

Abstract
Endoplasmic reticulum (ER) stress induces a variety of neuronal cell death pathways that play a critical role in the pathophysiology of Stroke. ER stress occurs when unfolded/misfolded proteins accumulate and the folding capacity of ER chaperones exceeds the capacity of ER lumen to facilitate their disposal. As a consequence, a complex set of signaling pathways will be induced that transmit from ER to cytosol and nucleus to compensate damage and to restore the normal cellular homeostasis, collectively known as unfolded protein response (UPR). However, failure of UPR due to severe or prolonged stress leads to cell death. Following acute CNS injuries, chronic disturbances in protein folding and oxidative stress prolong ER stress leading to sustained ER dysfunction and neuronal cell death. While ER stress responses have been well studied after stroke, there is an emerging need to study the association of ER stress with other cell pathways that exacerbate neuronal death after an injury. In this review we summarize the current understanding of the role for ER stress in acute brain injuries, highlighting the diverse molecular mechanisms associated with ER stress and its relation to oxidative stress and autophagy. We also discussed the existing and developing therapeutic options aimed to reduce ER stress to protect the CNS after acute injuries.
Keywords: ER stress, Oxidative stress, Autophagy, crosstalk, acute CNS injury
1. Introduction
Endoplasmic reticulum (ER) plays a role in many essential cellular processes that include maintenance of intracellular Ca2+ homeostasis, folding of the newly synthesized secretory and membranous proteins and post-translational modifications (1, 2). As ~30% of the newly synthesized proteins are rapidly degraded due to improper folding (3), any increase in protein translation leads to a potential buildup of misfolded/unfolded proteins that stresses the cell. If this is combined with perturbations in the ER microenvironment such as alterations in redox state, depletion of Ca2+ levels and failure of posttranslational modifications, cells will be further stressed. As a result, the protein folding capacity of ER will be compromised, resulting in further accumulation of misfolded/unfolded proteins leading to unfolded protein response (UPR) generally referred as ER stress (4). The major goals of UPR/ER stress are 1) to shutdown protein translation to reduce the newly synthesized protein load, 2) to induce ER chaperones that promote protein folding, and 3) to activate ubiquitylation and proteasomal degradation of the misfolded/unfolded proteins. However, if stress is severe and persistent, UPR signaling switches from pro-survival to pro-apoptotic. ER stress is associated with numerous pathophysiological conditions including diabetes, stroke, traumatic injury to CNS and many neurodegenerative disorders (5).
ER stress precipitates neuronal death by multiple synergistic mechanisms. A major mechanism is the disruption of Ca2+ homeostasis that plays an important role in neuronal function and survival (2). ER is the major store for cellular Ca2+ and disruption of ER-associated Ca2+ channels including ryanodine receptors (RyRs) due to energy failure after stroke releases intracellular Ca2+ that induces proteases and nucleases leading to necrotic cell death. Depletion of Ca2+ stores in ER, activation of ER-associated Ca2+ -ATPases and failure of endoplasmic reticulum oxidoreductin-1 alpha (ERO1α) leading to disrupted protein disulphide bond formation also decrease protein folding leading to further accumulation of unfolded proteins (2, 6–9). Furthermore, ER stress induces cell death pathways associated with autophagy and apoptosis (10). All the above pathways collaborate to precipitate the neuronal death due to ER stress following acute CNS injuries (Figure 1). Although, limited UPR/ER stress is needed to induce neuroprotective mechanisms, excess ER stress leads to cell death and the molecular mechanisms that facilitate the switch from protection to death are yet to be understood completely. This review will discuss the current understanding of the complex signaling events induced by ER stress, highlighting the roles of ER stress in the pathophysiology of acute CNS injuries and emerging therapeutic opportunities for drug discovery.
Figure 1.
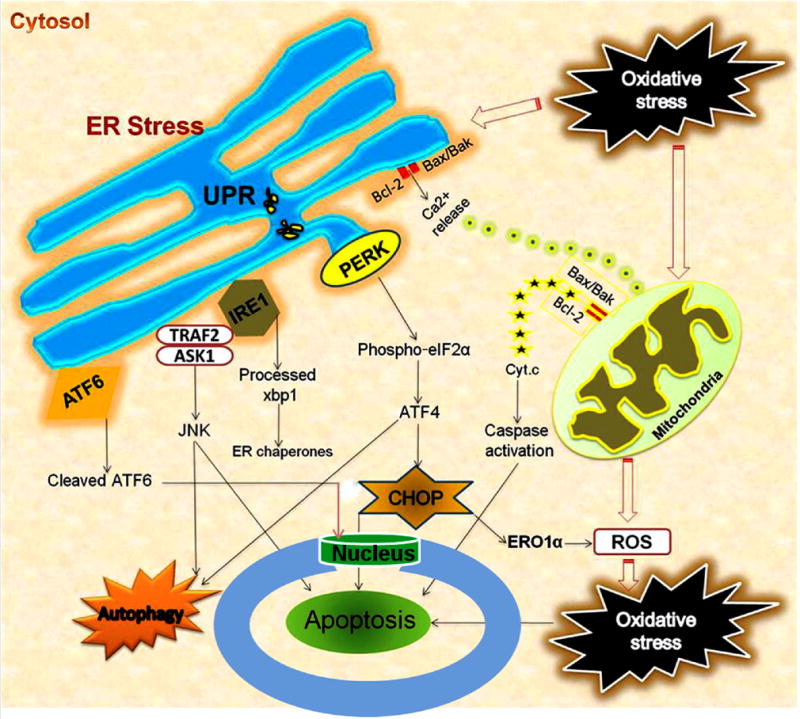
Interaction of ER stress, oxidative stress, mitochondrial dysfunction and autophagy following acute CNS injuries. Oxidative stress/ROS trigger ER stress, and ER stress exacerbates ROS production. UPR leads to activation of ER transmembrane kinases PERK, IRE1, and ATF6. PERK activates phosphorylation of eIF2α and halts protein translation, but can also induce expression of ATF4 due to presence of alternate ORFs in ATF4 mRNA. ATF4 induces CHOP which in turn induces many downstream genes leading to apoptosis and autophagy. Further, CHOP also induces ER oxidase ERO1α thus rendering the ER more oxidized. JNK activated by IRE1-TRAF2-ASK1 complex induces autophagy and apoptosis if unrestrained. The proapoptotic Bcl-2 family members residing on ER induces Ca2+ release from ER leading to mitochondrial dysfunction, ROS generation and apoptosis. ATF6 translocates to the nucleus and activates the transcription of ERAD genes and XBP1. In the nucleus, the cytosolic fragment of cleaved ATF6 binds to cis-acting ER stress response element and UPR element, and up-regulates major ER chaperones and ERAD components responsible for cell survival.
2. UPR-signaling pathways
UPR activates 3 major signaling pathways initiated by prototypical ER localized stress sensors: pancreatic ER kinase (PKR)-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF-6) (11). Under normal physiological conditions, all 3 effectors bind to the ER chaperone 78KDa glucose-regulated protein/binding immunoglobulin protein (GRP78/BIP) on their luminal domains; thus GRP78 suppress their activity (12). Under conditions of ER stress, when misfolded proteins accumulate in the ER lumen, GRP78 dissociates from the PERK, ATF-6 and IRE1, allowing their activation (13). Activation of the ER signaling pathways help to fight the cellular stress due to UPR by suppressing the translation of new proteins and thus reduce the load of unfolded/misfolded proteins, by inducing the ER chaperones that promote protein refolding and by activation of proteasome that degrade the misfolded/unfolded proteins. Thus, the primary function of the UPR/ER stress signaling is to promote the cell survival under hostile conditions.
