might be generated by splicing. This database, concatenated to its reverse database, was used during the SEQUEST search of all the runs of pulldown experiment number 2 and the search results (Supplementary Table 4) were used to generate Figures 4 and 5. Box-and-Whisker plots in Figure 4 were generated in JMP Pro10 following a one way ANOVA analysis (each pair Students-t test with p < 0.05). RNA-seq reads were collected from the mm10 build of the mouse ENCODE project from the University of California at Santa Cruz Genome Bioinformatics website (https://genome.ucsc.edu/ENCODE/) and were originally generated from both E15.5 and E18.5 murine brain and liver. From total reads we tabulated the exon-exon junction spanning reads.
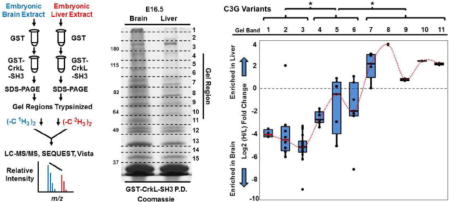
Fig. 4. C3G protein species variants of distinct molecular weights are variably enriched in embryonic murine brain and liver extracts and in CrkL-SH3 pulldowns from the same.
(A) Immunoblotting using a polyclonal anti-C3G antibody (raised with an immunogen to approximately the most N-terminal 300 amino acids) was used to blot the whole cell (tissue) extracts (WCE) and GST-CrkL-SH3 pulldowns from embryonic murine brain and liver. Indicated are both the approximate molecular weights in kDa and the gel region used for the quantitative mass spectrometry. (B) Quantitative mass spectrometry exemplified by a mass spectrum of the indicated light and heavy precursor ion pair of the indicated peptide from C3G identified from gel region three. (C) Quantitative mass spectrometry results of all tryptic C3G peptides in biological replicate number two from the indicated gel regions that identified two or more C3G peptides. Quantifications are the Log2 ratio of the heavy/light (liver/brain) relative abundances from peptides in the CrkL-SH3 pulldowns. Box-and-Whisker plots for the peptides in each gel region are provided with means that are connected with a dotted red line. One way ANOVA analysis using an each pair Students-t test was performed and the statistical significance (*) is indicted between means of quantified C3G peptides in distinct sets of gel regions (p < 0.05).
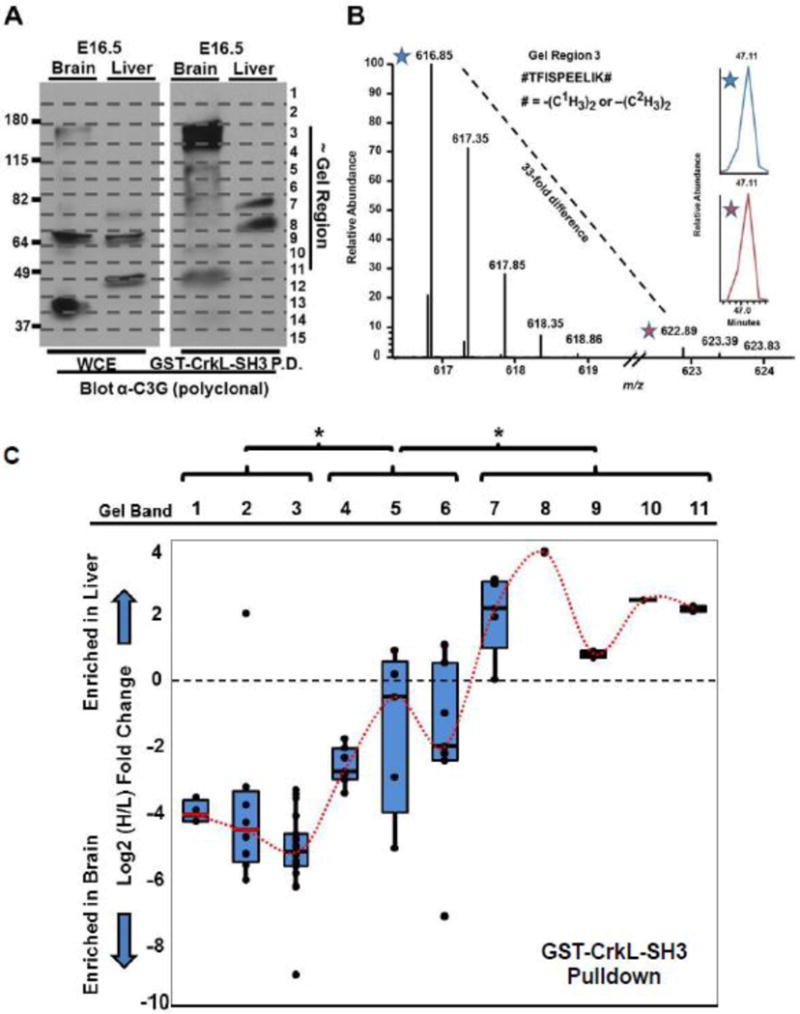
Fig. 5. Mapping of MS-identified C3G peptides from GST-CrkL-SH3 pulldowns and RNA-seq data to functional domains of C3G.
