

Abstract
Nonclassical protein secretion is of major importance as a number of cytokines and inflammatory mediators are secreted via this route. Current evidence indicates that there are several mechanistically distinct methods of nonclassical secretion. We have shown recently that peroxiredoxin (Prdx) 1 and Prdx2 are released by various cells upon exposure to inflammatory stimuli such as lipopolysaccharide (LPS) or tumor necrosis factor alpha (TNF-α). The released Prdx then acts to induce production of inflammatory cytokines. However, Prdx1 and 2 do not have signal peptides and therefore must be secreted by alternative mechanisms, as has been postulated for the inflammatory mediators interleukin-1β (IL-1β) and high mobility group box-1 (HMGB1). We show here that circulating Prdx1 and 2 are present exclusively as disulfide-linked homodimers. Inflammatory stimuli also induce in vitro release of Prdx1 and 2 as disulfide-linked homodimers. Mutation of cysteines Cys51 or Cys172 (but not Cys70) in Prdx2, and Cys52 or Cys173 (but not Cys71 or Cys83) in Prdx1 prevented dimer formation and this was associated with inhibition of their TNF-α-induced release. Thus, the presence and oxidation of key cysteine residues in these proteins are a prerequisite for their secretion in response to TNF-α, and this release can be induced with an oxidant. By contrast, the secretion of the nuclear-associated danger signal HMGB1 is independent of cysteine oxidation, as shown by experiments with a cysteine-free HMGB1 mutant. Release of Prdx1 and 2 is not prevented by inhibitors of the classical secretory pathway, instead, both Prdx1 and 2 are released in exosomes from both human embryonic kidney (HEK) cells and monocytic cells. Serum Prdx1 and 2 also are associated with the exosomes. These results describe a novel pathway of protein secretion mediated by cysteine oxidation that underlines the importance of redox-dependent signaling mechanisms in inflammation.
INTRODUCTION
Peroxiredoxins (Prdxs) are a family of antioxidant proteins that protects cells from oxidative damage via reduction of peroxides using thioredoxin (TXN) and, in some cases glutaredoxin, as the electron donor and also functions as molecular chaperones and regulators of signal transduction (1,2). A number of studies have shown that Prdxs are secreted from cancer or virus-infected cells (3), are present in biological fluids such as bronchiolar lavage (4) and serum (5) as well as on the membrane of lymphocytes from rheumatoid arthritis patients (6). Prdxs are also known as natural killer enhancing factors (NKEF) due to early observations that these proteins could enhance natural killer cell cytotoxicity against tumor cells (7). Soon after, the antioxidant properties of both NKEF-A and B (Prdx I and II respectively) were demonstrated in erythrocytes (8). The association of oxidative stress with inflammation has been known for some time, but the precise relationship and mechanisms by which these two phenomena are related are not well understood.
In a previous study using redox proteomics, a technique originally developed to identify intracellular glutathionylated proteins (9), we identified Prdx2 among the proteins released from mouse macrophages in response to LPS under specific forms of cysteine oxidation, including formation of disulfide-linked homodimers and mixed disulfides with glutathione (glutathionylation) (10). The released Prdx2 then acts to induce production of inflammatory cytokines, like a classical inflammatory danger signal (11). Studies by others also have shown that Prdx1, 5 and 6 also induce inflammatory cytokines in vitro (12,13).
However, Prdxs (except Prdx4) do not have signal peptides and, therefore, must be secreted by alternative, nonclassical mechanisms as postulated for IL-1β (14) and HMGB1 (15). Here we test the hypothesis that cysteine oxidation is a prerequisite for the release of Prdx1 and 2. For this purpose we set up an experimental model where Prdx release is induced from HEK 293T cells upon stimulation with TNF-α. Previous studies have shown that HEK 293T cells respond to TNF-α (but not LPS) by secreting IL-8 (16); hence this system was used as a model of a proinflammatory stimulus. We then expressed different cysteine mutants of Prdx1 and 2 to investigate the role of specific cysteines in the secretion of these proteins, both in response to LPS and an oxidant, menadione. Release of a classical cytokine, IL-8, was used for comparison. We also investigated whether Prdx1 and 2 were released in exosomes and whether an inhibitor of the classical secretory pathway, brefeldin A (BFA), affected their secretion. The results presented here confirm that the presence and oxidation of specific cysteines in Prdx1 and 2 are an essential requirement for their secretion through the exosomal pathway.
MATERIALS AND METHODS
Reagents
LPS from E. coli 055:B5, glyburide, nocodazole, brefeldin A (BFA), menadione and N-ethylmaleimide (NEM) were all purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cloning and Mutation of Human Prdx2 and Human Prdx1 in pcDNA6
Human Prdx1 and 2 were initially cloned from human cDNA into the pGEM-T plasmid (Promega, Southampton, UK) using specific oligonucleotides (Table 1). Inserts were restricted using specific endonucleases and were ligated into the expression plasmid pet15b (Novagen; Millipore, Watford, Hertford-shire, UK). All cysteines (hPrdx1: C52S, C71S, C83S and C172S; hPrdx2: C51S, C70S and C172S) were changed to serine residues by rolling circle PCR using specific oligonucleotides as shown in Table 1. Sequences were then amplified using the following oligonucleotides: Prdx2 sense: 5′–CAG AAG CTT ATG GCC TCC GGT AAC GCG CGC ATC–3′ and anti-sense: 5′-CAG CTC GAG AT ATT GTG TTT GGA GAA ATA TTC CTT–3′; Prdx1 sense 5′–CTA AAGCTT ATG TCT TCA GGA AAT GCT AAA ATT–3′ and anti-sense 5′–CTAG CTCGAG CG CTT CTG CTT GGA GAA ATA TTC TTT–3′ for cloning into the mammalian expression plasmid pcDNA6 (Life Technologies [Thermo Fisher Scientific Inc., Waltham, MA, USA]). For expression of GFP-Prdx2 fusion proteins, sequences coding for W/T or C51S Prdx2 were cloned downstream of the eGFP sequence in pcDNA3 using HindIII and EcoRI restriction sites. The sequence of all constructs was verified by sequencing.