2-1. PERK pathway
The primary cellular response to ER stress is transient global translation attenuation (13). Dissociation of GRP78 initiates dimerization and autophosphorylation of PERK leading to its activation. PERK activation is the first indicator of UPR that is evident during the early hours of reperfusion after cerebral ischemia (14). Once activated, PERK phosphorylates serine-51 residue of eukaryotic translation initiation factor 2 subunit α (eIF2α) preventing the 80S ribosomal assembly and thus curtails global protein synthesis (15–16). However, certain PERK downstream proteins like activating transcription factor 4 (ATF4) continued to be translated due to the presence of specific arrangement in the open reading frame-2 (ORF2) of ATF4 mRNA (17). ATF4 is known to promote cell survival by inducing ER stress target genes that control amino acid metabolism, redox reactions, stress response and protein secretion (18). However, prolonged activation of ATF4 induces its down-stream proinflammatory transcription factor C/EBP homologous protein/growth arrest and DNA damage-inducible gene 153 (CHOP/GADD153) and further down-stream growth arrest and DNA damage-inducible gene 34 (GADD34) which forms a complex with protein phosphatase 1 (PP1) that mediates translational recovery by dephosphorylation of p-eIF2α (19). The importance of PERK pathway in curtailing ER stress has been well documented. Cells lacking PERK and knock-in cells that express a non-phosphorylatable form of eIF2α (S51A) showed significant hypersensitivity to ER stress (20). Furthermore, attenuation of translational recovery by pharmacological inhibition of p-eIF2α dephosphorylation using salubrinal protects cells from ER stress-induced apoptosis (21). Salubrinal prevents formation of GADD34/PP1 complex and thus attenuates dephosphorylation of p-eIF2α (21). Salubrinal administration was shown to decrease neuronal death in experimental rodent models of epilepsy, excitotoxicity and focal ischemia (22–23). Thus, activation of PERK-eIF2α pathway is critical for survival of neurons during the acute phase after a brain insult. However, uncontrolled activation of PERK pathway promotes cell death via transcriptional responses mediated by ATF4 and CHOP, which inhibit the expression of pro-survival B-cell lymphoma 2 (BCL-2) and anti-apoptotic gene Bcl-2-associated X protein (BAX) down-stream to CHOP, and activate BCL-2-interacting mediator of cell death (BIM), p53 up-regulated modulator of apoptosis (PUMA) and Tribbles homolog 3 (TRB3) (24–25).
2-2. IRE1 pathway
IRE1α is a 100 kDa type I transmembrane protein that has a Ser/Thr kinase domain and an endoribonuclease domain (60). During ER stress, GRP78 dissociation leads to dimerization and activation of IRE1 (26). Upon activation, the endonuclease activity of IRE1 specifically cuts a 26-nucleotide intron from the transcription factor X-box binding protein-1 (xbp1) mRNA leading to a shift in its open reading frame (26). Processed xbp1 mRNA is translated into a 54 KDa protein that induces the expression of glucose-regulated proteins such as GRP78 and GRP94 (27). Once, adequate new GRP78 protein has been synthesized, it binds to unfolded proteins (enabling them to refold) and to PERK and IRE1 to restore the normal ER function. IRE1α activation and xbp-1 processing are early neuroprotective events after acute brain insults like ischemia (28–29, 23). However, depending on ER stress levels, the IRE1α promotes either adaptation or apoptosis. Under chronic ER stress, IRE1α’s RNase relaxes its endonucleolytic activity to cleave ER localized mRNAs and non-coding RNAs, leading to apoptosis (30–33). Interestingly, kinase-inhibiting RNase attenuators (KIRAs) allosterically inhibit IRE1α’s kinase/RNase activity by breaking oligomers leading to inhibition of apoptosis (34). Furthermore, blocking IRE1α with KIRA6 (an optimized KIRA), promotes cell survival and preserves physiological functions against chronic ER stress in vivo (34).
Activation of ER-localized caspase-12 is known to occur following acute brain injuries (35–37). Indeed, mice lacking capase-12 are resistant to ER stress induced apoptosis (38). In unstressed conditions GRP78 binds to both IRE1α (12) and procaspsae-12 (39), whereas during ER stress GRP78 dissociate from IRE1α and procaspsae-12, and binds to unfolded proteins. The dissociation of IRE1α and procaspsae-12 from GRP78 was observed in cultured astrocytes after acidosis-induced ER stress (40). Although, direct evidence is lacking on the interaction between IRE1α and procaspase-12, the IRE1α binding partner TRAF-2 (tumor necrosis factor-receptor associated factor 2) is known to interact with procapase-12 to promote clustering and activation of procaspase-12 during ER stress (41). Activated IRE1α recruits TRAF2 to the ER membrane, which is regulated by c-Jun NH2-terminal inhibitory kinase (JIK) (42, 41). The IRE1α/TRAF2 complex then recruits apoptosis signal-regulating kinase 1 (ASK1) and activates the downstream JNK pathway, which promotes neuronal death (43–44). Furthermore, the dominant-negative TRAF-2 inhibits the activation of JNK by IRE1 (42). Post-ischemic inhibition of JNK with a cell penetrating, protease inhibitor peptide called D-JNK-1 was shown to result in a robust and long-term neuroprotection and improved neurological function after focal and global ischemia (45). Small molecule inhibitors of JNK like SP600125 reduce cerebral infarct volume after experimental ischemia in mice (46). Moreover, JNK inhibition also prevents mitochondrial translocation of BAX and BIM, release of cytochrome c and second mitochondrial-activated factor (Smac), and subsequent activation of caspase-9 and caspase-3 (46). Overall, IRE1α signaling effectively controls cell fate, but can be controlled pharmacologically to reduce the cell death under sustained ER stress.
2-3. ATF6 pathway
Dissociation of transcription factor ATF-6 from GRP78 leads to translocation of ATF6 to Golgi, where it is cleaved by site-1 and site-2 proteases to yield an active N-terminal 50kDa domain (N-ATF-6/p50ATF-6) that translocate to the nucleus (47). In the nucleus, ATF6 binds to cis-acting ER stress response element and UPR element, and up-regulates major ER chaperones and ER-associated protein degradation (ERAD) components (47). Studies with mice lacking ATF6 suggested that ATF6α is required to optimize ER functions such as protein folding, secretion and degradation to protect cells from chronic ER stress (48). Further, ATF6α ablation protected neurons against 1-Methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) induced neurotoxicity by increasing ER chaperones and ERAD (49). Thus, ATF6 activation appears to be a pro-survival endogenous adaptation to counteract ER stress.
3-1. Synergy of ER stress and oxidative stress
Reactive oxygen species (ROS) are produced at low levels during normal physiological conditions and are scavenged by endogenous antioxidant enzymes that include superoxide dismutase, glutathione peroxidase and catalase (50). Following insults like ischemia, generation of ROS overrides the ability of the endogenous antioxidant system leading to oxidative stress and neuronal cell death (51). On the other hand, production of ROS also increases after CNS insults. Postmortem brains of patients suffered from neurodegenerative disorders display increased ROS in affected brain regions (52). Excessive Ca2+ influx into the cell after an insult due to NMDA receptor activation leads to increased superoxide production by cytosolic NADPH oxidase and mitochondrial electron transport chain (53–54). NADPH oxidase is known to be the primary source of superoxide production via NMDA activation (55). NADPH oxidase is composed of catalytic and regulatory subunits, and when they get activated, they translocate to plasma membrane and combine with an assembly subunit (55). Neurons predominantly express the NOX2 isoform of NADPH oxidase that contain the gp91 catalytic subunit and requires the p47phox assembly subunit (53) and inhibition or genetic deletion of NOX2 and/or p47phox was shown to reduce neuronal damage after stroke experimental stroke (56). Alleviation of either ER stress or oxidative stress protects neurons from apoptosis in neurodegenerative disorders (57). Many studies further indicate that ER stress and oxidative stress potentiate each other in several conditions including diabetes, atherosclerosis, renal dysfunction and neurodegenerative diseases (58–60). Interestingly, NOX-mediated oxidative stress was shown to be induced by UPR/ER stress, and ER-stress mediated apoptosis was shown to be blocked by genetic deletion of NOX2 or treatment with the antioxidant N-acetylcysteine (61).