(A) The longest known murine species variant of C3G (1218 amino acids) is diagrammatically displayed highlighting its major functional domains and the amino acid residue numbers that comprise them (See Supplementary Figure 1 for additional details). Also indicated is the region of the immunogen used to generate the anti-C3G polyclonal antibody used in Figure 4. (B) All peptides identified in biological replicate number two are mapped in relationship to these domains. The gel regions in which the peptides were identified are indicated. Also indicated is whether or not the MS/MS spectrum for the peptide was found from light (brain), heavy (liver), or both light and heavy precursor ions. Vertical lines connect peptides from different gel regions with overlapping amino acid sequences. (C) RNA-seq data reveal differences in the relative abundances of discrete exons when examining exon-exon junction reads between embryonic murine brain and liver. Differences in RNA-seq data are consistent the protein analyses showing greater representation in embryonic brain of C3G variants harboring the N-terminal cadherin binding domain. Above the exon numbers are the schematics of the domains from (A) which are encoded by the indicated exons. See Supplementary Table 5 and Supplementary Figure 1 for more details.
2.5 Pulldown assays coupled with immunoblotting
E16.5 brain and liver were lysed in BCLB as described above. After pre-clearing the extracts with glutathione agarose-GST for two hours, glutathione agarose-bound GST-CrkL-SH3 was incubated with the pre-cleared brain and liver extracts overnight. The beads were washed three times with BCLB and proteins were eluted with protein sample buffer and separated by SDS-PAGE. Proteins were transferred to 0.2 μm nitrocellμlose membranes, and membranes were blocked for one hour in 5% dry milk, Tris-buffered saline with 0.05% Tween-20 (TBST). Primary antibody incubation was carried out in 1.5% BSA in TBST at 4°C overnight and the blots were then washed in TBST three times for 5–10 minutes each. Blots were incubated with horse radish peroxidase-conjugated secondary antibodies diluted in TBST for 1–2 hours followed by three 5–10 minute washes with TBST and detection by enhanced chemiluminescence and exposure to x-ray film.
3. Results and discussion
3.1. CrkL-SH3 binding partners are enriched in murine brain over murine liver
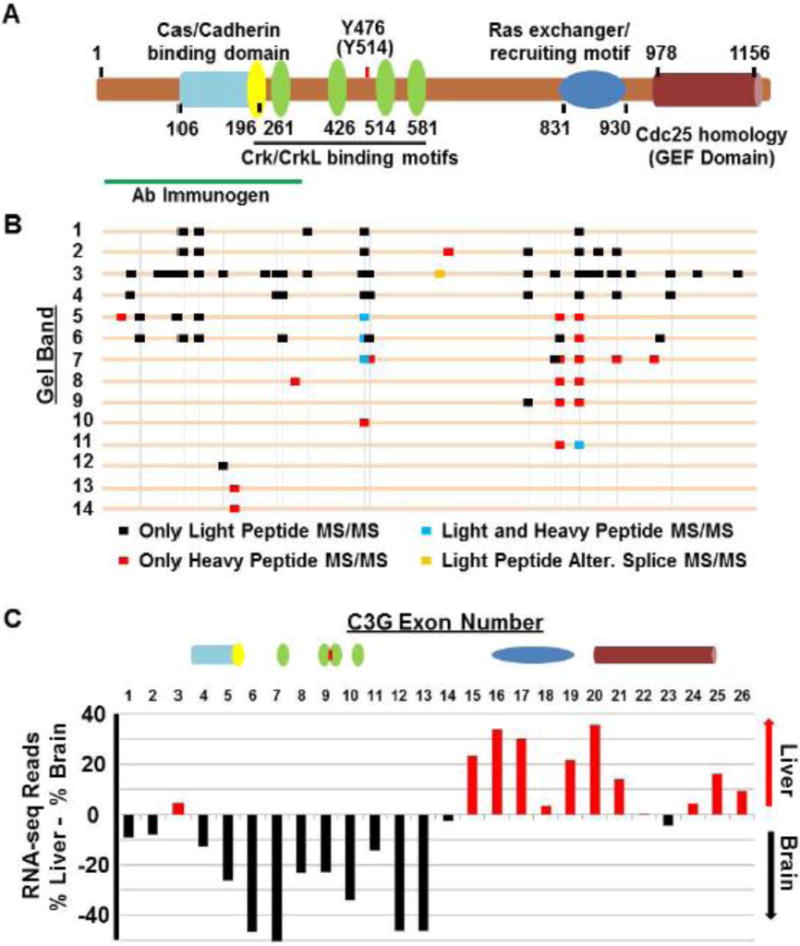
In order to determine if CrkL-SH3 binding partners were similar between embryonic murine brain and a distinctly different embryonic murine tissue, we harvested several embryonic brains and livers from E16.5 mouse embryos. Embryonic liver and brain were chosen for comparison given that at E16.5 these tissues are relatively easy to dissect, are not too different in size, and are differentiating into tissues with dramatically different physiological capacities. In two biological replicate experiments we performed GST-CrkL-SH3 pulldowns from each tissue. The protein complexes were separated using SDS-PAGE. Gel regions were then subjected to in-gel tryptic digestion and alkylation. Peptides were extracted and subjected to reductive amination to dimethylate primary amines and thereby introduce discriminatory stable isotope-based mass tags. The workflow and the coomassie-stained gel of the CrkL-SH3 pulldowns for one of the biological replicates are shown in Figure 1.