ER provides a unique oxidizing folding-environment that favors the formation of the disulfide bonds and hence, protein folding generates ROS as a byproduct in the ER lumen (62). Activation of the UPR in a controlled manner on exposure to oxidative stress might be an adaptive mechanism to preserve cell function and survival. However, excess ER stress promotes the accumulation of ROS and thus exacerbates the oxidative stress (62).
PERK pathway is a molecular mechanism that links oxidative stress and ER stress. Transcription factor Nrf2 (nuclear factor (erythroid-derived 2)-like 2) induces the expression of many anti-oxidant genes and hence considered as a master regulator of the antioxidant response of cell (63). Under physiological conditions, Kelch-like ECH-associated protein 1(Keap1) binds to Nrf2 to maintain in an inactive form. Whereas, PERK-dependent phosphorylation of Nrf2 dissociates Nrf2 from the Nrf2/keap1 complex allowing its translocation to the nucleus to promote the expression of antioxidant genes (64). Moreover, Nrf2 together with ATF4 induces antioxidant response element (ARE) dependent gene transcription, suggesting the convergence of ER stress and oxidative stress signaling pathways (65). The ER oxidoreductase ERO1α forms disulfide bonds that are essential for protein refolding and thus helps to relieve ER stress. However, as ERO1α transfers electrons to molecular oxygen during its activity, it forms ROS as a byproduct (66). The pro-apoptotic transcription factor CHOP (downstream to PERK) induces ERO1α expression, thus rendering the ER more oxidized and cells lacking CHOP attenuated ERO1α induction against ER stress (19). As a result, ER calcium channel inositol 1, 4, 5-triphosphate receptor (IP3R) releases intracellular Ca2+ from ER lumen to the cytosol (67). The IP3R-induced Ca2+ release is blocked by siRNA-mediated silencing of ERO1α or IP3R1 and by loss-of-function mutations in ERO1α or CHOP (67). Further, due to release from ER store, Ca2+ concentration within the mitochondria and eventually stimulates ROS production (68). The close proximity of ER and mitochondria leads to accumulation of Ca2+ in the mitochondrial microenvironment and further sensitizes the mitochondria to ROS that eventually causes the opening of mitochondrial permeability transition pore (69). During early stages of apoptosis, Cytochrome c released from the mitochondria translocate to ER and selectively binds to IP3R, resulting in a sustained increase of cytosolic Ca2+ (70). ER stress further triggers release of Cytochrome c from the mitochondria leading to more Ca2+ release from the ER in a feedback loop (70). Furthermore, mitochondrial ROS leads to thiol oxidation of ryanodine receptors (RyRs; another ER Ca2+ release channel), causing its activation and release of Ca2+ from the ER (71–72). Thus, Ca2+ acts as an input signal in activating IP3Rs or the RyRs, a process known as Ca2+ -induced Ca2+ release (73). The ER Ca2+ released through RyRs and IP3Rs may contribute to excitotoxicity of neurons (74). The involvement of glutamate-mediated excitotoxicity is well-documented after acute CNS injuries and chronic neurodegenerative disorders (75–76). Interestingly, the ER stress inhibitor salubrinal protects oligodendrocytes and neurons from glutamate receptor-mediated excitotoxicity (77, 22). Collectively, it appears that ER stress, oxidative stress and mitochondrial dysfunction are closely linked events.
3-2. ER stress and mitochondrial dysfunction
Cell death signaling pathways originating from membrane receptors, such as protein kinase A, serine/threonine kinase Akt (protein kinase B), protein kinase C (PKC), extracellular signal regulated protein kinases (ERK1/2), c-Jun N terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 mitogen activated protein kinase (p38 MAPK), cytosol, nucleus, lysosome, and ER converge leading to mitochondrial membrane permeabilization (78). As a result, the inner mitochondrial components such as cytochrome c, caspase-9, Apoptotic protease activating factor 1 (APAF-1) and apoptosis-inducing factor (AIF) release into cytosol to initiate both the caspase-dependent and – independent apoptotic pathways (79). Ample evidence suggests the activation of these pathways in rodent models of stroke (80–84). The principal proteins involved with mitochondrial membrane permeabilization are the proapoptotic BCL-2 family members including BAX, BCL-2 homologous antagonist killer (BAK), BH3 interacting-domain death agonist (BID), BCL-2-associated death promoter (BAD), BIM, and PUMA (85). Among them BAX and BAK cause mitochondrial membrane disruption via channel formation in the outer mitochondrial membrane interacting with the mitochondrial adenine nucleotide translocator and the voltage-dependent anion channel (83, 86). The BH3-only proteins BID and PUMA facilitate BAX and BAK channel formation, whereas BAD and BIM acts to inhibit pro-survival BCL-2 and BCL-xL (87, 85). The post-stroke alterations in the expression of both pro-apoptotic and anti-apoptotic Bcl-2 family members were observed in rodent models (88). Although Bcl-2 family members have a direct effect on mitochondrial membrane and at the same time they are also located in ER and influences ER function (89). It has been estimated that 5–20% of the mitochondrial surface is in close appositions with the ER (90). ER stress leads to up-regulation of the expression of proapoptotic BAX, BIM and PUMA, and activation of caspase-2 and caspase-9 and dissipation of mitochondrial transmembrane potential (Δψm) (91–92). Interestingly, BIM and PUMA induce cytochrome c release and apoptosis exclusively in the presence of ER localized BAK (92). The post-stroke temporal profile of markers for ER and mitochondrial dysfunction suggests that ER dysfunction may be upstream of mitochondrial dysfunction (14, 93–95). Phosphorylation of PERK and eIF2α was observed much earlier than cytochrome c release during reperfusion after transient cerebral ischemia, implying that ER dysfunction precedes mitochondrial impairment (96). A major consequence of over-activation of PERK phosphorylation is the induction of its down-stream transcription factor ATF4 that induces the expression of BAX, BIM and PUMA that concurrently inhibits BCL-2 expression. This tips the balance towards apoptosis. ER stress inducing agents are known to activate AIF and caspase-12 and their subsequent re-distribution to the nucleus (97). Knockdown of either AIF or caspase-12 showed that AIF primarily controls apoptosis caused by disrupted Ca2+ homeostasis; whereas caspase-12 regulates both AIF and other apoptotic mechanisms (97). Thus, ER stress and mitochondrial dysfunction collaborate to modulate apoptotic after stroke and other CNS insults.
3-3. ER stress and autophagy
As discussed above, PERK over activation leading to the induction of CHOP and its down-stream proapoptotic genes is shown to induce neuronal death by apoptosis. Some studies also thought that neuronal death due to autophagy is also a major consequence of ER stress (98–100). In support, oxidative stress potentiated by ER stress also triggers autophagy (101). Furthermore, ER stress increases the formation of autophagosome via IRE1-JNK signaling pathway (102) and dysfunction of ERAD and autophagy and the resulting failure of protein folding render cells vulnerable to ER stress (102).
Autophagy is constitutively active in healthy neurons and is vital to cell survival (103). Mice lacking essential autophagy genes such as Atg 5 and Atg7 result in neurodegeneration, suggesting that it is important for normal neuronal function (104). In addition, mitophagy (autophagic removal of mitochondria) is considered to be an adaptive mechanism in response to hypoxia that is necessary to maintain redox homeostasis and cell survival (105). Previous studies showed that both mitochondrial and ER fragments damaged by ROS are sequestered in autophagolysosomes to prevent leakage of calcium into the cytosol from these organelles and subsequent activation of apoptosis (106). While limited amount of autophagy is essential, abnormal activation of autophagic pathway leads to secondary brain damage as seen after both chronic and acute insults to CNS (107).
Controlled activation of autophagy due to is mild physiological stress is beneficial for recycling the contents of the cell, but excessive activation of autophagy under severe pathological stress can be detrimental and kills the cells. Previous studies showed that the autophagy markers like beclin-1 and LC3-II are induced following cerebral ischemia (10, 104). It seems that BCL-2 interacting domain of Beclin-1 serves as a point of crosstalk between autophagy and apoptosis (79).
The hippocampal neuronal death after hypoxic-ischemic injury was shown to be mitigated in mice lacking the autophagy modulator Atg7 indicating the significance of autophagy after cerebral ischemia (108). Pharmacological inhibition of autophagy using 3-methyl-adenine (3-MA) and bafliomycin A1 (BFA) or the cathepsin B inhibitor Z-FA-fmk reduced infarct volume after focal ischemia. The neuroprotective effects of 3-MA and Z-FA-fmk associated with the inhibition of LC3-II and cathepsin B, and increased expression of BCL-2 (109). In addition, preconditioning- induced ischemic tolerance was observed to be mimicked by inducers of autophagy (110). Although oxidative stress and ER stress that precipitate neuronal death after ischemia were shown to potently stimulate autophagy (111), the precise molecular mechanisms by which ER is selected as autophagic cargo and the crosstalk between ER stress-induced autophagy and activation of cell death pathways after stroke are not yet clearly understood.
Autophagy is beneficial after ER stress to clear the unfolded proteins independent of the ubiquitin-proteasomal system (60). However, increased cytosolic Ca2+ and PERK and IRE1 pathways induced after UPR has been implicated as mediators of ER stress-induced autophagy in mammalian cells (99, 112–113). It is not clear how PERK-eIF2α regulates autophagy, but the autophagy mediator Atg12 was thought to be induced downstream of ATF4 (112). Mutations in the PERK phosphorylation site of eIF2α prevents Atg2 up-regulation and conversion of LC3-1(free form) to LC3-II (lipidated form) further supports that PERK pathway is a mediator of autophagy (112). IRE1 pathway downstream of UPR/ER stress is also thought to play an essential role in ER stress mediated autophagy. Accumulations of LC3-positive vesicles were shown to decrease in mouse embryonic fibroblasts lacking IRE1α indicating that it is a major mediator autophagy (10). Furthermore, XBP1 ablation also induces autophagy and protects against amyotrophic lateral sclerosis and Huntington’s disease (114–115). Although both PERK and IRE1 are induced after stroke, their involvement in post-ischemic autophagy is not yet evaluated. It appears that ER stress-induced autophagy could be neuroprotective based on the observation that ER stress inhibitor salubrinal inhibited the activation of autophagy and neuroprotection after ischemic preconditioning (116).
4-1. ER stress and cerebral ischemia
Cerebral ischemia/stroke is a major cause of death and disability worldwide. The pathology of ischemic stroke is very complex that involves multiple cell-signaling pathways leading to neuronal loss (79). Due to energy depletion during cerebral ischemia, neurons in the ischemic zone are unable to maintain the imbalance between ionic gradients, which eventually results in increased neuronal depolarization followed by excessive glutamate release (117). Further, neurons in the surrounding areas also release glutamate and spreads ischemic depolarization from the site of initial damage, which leads to widespread disturbance of Ca2+ homoeostasis. Excessive intracellular Ca2+ release leads to activation of signaling processes that kills neurons and impairs CNS functions (117). Following cerebral ischemia, low energy levels can disrupt normal protein folding leading to activation of UPR/ER stress, which plays a critical role in the ischemic brain damage (23). Chronic disturbances in protein folding and oxidative stress prolong ER stress leading to sustained ER dysfunction and neuronal cell death after stroke (118). Good evidence exists to suggest that focal ischemia leads to depletion of Ca2+ from ER stores (2), accumulation of unfolded proteins in the ER lumen (6), inhibition of protein synthesis (119), activation of ER stress down-stream pro-apoptotic genes such as CHOP/GADD153 (120, 23) and further down-stream PUMA (121), BIM (122), indicating a role for UPR/ER stress in post-ischemic brain damage. Cerebral ischemia also leads to oxidative stress which integrates with ER stress/UPR (if they are limited) as an adaptive mechanism to preserve cell function and survival (62, 123–124). In addition, ROS can trigger ER stress (125), and ER stress can exacerbate ROS production (19, 62, 126). Therefore, the post-ischemic neuronal death is mediated in part by the cooperative action of oxidative stress and ER stress. Further, as discussed in the earlier sections, ER stress and mitochondrial dysfunction also collaborate to promote apoptosis after stroke. Therefore, controlling ER stress exerts a significant protective effect on the ischemic brain, thus offers the prospect of new strategies for stroke therapies. The neuroprotective effect of ischemic preconditioning has been attributed to attenuation of ER stress response after ischemic insults (127). Mice lacking PERK did not show eIF2α phosphorylation or reduced protein translation during transient cerebral ischemia (128). Downstream to PERK-eIF2α, ATF-4 and CHOP knockout mice showed less brain damage, improved behavioral outcome and decreased neuronal cell death after ischemia (29, 129). Further, GADD34 induction is known to accompany ischemic penumbra and accounts for the translational recovery via dephosphorylation of eIF2α during stroke (128). Attenuation of translational recovery protects cells from accumulation of misfolded proteins and thus treatment with salubrinal sustained eIF2α phosphorylation and limited infarct size in a rat model of cerebral ischemia (23). Interestingly, a chemical chaperone sodium 4-phenylbutyrate (4PBA) ameliorated ischemic brain injury associated with diabetes by reducing ER stress and apoptosis (130). Further, (−)-epigallocatechin-3-gallate, which is an abundant constituent of green tea also provided neuroprotection via inhibition of ER stress after transient focal ischemia (131). Together, these findings strongly suggest that ER stress is induced by cerebral ischemia and that pharmacological manipulation of ER stress signaling could have important therapeutic effects as an acute intervention.