Fig. 1. Workflow to identify and quantify the relative abundance of CrkL-SH3 binding proteins in murine embryonic brain and liver extracts.
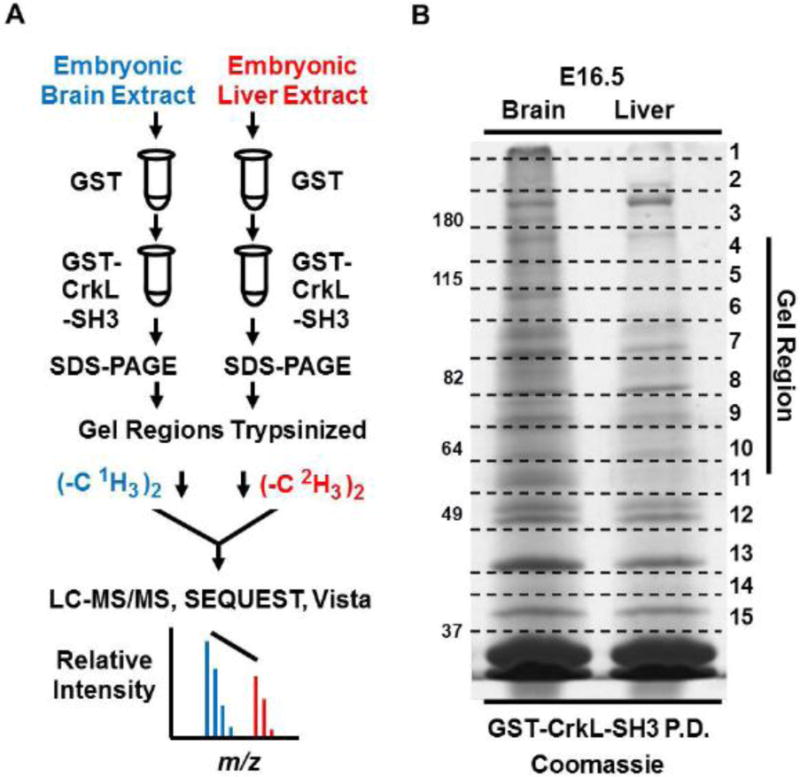
(A) Tissue extracts were pre-cleared with GST resin in batch format. The supernatants were then mixed with GST-CrkL-SH3 resin. Proteins in the pulldowns were subjected to SDS-PAGE. Gel regions were subjected to in-gel digestion and peptides originating from embryonic brain and liver were dimethylated using reagents without or with deuterium respectively. Peptides from the two samples were combined, desalted and subjected to LC-MS/MS. MS data were searched using SEQUEST, and MS1-based relative peptide quantification was performed using Vista. (B) Coomassie-stained gel of experiment one showing GST-CrkL-SH3pulldowns from embryonic murine brain and liver. Gel regions cut for in-gel tryptic digestion are indicated.
Combining proteins identified from both brain and liver extracts and subtracting proteins identified in the GST only pre-clearing step, the two biological replicates identified 41 and 54 GST-CrkL-SH3 binding proteins respectively. Common to the two experiments were 40 proteins (Fig. 2A). All of the peptides and proteins identified from these two experiments are presented in Supplementary Tables 1–3. Out of the 40 proteins common to both experiments we previously reported 30 to be CrkL-SH3 binding proteins identified from embryonic murine brain extracts, while ten were novel (Supplementary Table 3). 75% of the 40 proteins were found to contain CrkL-SH3 binding sites based on Scansite [15] (Supplementary Table 3). Of the 40 CrkL-SH3 binding proteins found common to both experiments 30 were enriched at least two-fold in the brain pulldowns and three were enriched at least two-fold in the liver pulldowns (Fig. 2B). The increased abundance of CrkL-SH3 binding partners in the embryonic brain is consistent with the increased signal from the brain pulldown observed in the coomassie-stained gel (Fig. 1B). Seven proteins did not show an overall enrichment in either tissue. However, when we further examined these seven proteins it was surprising to see that four of them showed variant-specific enrichment in one tissue or the other; four proteins showed enrichment in one tissue from one region of the gel but this enrichment was reversed in another region of the gel.
Fig. 2. Comparison of CrkL-SH3 binding proteins identified from E16.5 murine brain and liver in two biological replicates.