4-2. ER stress and traumatic brain injury (TBI)
Similar to stroke, secondary neuronal death that starts immediately after the insult is a leading cause of morbidity and mortality following acute injuries to CNS like TBI and spinal cord injury (SCI) with limited available therapeutic options (132, 133). Many stress genes related to ER and mitochondrial function were shown to be induced quickly after TBI (37, 134–136). A rapid increase in the expression of molecular chaperones specific to ER (GRP78), mitochondria (HSP60) and cytosol (HSP70) were observed by 4h after experimental TBI combined with hypoxia (136). A sustained expression of potent ER stress markers such as peIF-2α, ATF4, IRE1, and CHOP was observed in the cerebral cortex days after induction of experimental TBI (102). Furthermore, treatment with docosahexaenoic acid (an Omega 3 fatty acid) attenuates ER stress, reduces the accumulation of ubiquitinated proteins and promotes early recovery of sensorimotor function after TBI (134). The anti-apoptotic Bax-inhibitor-1 (BI-1), which is an ER resident protein modulates UPR signaling via regulating the release of Ca2+ (137). Transgenic mice that constitutively express BI-1 protein exhibit decreased expression of ER stress markers reduced brain damage and improved behavioral outcome in rodent models of acute brain injury (132). Mice lacking BI-1 showed increased vulnerability to experimental chronic mild stress accompanied by changes in the size and morphology of hippocampus; enhanced ROS production and ER stress response (137). Previous studies also showed that selective inhibition of eIF2α dephosphorylation with Salubrinal reduces neuronal damage in animal models of stroke and epilepsy (22–23). We also observed that Salubrinal induces neuroprotection after experimental TBI (138). Overall, a combined strategy of reducing ER stress and oxidative stress might lead to a better outcome after TBI.
4-3. ER stress and subarachnoid hemorrhage (SAH)
Recent reports indicate that ER stress also plays a role in regulating early brain damage after experimental SAH (108). An interesting observation is that enhancing ER stress by treating with tunicamycin significantly improved the neurological deficits, attenuated the expression of caspase-3, reduced the number of apoptotic neurons and enhanced the expression of autophagy markers after endovascular perforation model of SAH (139). In contrast, treatment with the ER stress inhibitor TUDCA after SAH aggravated neurological deficits and apoptotic cell death accompanied by decreased autophagy (139). The autophagy inducer rapamycin (RAP) administration decreased translocation of cytosolic Bax to the mitochondria and release of cytochrome c to the cytosol after experimental SAH in rats (140). Endothelial apoptosis plays an important role in the development of cerebral vasospasm after SAH (141). The pro-apoptotic transcription factor CHOP, which is implicated in post-stroke secondary brain damage, also plays an important role in mediating the cerebral vasospasm after SAH (141). SiRNA-mediated knockdown of CHOP was shown to mitigate apoptosis associated with cerebral vasospasm by reducing the expression of pro-apoptotic genes Bim and caspase-3 after SAH (141). CHOP siRNA treatment reduced the number of apoptotic endothelial cells in basilar artery as well after SAH (141). Further studies on ER stress and interconnected autophagy mechanisms will help to develop new therapeutic targets to minimize vasospasm and brain damage after SAH.
4-4. ER stress and Spinal Cord Injury (SCI)
ER stress was also shown to play an important role in apoptosis following SCI (142). Weight drop-induced SCI in mice was shown to rapidly induce PERK, ATF6, and IRE1α signaling pathways at the injury epicenter (143). Although, ER stress response seems to be protective after mild SCI, it leads to apoptosis after severe SCI in a rat contusive model (144). Following SCI, ER stress response varies depending on specific cell type. A recent study showed that astrocytes are more vulnerable to ER stress than oligodendrocyte precursor cells after SCI (144). In contrast, increased expression of proapoptotic CHOP was observed in neurons and oligodendrocytes, but not in astrocytes after SCI; and mice lacking CHOP showed attenuation of UPR and apoptosis after SCI (143, 145). Furthermore, CHOP null mice showed significant functional recovery with increased white matter sparing and higher levels of myelin basic protein and claudin 11 after SCI (143). Thus, attenuation of ER stress is protective after SCI as well.
5. Therapeutic opportunities for neuroprotection by modulating ER stress
Recent studies showed that ER stress can be modulated to alter the pathological outcome by either inhibiting or potentiating various proteins of PERK, ATF6 and IRE1 pathways. Some of these drugs are shown in Table 1. Pretreatment of mice with BIP-inducible factor X (BIX) that selectively induces GRP78 expression reduces infarct volume and apoptotic neuronal death in the ischemic penumbra (146). Acting upstream of CHOP and ATF4, pharmacological inhibition of ERO1α and thus eIF2α dephosphorylation by small molecule inhibitor salubrinal protects neurons from cell death in vitro and in vivo models of epilepsy and stroke by prolonging the protein synthesis inhibition and thus reducing the load of unfolded proteins (21–23). Inducible nitric oxide synthase (iNOS) knockout mice show reduced CHOP induction followed by delayed secondary brain damage after stroke (147). Hence, many NOS inhibitors can also induce neuroprotection after acute CNS insults (Table 1). Crosstalk between ER stress and oxidative stress potentiate each other and hence activating endogenous antioxidant pathways is another feasible strategy for protecting brain after ischemic and traumatic injuries. Many compounds that induce the endogenous Nrf2-KEAP1 anti-oxidant pathway show neuroprotection in ischemia and TBI animal models (148–151). Furthermore, combination therapies that inhibit both ER stress and oxidative stress might be a potentially better therapeutic strategy for preventing post-stroke and post-TBI secondary brain damage. Many antioxidant drugs like Edaravone and apocynin were shown to curtail ischemic and traumatic brain damage effectively (152–156).
Table 1.
Therapeutic opportunities based on modulating UPR/ER stress and its associated pathways
| Target | Compound | Mode of action/ Outcome | Reference |
|---|---|---|---|
| GRP78 (ER chaperone) | BIX | Induces GRP78 expression; neuroprotective after stroke | 145 |
| PERK-eIF2α pathway | Salubrinal | Inhibits eIF2α dephosphorylation; neuroprotective after stroke | 21, 22, 23 |
| NO signaling pathway | L-NNA, ONO-1714, L-NAME, L-NMMA | Inhibits NOS; neuroprotective after Stroke | 9, 157, 158, 159 |
| Nrf2–KEAP1 pathway | Carnosic acid, tri- terpenoids, sulphoraphane, Tertbutylhydroquino ne, Melatonin | Activates Nrf2 antioxidant pathway; neuroprotective after stroke and TBI | 148, 149, 150 |
| ROS/ER stress | Edaravone | Free radical scavenger; neuroprotective after hypoxia/ischemia and TBI | 155, 156 |
| TUDCA, 4PBA | Reduces ER stress; neuroprotective | 130, 160, 161 | |
| NADPH oxidase | Apocynin | Inhibits ROS generation; neuroprotective after stroke and TBI | 152, 153, 154 |
| ASK1 | Fused heterocyclic compounds | Inhibits ASK1 activity; neuroprotective | 162 |
| Oxidative stress | U83836E, Resveratrol, Curcumin, OPC-14177, Lipoic acid | Inhibits lipid peroxidation; neuroprotective after TBI | 163 |
| ER Stress | Docosahexaenoic acid | Reduces ER stress and abnormal protein accumulation; recovers neuronal function after TBI | 133 |
BIX, BiP protein Inducer X; ROS, reactive oxygen species; NOS, nitric oxide synthase; L-NNA, nitro-L-arginine; 0N01714, inhibitor of inducible NOS; L-NAME, L-NG-Nitroarginine methyl ester; L-NMMA, N(G)-monomethyl-L-arginine; TUDCA, Taurine-conjugated ursodeoxycholic acid; GRP78, Glucose regulatory protein 78 KDa; PERK, Protein kinase RNA-like endoplasmic reticulum kinase; eIF2, Eukaryotic translational initiation factor 2; KEAP1, Kelch-like ECH-associated protein 1; ASK1, Apoptosis signal regulating kinase 1; NRF2, Nuclear factor (erythroid-derived 2)-like 2.