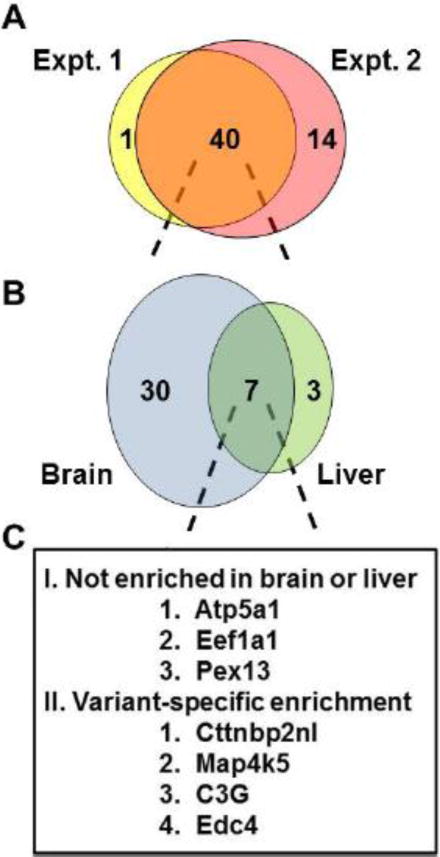
(A) Venn diagram showing the number of identified CrkL-SH3 binding proteins from biological replicates. Proteins from brain and liver were pooled for each replicate. (B) 40 CrkL-SH3 binding proteins were common to the two biological replicates and these are displayed as a function of their relative enrichment (>2 fold) from their source of embryonic brain or liver. (C) Proteins that showed no enrichment or that showed variable enrichment dependent on molecular weight were tabulated.
As a follow-up to the mass spectrometry analysis, we conducted CrkL-SH3 pulldowns from embryonic murine brain or liver extracts followed by immunoblotting for ASAP2, N-WASP, PEX13 and C3G. Consistent with the mass spectrometry results the anticipated full-length ASAP2, N-WASP and C3G showed major enrichments in CrkL-SH3 pulldowns from embryonic brain (Figure 3A). Also consistent with the mass spectrometry results, the novel CrkL-SH3 binding protein PEX13 showed no apparent enrichment from CrkL-SH3 pulldowns comparing embryonic brain and liver extracts (Figure 3A). As a loading control, Figure 3B shows coomassie-stained protein bands in which equal amounts of the brain and liver whole cell lysate (corresponding to 30 μg of input protein) were separated by SDS-PAGE. Although these results importantly compare several CrkL-SH3 binding proteins from these embryonic tissues, the results might be considered predictable given the levels of the proteins in the tissue extracts themselves (Figure 3A). This is consistent with SH3-dependent protein-protein interactions not requiring posttranslational modifications. Therefore, it is assumed that the SH3-dependent interactions we identified were more dependent on relative protein abundances than posttranslational regulatory mechanisms. However, we have previously shown that not all CrkL-SH3 binding proteins bind in a manner proportional to their total levels in embryonic brain extract [3]. Furthermore, mechanisms for the regulated binding to a SH3 domain have been nicely described such as in Grb2’s interaction with SOS which is disrupted by phosphorylation [16].
Fig. 3. Confirmatory immunoblotting of select CrkL-SH3 binding proteins identified from E16.5 murine brain and liver.
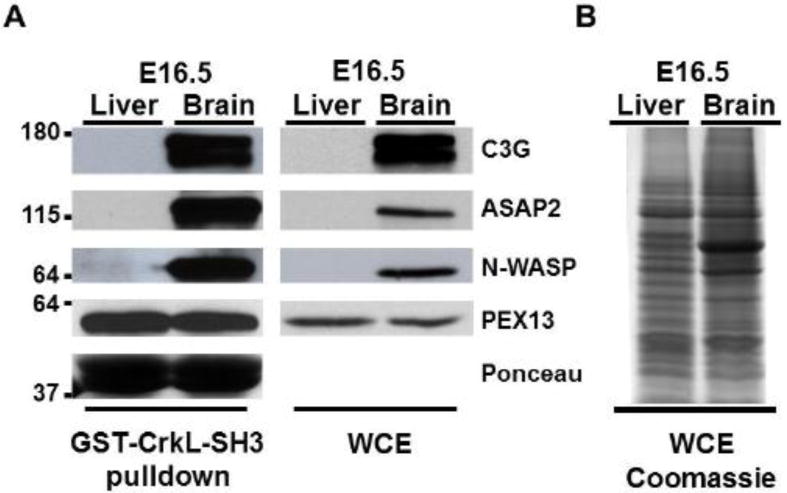
(A) Immunoblots of whole cell (tissue) extracts (WCE) and GST-CrkL-SH3 pulldowns using antibodies that recognize the indicated proteins. Approximate molecular weights in kDa are indicated at left. The Ponceau stain of the GST-CrkL-SH3 levels on the membrane prior to the blocking step is representative of all experiments. (B) Thirty micrograms of each tissue extract used in the pulldowns was separated by SDS-PAGE and the gel was stained with coommassie blue.
Given that the majority of CrkL-SH3 binding proteins were enriched in embryonic brain extracts, we asked if these thirty proteins had specific signaling signatures and we used them as input for functional evaluation using NIH DAVID (Database for Annotation, Visualization and Integrated Discovery)[17, 18]. There were four gene ontology annotation clusters that were found to be at least 2.5 fold enriched in the CrkL-SH3 binding protein dataset from embryonic brain. The most highly enriched (12.9 fold) functional annotation cluster involved SH3 domains, which was a nice confirmation of the screen itself. The next three annotation clusters involved the cytoskeleton/non-membrane-bound organelles, regulators of small GTPases, focal adhesions and PH domain-containing proteins. These results provide a specific list of CrkL-SH3 binding partners that together with CrkL may facilitate developmental roles in migratory cells of the embryonic brain.