6. Conclusions
Multiple cell death pathways may have common upstream initiators and their identification might help in the development of new strategies for stroke therapy. Hence, therapeutic strategies need to focus on combinatorial targets/mechanisms to inhibit alternate cell death pathways. Oxidative stress appears as a master regulator of cell death by influencing ER and mitochondria. Therefore, the molecular crosstalk between ER stress, mitochondria dysfunction, oxidative stress and autophagy represents a vicious cycle that can be pharmacologically targeted to minimize neuronal death after acute injuries to CNS.
References
- 1.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 2.Paschen W, Doutheil J. Disturbances Of The Functioning Of Endoplasmic Reticulum: A Key Mechanism Underlying Neuronal Cell Injury? J Cereb Blood Flow Metab. 1999;19:1–18. doi: 10.1097/00004647-199901000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 4.Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology. 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- 5.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu BR, Martone ME, Yones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+ -ATPase in skeletal muscle. Biochem J. 1999;340:657–669. [PMC free article] [PubMed] [Google Scholar]
- 9.Gotoh T, Mori M. Nitric oxide and endoplasmic reticulum stress. Arterioscler Thromb Vasc Biol. 2006;26:1439–1446. doi: 10.1161/01.ATV.0000223900.67024.15. [DOI] [PubMed] [Google Scholar]
- 10.Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–82. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 11.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 12.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 13.Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology. 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- 14.Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem. 2001;77:1418–21. doi: 10.1046/j.1471-4159.2001.00387.x. [DOI] [PubMed] [Google Scholar]
- 15.Prostko CR, Brostrom MA, Brostrom CO. Reversible phosphorylation of eukaryotic initiation factor 2 alpha in response to endoplasmic reticular signaling. Mol Cell Biochem. 1993;128:255–265. doi: 10.1007/BF01076776. [DOI] [PubMed] [Google Scholar]
- 16.Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–1388. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 18.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 19.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamanaka Robert B, Bennett Beth S, Cullinan Sara B, Diehl J Alan. PERK and GCN2 Contribute to eIF2α Phosphorylation and Cell Cycle Arrest after Activation of the Unfolded Protein Response Pathway. Mol Biol Cell. 2005;16:5493–5501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2a dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 22.Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–8. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- 24.Gorman AM, Healy SJ, Jäger R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther. 2012;134:306–16. doi: 10.1016/j.pharmthera.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–49. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- 26.Calfon M, Zeng H, Urano F, Till JH, Hubbart SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 27.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paschen W, Aufenberg C, Hotop S, Mengesdorf T. Transient cerebral ischemia activates processing of xbp1 messenger RNA indicative of endoplasmic reticulum stress. J Cereb Blood Flow Metab. 2003;23:449–461. doi: 10.1097/01.WCB.0000054216.21675.AC. [DOI] [PubMed] [Google Scholar]
- 29.Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 30.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–575. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 32.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. IRE1a induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Upton JP, Wang L, Han D, Wang ES, Huskey NE, Lim L, Truitt M, McManus MT, Ruggero D, Goga A, et al. IRE1a cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science. 2012;338:818–822. doi: 10.1126/science.1226191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014;158:534–48. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shibata M, Hattori H, Sasaki T, Gotoh J, Hamada J, Fukuuchi Y. Activation of caspase-12 by endoplasmic reticulum stress induced by transient middle cerebral artery occlusion in mice. Neuroscience. 2003;118:491–499. doi: 10.1016/s0306-4522(02)00910-7. [DOI] [PubMed] [Google Scholar]
- 36.Mouw G, Zechel JL, Gamboa J, Lust WD, Selman WR, Ratcheson RA. Activation of caspase-12, an endoplasmic reticulum resident caspase, after permanent focal ischemia in rat. Neuroreport. 2003;14:183–186. doi: 10.1097/00001756-200302100-00004. [DOI] [PubMed] [Google Scholar]
- 37.Larner SF, Hayes RL, McKinsey DM, Pike BR, Wang KK. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [DOI] [PubMed] [Google Scholar]
- 38.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 39.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–42. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 40.Aoyama K, Burns DM, Suh SW, Garnier P, Matsumori Y, Shiina H, Swanson RA. Acidosis causes endoplasmic reticulum stress and caspase-12-mediated astrocyte death. J Cereb Blood Flow Metab. 2005;25:358–70. doi: 10.1038/sj.jcbfm.9600043. [DOI] [PubMed] [Google Scholar]
- 41.Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 42.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 43.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- 45.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-jun n-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 46.Gao Y, Signore AP, Yin W, Cao G, Yin X, Sun F, Luo Y, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 47.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–64. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 49.Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. 2011;286:7947–7957. doi: 10.1074/jbc.M110.156430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moroa MA, Almeida A, Bolanos JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005;39:1291–1304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 51.Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Anderson JK. Oxidative stress in neurodegeneration: a cause or consequence? Nature Reviews Neuroscience. 2004;5:S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 53.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 54.Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Blood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 55.Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–63. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahles T, Brandes RP. NADPH oxidases as therapeutic targets in ischemic stroke. Cell Mol Life Sci. 2012;69:2345–63. doi: 10.1007/s00018-012-1011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei H, Kim SJ, Zhang Z, Tsai PC, Wisniewski KE, Mukherjee AB. ER and oxidative stresses are common mediators of apoptosis in both neurodegenerative and non-neurodegenerative lysosomal storage disorders and are alleviated by chemical chaperones. Hum Mol Genet. 2008;17:469–77. doi: 10.1093/hmg/ddm324. [DOI] [PubMed] [Google Scholar]
- 58.Martínez JA. Mitochondrial oxidative stress and inflammation: an slalom to obesity and insulin resistance. J Physiol Biochem. 2006;62:303–306. doi: 10.1007/BF03165759. [DOI] [PubMed] [Google Scholar]
- 59.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol. 2007;292:R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 60.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 61.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–25. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malhotra JD, Kaufman RJ. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxidants & Redox Signaling. 2007;9:2277–2294. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 63.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 64.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, Alam J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276:20858–20865. doi: 10.1074/jbc.M101198200. [DOI] [PubMed] [Google Scholar]
- 66.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–52. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 67.Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I. Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J Cell Biol. 2009;186:783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 69.Jacobson J, Duchen MR. Mitochondrial oxidative stress and cell death in astrocytes–requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci. 2002;115:1175–1188. doi: 10.1242/jcs.115.6.1175. [DOI] [PubMed] [Google Scholar]
- 70.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1, 4, 5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–1061. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 71.Bhandary B, Marahatta A, Kim HR, Chae HJ. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci. 2012;14:434–56. doi: 10.3390/ijms14010434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G, Terentyev D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol. 2013;591:5895–911. doi: 10.1113/jphysiol.2013.260521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- 74.Ruiz A, Matute C, Alberdi E. Endoplasmic reticulum Ca(2+) release through ryanodine and IP (3) receptors contributes to neuronal excitotoxicity. Cell Calcium. 2009;46:273–81. doi: 10.1016/j.ceca.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 75.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 76.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1998;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 77.Ruiz A, Matute C, Alberdi E. Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis. 2010;15(1):e54. doi: 10.1038/cddis.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: a matter of life or death. Free Radic Biol Med. 2005;38:2–11. doi: 10.1016/j.freeradbiomed.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 79.Nakka VP, Gusain A, Mehta SL, Raghubir R. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol neurobiol. 2008;37:7–38. doi: 10.1007/s12035-007-8013-9. [DOI] [PubMed] [Google Scholar]
- 80.Ouyang YB, Tan Y, Comb M, Liu CL, Martone ME, Siesjo BK, Hu BR. Survival and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome C, and activation of caspase-like proteases. J Cereb Blood Flow Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 81.Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic distribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;8:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- 82.Perez-Pinzon MA, Xu GP, Born J, Lorenzo J, Busto R, Rosenthal M, Sick TJ. Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J Cereb Blood Flow Metab. 1999;19:39–43. doi: 10.1097/00004647-199901000-00004. [DOI] [PubMed] [Google Scholar]
- 83.Cao G, Minami M, Pei W, Yan C, Chen D, O’Horo C, Graham SH, Chen J. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 84.Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient forebrain ischemia. J Neurosci. 1999;19:RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vosler PS, Graham SH, Wechsler LR, Chen J. Mitochondrial targets for stroke: focusing basic science research toward development of clinically translatable therapeutics. Stroke. 2009;40:3149–55. doi: 10.1161/STROKEAHA.108.543769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gogvadze V, Orrenius S, Zhivotovsky B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim Biophys Acta. 2006;1757:639–47. doi: 10.1016/j.bbabio.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 87.Youle RJ, Strasser A. The bcl-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 88.Ferrer I, Planas AM. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol. 2003;62:329–339. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- 89.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress–induced apoptosis. EMBO reports. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–6. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 91.Gupta S, Cuffe L, Szegezdi E, Logue SE, Neary C, Healy S, Samali A. Mechanisms of ER Stress-Mediated Mitochondrial Membrane Permeabilization. Int J Cell Biol. 2010;2010:170215. doi: 10.1155/2010/170215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klee M, Pallauf K, Alcalá S, Fleischer A, Pimentel-Muiños FX. Mitochondrial apoptosis induced by BH3-only molecules in the exclusive presence of endoplasmic reticular Bak. EMBO J. 2009;28:1757–68. doi: 10.1038/emboj.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mengesdorf T, Proud CG, Mies G, Paschen W. Mechanisms underlying suppression of protein synthesis induced by transient focal cerebral ischemia in mouse brain. Exp Neurol. 2002;177:538–46. doi: 10.1006/exnr.2002.8002. [DOI] [PubMed] [Google Scholar]
- 94.Owen CR, Kumar R, Zhang P, McGrath BC, Cavener DR, Krause GS. PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J Neurochem. 2005;94:1235–42. doi: 10.1111/j.1471-4159.2005.03276.x. [DOI] [PubMed] [Google Scholar]
- 95.Beresewicz M, Kowalczyk JE, Zablocka B. Cytochrome c binds to inositol (1, 4, 5) trisphosphate and ryanodine receptors in vivo after transient brain ischemia in gerbils. Neurochem Int. 2006;48:568–571. doi: 10.1016/j.neuint.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 96.Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I, Borner C. Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene. 2000;19:2286–2295. doi: 10.1038/sj.onc.1203592. [DOI] [PubMed] [Google Scholar]
- 97.Sanges D, Marigo V. Cross-talk between two apoptotic pathways activated by endoplasmic reticulum stress: differential contribution of caspase-12 and AIF. Apoptosis. 2006;11:1629–1641. doi: 10.1007/s10495-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 98.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–82. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 100.Kolattukudy PE, Niu J. Inflammation, endoplasmic reticulum stress, autophagy, and the monocyte chemoattractant protein-1/CCR2 pathway. Circ Res. 2012;6(110):174–89. doi: 10.1161/CIRCRESAHA.111.243212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harraz MM, Dawson TM, Dawson VL. Advances in neuronal cell death. Stroke. 2008;39:286–8. doi: 10.1161/STROKEAHA.107.511857. [DOI] [PubMed] [Google Scholar]
- 102.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boland B, Nixon RA. Neuronal macroautophagy: from development to degeneration. Mol Aspects Med. 2006;27:503–519. doi: 10.1016/j.mam.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 104.Rami A, Kogel D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: Two sides of the same coin? Autophagy. 2008;4:422–426. doi: 10.4161/auto.5778. [DOI] [PubMed] [Google Scholar]
- 105.Semenza GL. Mitochondrial autophagy: life and breath of the cell. Autophagy. 2008;4:534–536. doi: 10.4161/auto.5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xilouri M, Stefanis L. Autophagy in the central nervous system: implications for neurodegenerative disorders. CNS Neurol Disord Drug Targets. 2010;9:701–19. doi: 10.2174/187152710793237421. [DOI] [PubMed] [Google Scholar]
- 108.Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 110.Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–94. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- 111.Adhami F, Schloemer A, Kuan C. The roles of Autophagy in cerebral ischemia. Autophagy. 2007;3:42–44. doi: 10.4161/auto.3412. [DOI] [PubMed] [Google Scholar]
- 112.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 113.Fujita E, Kouroku Y, Isoai A, Kumagai H, Mizutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation systems (ERAD) for the novel variant of the mutant dysferlin; ubiquitin/proteasome ERAD (I) and Autophagy/Lysosome ERAD (II) Hum Mol Genet. 2007;16:618–629. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 114.Vidal RL, Figueroa A, Court FA, Thielen P, et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Mol Genet. 2012;21:2245–2262. doi: 10.1093/hmg/dds040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gao B, Zhang XY, Han R, Zhang TT, Chen C, Qin ZH, Sheng R. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharmacol Sin. 2013;34:657–66. doi: 10.1038/aps.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Szydlowska K, Tymianskia M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 118.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.DeGracia DJ, Kumar R, Owen CR, Krause GS, White BC. Molecular pathways of protein synthesis inhibition during brain reperfusion: implications for neuronal survival or death. J Cereb Blood Flow Metab. 2002;22:127–141. doi: 10.1097/00004647-200202000-00001. [DOI] [PubMed] [Google Scholar]
- 120.Paschen W, Gissel C, Linden T, Althausen S, Doutheil J. Activation of gadd153 expression through transient cerebral ischemia: evidence that ischemia causes endoplasmic reticulum dysfunction. Brain Res Mol Brain Res. 1998;60:115–122. doi: 10.1016/s0169-328x(98)00180-6. [DOI] [PubMed] [Google Scholar]
- 121.Niizuma K, Endo H, Nito C, Myer DJ, Chan PH. Potential role of PUMA in delayed death of hippocampal CA1 neurons after transient global cerebral ischemia. Stroke. 2009;40:618–25. doi: 10.1161/STROKEAHA.108.524447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ness JM, Harvey CA, Strasser A, Bouillet P, Klocke BJ, Roth KA. Selective involvement of BH3-only Bcl-2 family members Bim and Bad in neonatal hypoxia-ischemia. Brain Res. 2006;1099:150–159. doi: 10.1016/j.brainres.2006.04.132. [DOI] [PubMed] [Google Scholar]