3.2. C3G exhibits protein species variants with tissue-specific CrkL-SH3 binding
The results in Figure 3 don’t address how four (ten percent) of the CrkL-SH3 binding proteins in our dataset showed tissue-specific and size-dependent variation in their relative CrkL-SH3 binding. We reasoned it was important to characterize this phenomenon as it suggested protein species variants that might play distinct roles in the two tissue types. C3G was one of the first-identified, and is perhaps the best-characterized, CrkL-SH3 binding protein. We focused our attention on C3G which is a prominent guanine nucleotide exchange factor for members of the Ras superfamily of G-proteins, most-specifically Rap1. C3G plays important roles in a number of pathways such as Reelin, growth factor and integrin signaling [2, 4, 8, 19]. Figure 4A compares anti-C3G immunoblots of the tissue extracts and the CrkL-SH3 pulldowns from the same while demarcating the regions of the gel that were subjected to mass spectrometry analysis. The immunogen used for the polyclonal rabbit antibody employed for Figure 4A was ~300 amino acids long and corresponded to sequence near the extreme N-terminus of C3G. From the blots it is evident that the CrkL-SH3 pulldown enriches for certain protein species variants over others. For example, the protein species variants recognized by the antibody in gel region three are highly enriched in the pulldowns from the brain extracts relative to other protein species variants found in the whole brain extracts. On the other hand, the protein species variants recognized by the antibody in gel regions 7 and 9 are highly enriched in the pulldowns from the liver extracts relative to other protein species variants found in the whole liver extracts. Overall the immunoblots parallel well, but not exactly, the results obtain by mass spectrometry. An example of the mass spectrometry-based quantification of peptides is shown in Figure 4B for a tryptic C3G peptide found in gel region three that was found to be 33-fold enriched in the CrkL-SH3 pulldown from embryonic brain. In Figure 4C the relative quantifications between embryonic brain and embryonic liver for all of the peptides from experiment two for C3G from each gel region with at least two different quantified peptides are plotted in Box-and-Whisker plots. We chose experiment number two to display given it yielded many more total peptides and would provide a richer comparison. Peptide quantifications in gel regions 1–3 were significantly different than those in gel regions 4–6, which were again significantly different from peptide quantifications from gel regions 7–11. Note that in this analysis we re-searched the original data using a custom database specifically for C3G but which was composed of fasta entries of the amino acid sequences of all possible two exon splices. For example the amino acid sequence for exon one was fused to each of three possible frames for exon two to generate three distinct fasta entries. This was continued to generate three more entries with exon one fused to exon three and so on. We reasoned that by this in silico splicing all possible tryptic peptides would be available for identification, including peptides that straddled known and potentially novel splice junctions. Our search results found several peptides that covered splice junctions, but only one that was an alternative splicing event that joined exon 11 with exon 15 to give the tryptic peptide QLEPPSGK. We anticipate that our inability to detect additional splice variants was due to a low sample abundance issue in addition to the fact that some splice junctions will simply not be amenable to detection using trypsin. This approach is helpful in identifying splice variants in both large-scale and single protein analyses as a companion to searches that make use of important splice variant databases [20, 21].
3.3. Functional inference of CrkL-SH3 binding C3G protein species variants based on peptide mapping
When alternative splicing or proteolysis leads to a protein deficient in a given functional domain, the outcome can be variable, including the deficient molecule acting as a dominant negative, or the molecule having increased or gain of functionality, or having altered subcellular localization. C3G has three main regions of function. At the N-terminus is a Cas and Cadherin binding region, followed by the proline-rich region with at least four CrkL-SH3 binding motifs. The C-terminal third of the protein has two domains that function together to execute guanine nucleotide exchange [22] for the small G-protein on which they act. Loss of the Cas and Cadherin binding region would primarily alter subcellular location. Loss of various CrkL-SH3 binding motifs would lower affinity/avidity for CrkL and reduce its localization to proteins where the CrkL-SH2 domain would recruit it. Finally, compromising the guanine nucleotide exchange activity would have implications on the molecules ability to participate in signal transduction. Interestingly, it is known that loss of functional domains by alternative splicing occurs more often than by chance alone [20] reflecting evolved cellular capacities to use splicing as a means to alter protein function.