- 123.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 124.Cherubini A, Ruggiero C, Polidori MC, Mecocci P. Potential markers of oxidative stress in stroke. Free Radic Biol Med. 2005;39:841–52. doi: 10.1016/j.freeradbiomed.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 125.Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PK. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab. 2005;25:41–53. doi: 10.1038/sj.jcbfm.9600005. [DOI] [PubMed] [Google Scholar]
- 126.Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal. 2009;11:2409–27. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- 127.Lehotský J, Urban P, Pavlíková M, Tatarková Z, Kaminska B, Kaplán P. Molecular mechanisms leading to neuroprotection/ischemic tolerance: effect of preconditioning on the stress reaction of endoplasmic reticulum. Cell Mol Neurobiol. 2009;29:917–25. doi: 10.1007/s10571-009-9376-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105–18. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- 129.Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2007;205:1227–42. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Srinivasan K, Sharma SS. Sodium phenylbutyrate ameliorates focal cerebral ischemic/reperfusion injury associated with comorbid type 2 diabetes by reducing endoplasmic reticulum stress and DNA fragmentation. Behav Brain Res. 2011;225:110–6. doi: 10.1016/j.bbr.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 131.Yao C, Zhang J, Liu G, Chen F, Lin Y. Neuroprotection by (−)-epigallocatechin-3-gallate in a rat model of stroke is mediated through inhibition of endoplasmic reticulum stress. Mol Med Rep. 2014;9:69–76. doi: 10.3892/mmr.2013.1778. [DOI] [PubMed] [Google Scholar]
- 132.Shoichet MS, Tate CC, Baumann MD, LaPlaca MC. Indwelling neural implants: strategies for contending with the in vivo environment. Boca Raton (FL): CRC Press; 2008. Strategies for regeneration and repair in the injured central nervous system. Chapter 8. [PubMed] [Google Scholar]
- 133.Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, Yang L, Irie F, Yamaguchi Y, Lipton SA, Reed JC. Endoplasmic Reticulum Protein BI-1 Modulates Unfolded Protein Response Signaling and Protects Against Stroke and Traumatic Brain Injury. Brain Res. 2011;1370:227–37. doi: 10.1016/j.brainres.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic Acid Reduces ER Stress and Abnormal Protein Accumulation and Improves Neuronal Function Following Traumatic Brain Injury. J Neurosci. 2014;34:3743–55. doi: 10.1523/JNEUROSCI.2872-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol. 2012;167:699–719. doi: 10.1111/j.1476-5381.2012.02025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Truettner JS, Hu B, Alonso OF, Bramlett HM, Kokame K, Dietrich WD. Subcellular stress response after traumatic brain injury. J Neurotrauma. 2007;24:599–612. doi: 10.1089/neu.2006.0186. [DOI] [PubMed] [Google Scholar]
- 137.Lee HY, Lee GH, Marahatta A, Lin SM, Lee MR, Jang KY, Kim KM, Lee HJ, Lee JW, Bagalkot TR, Chung YC, Lee YC, Kim HR, Chae HJ. The protective role of Bax inhibitor-1 against chronic mild stress through the inhibition of monoamine oxidase A. Sci Rep. 2013;3:3398. doi: 10.1038/srep03398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nakka VP, Bodden LO, Vemuganti R. ER stress inhibitor salubrinal is neuroprotective after TBI. J Neurotrauma. 2014;31(12):A107–A107. [Google Scholar]
- 139.Yan F, Li J, Chen J, Hu Q, Gu C, Lin W, Chen G. Endoplasmic reticulum stress is associated with neuroprotection against apoptosis via autophagy activation in a rat model of subarachnoid hemorrhage. Neurosci Lett. 2014;563:160–5. doi: 10.1016/j.neulet.2014.01.058. [DOI] [PubMed] [Google Scholar]
- 140.Jing CH, Wang L, Liu PP, Wu C, Ruan D, Chen G. Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience. 2012;213:144–53. doi: 10.1016/j.neuroscience.2012.03.055. [DOI] [PubMed] [Google Scholar]
- 141.He Z, Ostrowski RP, Sun X, Ma Q, Tang J, Zhang JH. Targeting C/EBP homologous protein with siRNA attenuates cerebral vasospasm after experimental subarachnoid hemorrhage. Exp Neurol. 2012;238:218–24. doi: 10.1016/j.expneurol.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang HY, Wang ZG, Lu XH, Kong XX, Wu FZ, Lin L, Tan X, Ye LB, Xiao J. Endoplasmic Reticulum Stress: Relevance and Therapeutics in Central Nervous System Diseases. Mol Neurobiol. 2014 Jul 22; doi: 10.1007/s12035-014-8813-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 143.Ohri SS, Maddie MA, Zhao Y, Qiu MS, Hetman M, Whittemore SR. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia. 2011;59:1489–502. doi: 10.1002/glia.21191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Matsuyama D, Watanabe M, Suyama K, Kuroiwa M, Mochida J. Endoplasmic reticulum stress response in the rat contusive spinal cord injury model-susceptibility in specific cell types. Spinal Cord. 2014;52:9–16. doi: 10.1038/sc.2013.118. [DOI] [PubMed] [Google Scholar]
- 145.Penas C, Guzmán MS, Verdú E, Forés J, Navarro X, Casas C. Spinal cord injury induces endoplasmic reticulum stress with different cell-type dependent response. J Neurochem. 2007;102:1242–55. doi: 10.1111/j.1471-4159.2007.04671.x. [DOI] [PubMed] [Google Scholar]
- 146.Kudo T, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008;15:364–375. doi: 10.1038/sj.cdd.4402276. [DOI] [PubMed] [Google Scholar]
- 147.Ladecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci. 1997;17:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 149.Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S.-alkylation of targeted cysteines on Keap1. J Neurochem. 2008;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shih AY, Li P, Murphy TH. A small-molecule inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mesenge C, Margaill I, Verrecchia C, Allix M, Boulu RG, Plotkine M. Protective effect of melatonin in a model of traumatic brain injury in mice. J Pineal Res. 1998;25:41–6. doi: 10.1111/j.1600-079x.1998.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 152.Lu XY, Wang HD, Xu JG, Ding K, Li T. NADPH oxidase inhibition improves neurological outcome in experimental traumatic brain injury. Neurochem Int. 2014;69:14–9. doi: 10.1016/j.neuint.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 153.Ferreira AP, Rodrigues FS, Della-Pace ID, Mota BC, Oliveira SM, et al. The effect of NADPH-oxidase inhibitor apocynin on cognitive impairment induced by moderate lateral fluid percussion injury: Role of inflammatory and oxidative brain damage. Neurochem Int. 2013;63:583–593. doi: 10.1016/j.neuint.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 154.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Itoh T, Satou T, Nishida S, Tsubaki M, Hashimoto S, Ito H. The novel free radical scavenger, edaravone, increases neural stem cell number around the area of damage following rat traumatic brain injury. Neurotox Res. 2009;16:378–89. doi: 10.1007/s12640-009-9081-6. [DOI] [PubMed] [Google Scholar]
- 156.Qi X, Okuma Y, Hosoi T, Nomura Y. Edaravone protects against hypoxia/ischemia-induced endoplasmic reticulum dysfunction. Pharmacol Exp Ther. 2004;311:388–93. doi: 10.1124/jpet.104.069088. [DOI] [PubMed] [Google Scholar]
- 157.Kohno K, Higuchi T, Ohta S, Kumon Y, Sakaki S. Neuroprotective nitric oxide synthase inhibitor reduces intracellular calcium accumulation following transient global ischemia in the gerbil. Neurosci Lett. 1997;224:17–20. doi: 10.1016/s0304-3940(97)13459-0. [DOI] [PubMed] [Google Scholar]
- 158.Sekiguchi F, et al. The potent inducible nitric oxide synthase inhibitor ONO-1714 inhibits neuronal NOS and exerts antinociception in rats. Neurosci Lett. 2004;365:111–115. doi: 10.1016/j.neulet.2004.04.069. [DOI] [PubMed] [Google Scholar]
- 159.Terpolilli NA, Moskowitz MA, Plesnila N. Nitric oxide: considerations for the treatment of ischemic stroke. J Cereb Blood Flow Metab. 2012;32:1332–46. doi: 10.1038/jcbfm.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rodrigues CM, Spellman SR, Solá S, Grande AW, Linehan-Stieers C, Low WC, Steer CJ. Neuroprotection by a bile acid in an acute stroke model in the rat. Cereb Blood Flow Metab. 2002;22:463–71. doi: 10.1097/00004647-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 162.Uchikawa O, Sakai N, Terao Y, Suzuki H. WO2008016131 Fused heterocyclic compound. 2008
- 163.Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurotherapeutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]