In Figure 5 we map all the C3G peptides that we identified from the CrkL-SH3 pulldown in experiment number two, using a concept similar to mapping approaches by others [23]. By color-coding we retained the information regarding whether MS/MS events were triggered from light-labeled peptides, heavy-labeled peptides or both. Consistent with the relative abundances plotted in Figure 4C, MS/MS events from light-labeled peptides were triggered more often in higher molecular weight gel regions compared to MS/MS events from heavy peptides. Furthermore the general trend is for the higher regions of the gel to contain peptides scattered across all functional domains. The middle-regions of the gel show a trend toward C3G molecules that lack the N-terminal Cas/Cadherin binding region. Interestingly, the few C3G peptides found in the lowest molecular weight bands map to the N-terminus, possibly a reflection of a proteolytic truncation. Presumably all identified peptides originated from C3G molecules bound directly to the CrkL-SH3 domain and thus contain at least a few of the CrkL-SH3 binding motifs even if they weren’t identified by MS/MS. The overall pattern suggest that the lack of the Cas/Cadherin binding region is the major different in the E16.5 liver protein species variants compared to the E16.5 brain protein species variants. Cadherin’s play important roles in cell adherence and serve as β-catenin-dependent sites of actin nucleation [24]. Cas plays important roles in cell adhesion as well, particularly regarding focal adhesions [25, 26]. Interestingly, Cas is a major interacting protein with the SH2-domain of Crk and CrkL following tyrosine kinase signaling [26]. Perhaps the roles of C3G in the embryonic liver, while likely still important for regulating Rap1, are conducted primarily by recruiting C3G to CrkL-SH2 binding partners, whereas in embryonic brain C3G is more directly and constitutive localized to focal adhesions and Cadherins. Of note, however, is that for robust activation of C3G, its phosphorylation at Y504 is needed by tyrosine kinases such as SFKs [27]. Furthermore, tyrosine phosphorylation of C3G at Y504 directs its subcellular localization [28]. Therefore, localization of C3G to Cadherins and focal adhesions may serve as a priming event to facilitate more rapid regulation of actin dynamics. If this priming were coupled to avidity differences held by molecules having different numbers of CrkL-SH3 binding motifs, the cell would conceivable have a very large differential in its ability to concentrate C3G to various locals, even perhaps allowing it to more readily activate G-proteins in addition to Rap1 [19]. Additionally, given that tyrosine 504 lies within the proline-rich region that interacts with the Crk/CrkL SH3 domain, it is conceivable that protein-protein complexes that dock to phosphorylated tyrosine 504 of C3G could block the ability of C3G to bind to the Crk/CrkL-SH3 domain. While the stoichiometry of phosphorylation at tyrosine 504 is unlikely to be universally high in a heterogeneous tissue extract, differential posttranslation modification at this site or others remain formal reasons for our observed differences in the relative binding interactions of proteins from different tissues or from different regions of the gel.
3.4. The profile of embryonic murine C3G liver and brain RNA-seq splice junction reads are consistent with identified C3G protein species variants
It is well appreciated that the complexity of proteomes is enormous, in part due to the complexity of alternative splicing [29]. While a few transcript variants are known to exist for C3G in different organisms [19, 30–32], our protein data suggested increased variability than previously predicted, at least when examining the embryonic tissues of the mouse brain and liver. We therefore examined publically-available RNA-seq reads measured from E15.5–E18.5 murine brain and liver. We specifically counted and tabulated RNA-seq reads that spanned any exon-exon junction (Supplementary Table 5). We summed individually the junction reads involving each exon and set the highest number of reads as 100% for each tissue type. We then normalized all reads for that tissue and examined the difference in exon coverage between the two tissues. All things being otherwise equal the difference between the number of reads from one exon in the brain to the number of reads from the same exon in the liver would be zero. However, the profile obtained showed a striking relative increase in splice junction reads encoding the N-terminal half or the C-terminal half of C3G from embryonic brain and embryonic liver respectively (Figure 5C). We mapped the functional domains of C3G to these exons (Figure 5C and Supplementary Figure 1) and found the results strikingly similar to the profiles observed in the immunoblotting (Figure 4) and the identified peptides from the specific gel regions of proteins bound to the CrkL-SH3 domain (Figure 5B). Together these data point to an increase in the relative abundance of C3G protein species variants as a consequence of differential RNA processing. However, we cannot rule out that some of these bands are the result of various post-translational modifications or even some proteolysis.
3.5. Increased variance in protein quantifications obtained through bottom-up proteomics suggests increased richness in protein species variants
The variability in abundance that we observed in C3G variants suggested that we might find a correlation between increased protein species number and overall protein quantification variance. That is to say, if a high standard deviation were found around the mean protein abundance, as determined by the collection of individual peptide quantifications, this would suggest the existence of at least two protein species variants of different relative abundance. We emphasize that we are discussing protein variance within a single experiment type and that such variance would be observed in, and hold up across, biological replicates. However, to be clear, we are not describing variance that might emerge simply as a function of biological replicates. We therefore predicted that we would find increased overall protein quantification variance when comparing proteins in our CrkL-SH3 pulldown that were identified in several gel regions in the same experiment given that proteins in different gel regions likely represented different protein species variants and may be found at different relative abundances. On the other hand, we predicted that proteins identified in a single gel region would have fewer overall species variants and therefore less variance in their calculated abundance. Indeed we found this to be the case when comparing the brain to liver quantifications for proteins identified in a single gel region compared to proteins identified in more than four gel regions (Figure 6). This is an indication of the known problem in bottom-up proteomics that for many proteins their “identification” is subjective [33] given the huge potential variability in protein species variants. In experiments where molecular weight information is retained we recommend re-examining the proteins with high variance in their quantifications to determine if indeed there might be multiple protein species variants with distinct abundances dependent on their observed molecular weights. In gel-free experiments we propose that high variance in a subset of proteins should be considered to be an indicator of potential source of biological richness and not simply as inappropriate quantification.
Fig. 6. Increased standards deviations in peptide quantifications for proteins found in multiple gel regions.
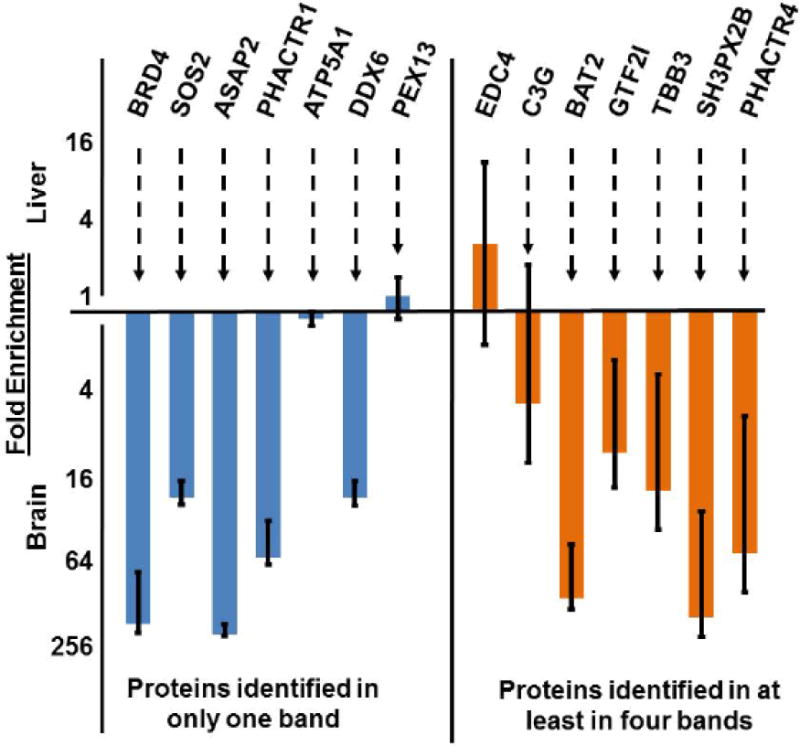
The means and standard deviations of Embryonic brain versus liver quantifications of peptides from CrkL-SH3 binding proteins that were identified in only one gel region (left side) versus peptides from proteins that were identified in at least four bands (right side) are indicated. Note that the y-axis is logarithmic.
4. Conclusions
We have identified major differences in the quantitative profiles of CrkL-SH3 binding proteins between embryonic murine brain and liver, suggesting distinct differences in signaling capacity between these two tissues. Furthermore, we have identified a few CrkL-SH3 proteins with relative abundances which do not differ uniformly between liver and brain but differ dependent on the protein species quantified. We took an in-depth look at one of these proteins, C3G, and found that the variance is in part due to differences in RNA processing, leading to the prediction that C3G has different functional capacities between embryonic liver and brain. Together our results suggest that high variance when using bottom-up proteomic quantification, particularly when obtained without molecular weight information, could be an indication of protein species variants with differential levels between the states or tissues being quantified. Finally, while the power and utility of bottom-up proteomics is unquestioned, our results suggest that high protein quantification variance should be examined cautiously and can be used as a signature to explore differences in protein species variants and their functional capacity between biological samples.
Supplementary Material
Significance.
Crk and CrkL are essential players in a number of signaling pathways including Reelin signaling which regulates neuronal positioning during brain development. This study identifies and comparatively quantifies CrkL-SH3 binding proteins between murine embryonic brain and liver. Furthermore it highlights potential caveats in quantifying protein species variants by bottom-up proteomics. Nevertheless, even without molecular size information, signatures of major variants of different relative abundances could still be inferred due to large standard deviations in peptide quantifications for a given protein. This is important when considering that the bioactivities of protein species variants can be dramatically different and may be decoupled from the protein’s total abundance.
Highlights.
Quantitative Proteomics Reveals Tissue-Specificity in CrkL-SH3 Binding Proteins.
High Standard Deviations in Peptide Quantification Suggest Hidden Protein Variants.
CrkL-SH3-binding C3G Variants Exhibit Functional Domain-Specific Variability
Acknowledgments
This work was supported by NSF grant IOS 1021795, the Vermont Genetics Network through NIH Grant 8P20GM103449 from the INBRE program of the NIGMS, and NIH Grant 5 P20 RR016435 from the COBRE (neuroscience) program of the NIGMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that they have no competing or conflicting interests including financial, institutional or personal.
References
- 1.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–5. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 2.Feller SM. Crk family adaptors-signalling complex formation and biological roles. Oncogene. 2001;20:6348–71. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 3.Cheerathodi M, Ballif BA. Identification of CrkL-SH3 binding proteins from embryonic murine brain: implications for Reelin signaling during brain development. Journal of proteome research. 2011;10:4453–62. doi: 10.1021/pr200229a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell communication and signaling: CCS. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guris DL, Fantes J, Tara D, Druker BJ, Imamoto A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat Genet. 2001;27:293–8. doi: 10.1038/85855. [DOI] [PubMed] [Google Scholar]
- 6.Park TJ, Boyd K, Curran T. Cardiovascular and craniofacial defects in Crk-null mice. Molecular and cellular biology. 2006;26:6272–82. doi: 10.1128/MCB.00472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park TJ, Curran T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J Neurosci. 2008;28:13551–62. doi: 10.1523/JNEUROSCI.4323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballif BA, Arnaud L, Arthur WT, Guris D, Imamoto A, Cooper JA. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr Biol. 2004;14:606–10. doi: 10.1016/j.cub.2004.03.038. [DOI] [PubMed] [Google Scholar]
- 9.Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, et al. Receptor clustering is involved in Reelin signaling. Molecular and cellular biology. 2004;24:1378–86. doi: 10.1128/MCB.24.3.1378-1386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takagi J. Crystal Structure of Reelin Repeats. In: Fatemi SH, editor. Reelin Glycoprotein. New York, NY: Springer; 2008. pp. 57–67. [Google Scholar]
- 11.Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, et al. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol. 2007;3:339–48. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- 12.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakalarski CE, Elias JE, Villen J, Haas W, Gerber SA, Everley PA, et al. The impact of peptide abundance and dynamic range on stable-isotope-based quantitative proteomic analyses. Journal of proteome research. 2008;7:4756–65. doi: 10.1021/pr800333e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doubleday PF, Ballif BA. Developmentally-Dynamic Murine Brain Proteomes and Phosphoproteomes Revealed by Quantitative Proteomics. Proteomes. 2014;2:197–207. doi: 10.3390/proteomes2020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic acids research. 2003;31:3635–41. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corbalan-Garcia S, Yang SS, Degenhardt KR, Bar-Sagi D. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Molecular and cellular biology. 1996;16:5674–82. doi: 10.1128/mcb.16.10.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radha V, Mitra A, Dayma K, Sasikumar K. Signalling to actin: role of C3G, a multitasking guanine-nucleotide-exchange factor. Bioscience reports. 2011;31:231–44. doi: 10.1042/BSR20100094. [DOI] [PubMed] [Google Scholar]
- 20.Resch A, Xing Y, Modrek B, Gorlick M, Riley R, Lee C. Assessing the impact of alternative splicing on domain interactions in the human proteome. Journal of proteome research. 2004;3:76–83. doi: 10.1021/pr034064v. [DOI] [PubMed] [Google Scholar]
- 21.Tavares R, de Miranda Scherer N, Pauletti BA, Araujo E, Folador EL, Espindola G, et al. SpliceProt: a protein sequence repository of predicted human splice variants. Proteomics. 2014;14:181–5. doi: 10.1002/pmic.201300078. [DOI] [PubMed] [Google Scholar]
- 22.Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, et al. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16692–7. doi: 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Hultin-Rosenberg L, Forshed J, Branca RM, Orre LM, Lehtio J. SpliceVista, a tool for splice variant identification and visualization in shotgun proteomics data. Molecular & cellular proteomics: MCP. 2014;13:1552–62. doi: 10.1074/mcp.M113.031203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, et al. Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science. 2014;346:1254211. doi: 10.1126/science.1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. Journal of cellular biochemistry. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 26.Bouton AH, Riggins RB, Bruce-Staskal PJ. Functions of the adapter protein Cas: signal convergence and the determination of cellular responses. Oncogene. 2001;20:6448–58. doi: 10.1038/sj.onc.1204785. [DOI] [PubMed] [Google Scholar]
- 27.Ichiba T, Hashimoto Y, Nakaya M, Kuraishi Y, Tanaka S, Kurata T, et al. Activation of C3G guanine nucleotide exchange factor for Rap1 by phosphorylation of tyrosine 504. The Journal of biological chemistry. 1999;274:14376–81. doi: 10.1074/jbc.274.20.14376. [DOI] [PubMed] [Google Scholar]
- 28.Radha V, Rajanna A, Mitra A, Rangaraj N, Swarup G. C3G is required for c-Abl-induced filopodia and its overexpression promotes filopodia formation. Experimental cell research. 2007;313:2476–92. doi: 10.1016/j.yexcr.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 29.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–63. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shivakrupa, Singh R, Swarup G. Identification of a novel splice variant of C3G which shows tissue-specific expression. DNA and cell biology. 1999;18:701–8. doi: 10.1089/104454999314980. [DOI] [PubMed] [Google Scholar]
- 31.Zhai B, Huo H, Liao K. C3G, a guanine nucleotide exchange factor bound to adapter molecule c-Crk, has two alternative splicing forms. Biochemical and biophysical research communications. 2001;286:61–6. doi: 10.1006/bbrc.2001.5348. [DOI] [PubMed] [Google Scholar]
- 32.Knudsen BS, Feller SM, Hanafusa H. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. The Journal of biological chemistry. 1994;269:32781–7. [PubMed] [Google Scholar]
- 33.Rappsilber J, Mann M. What does it mean to identify a protein in proteomics? Trends in biochemical sciences. 2002;27:74–8. doi: 10.1016/s0968-0004(01)02021-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.