Table 1.
Oligonucleotides used in the cloning and mutagenesis of human Prdx1, human Prdx2 and rat HMGB-1.
| Protein | Mutagenesis | Primer forward | Primer reverse |
|---|---|---|---|
| Prdx1 WT | — | CATATGTCTTCAGGAAATGCTAAAATTGG | GGATCCTCACTTCTGCTTGGAGAAATATTC |
| Prdx1 | C52S | CACCTTTGTGTCCCCCACG | CGTGGGGGACACAAAGGTG |
| Prdx1 | C71S | GAAACTCAACTCCCAAGTGATTG | CAATCACTTGGGAGTTGAGTTTC |
| Prdx1 | C83S | GATTCTCACTTCTCTCATCTAGCATG | CATGCTAGATGAGAGAAGTGAGAATC |
| Prdx1 | C173S | GAAGTGTCCCCAGCTG | CAGCTGGGGACACTTC |
| Prdx2 WT | — | CATATGGCCTCCGGTAACGCG | GGATCCCTAATTGTGTTTGGAGAAATATTCCTTG |
| Prdx2 | C51S | CACTTTTGTGTCCCCCACCGAGATC | GATCTCGGTGGGGGACACAAAAGTG |
| Prdx2 | C70S | CAAAGCTGGGCAGTGAAGTGCTG | CAGCACTTCACTGCCCAGCTTTG |
| Prdx2 | C172S | CATGGGGAAGTTAGTCCCGCTGCTG | CAGCCAGCGGGACTAACTTCCCCATG |
| HMGB-1 W/T | — | CTAAAGCTTATGGGCAAAGGAGATCCTAAGAAG | CTAGCTCGAGCGTTCATCATCATCATCTTCTTCTTC |
| HMGB-1 | C23S | TCATATGCATTTTTTGTGCAAACTTCTCGGGAGGAGCAT | ATGCTCCTCCCGAGAAGTTTGCACAAAAAATGCATATGA |
| HMGB-1 | C45S | TCAGAGTTTTCTAAGAAGAGCTCAGAGAGGTGGAAG | CTTCCACCTCTCTGAGCTCTTCTTAGAAAACTCTGA |
| HMGB-1 | C106S | GCCTTCTTCCTCTTCAGCTCTGAGTATCGCC | GGCGATACTCAGAGCTGAAGAGGAAGAAGGC |
Cloning and Mutation of Rat HMGB-1 in pcDNA6
Rat HMGB-1 was amplified from rat cDNA (a generous gift from Huan Yang, The Feinstein Institute for Medical Research, Manhasset, NY, USA) by PCR using Pfu-DNA polymerase (Promega) using primers designed to insert restriction sites for HindIII and XhoI. Inserts were restricted using HindIII and XhoI and ligated into pcDNA6 (Life Technologies [Thermo Fisher Scientific]). All cysteines (C23S, C45S and C106S) were first changed to serine residues using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA) and the oligonucleotides shown in Table 1. Double and triple cysteine mutations were achieved by additional rounds of site-directed mutagenesis. The mutations of all constructs were verified by sequencing.
Culture and Transient Transfection of 293T Cells
293T cells were routinely cultured in-Dulbecco modified Eagle medium (DMEM) (Sigma-Aldrich) supplemented with 10% fetal bovine serum and penicillin and streptomycin. Transient transfections were performed using 25-kDa linear polyethylenimine (PEI) (purchased from Polysciences Inc., Warrington, PA, USA). Stock solutions of PEI were prepared in water at a concentration of 1 mg/mL, and the pH adjusted to 7.0. The transfection conditions used are essentially those as described previously (17). Briefly, HEK 293T cells were plated at a density of 0.5 × 106 cells/well in 6-well plates and transfected with 5 μg of DNA. The transfection complex was formed at a DNA-PEI ratio of 1:3 in OPTI-MEM (Life Technologies [Thermo Fisher Scientific]), with a 30 min incubation at room temperature prior to addition to the cells. Twenty-four hours later, culture media were replaced with 1 mL of serum-free DMEM containing 50 ng/mL of recombinant human TNF-α (PeproTech, Rocky Hill, NJ, USA). Cells were cultured for a further 24 h before collecting supernatants and lysing cells in 200 μL of lysis buffer (10 mmol/L Tris HCl, 150 mmol/L NaCl, 1 mmol/L ethylene glycol tetraacetic acid (EGTA), 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1% NP-40, pH 7.4). NEM was added to all supernatants and cell lysates to a final concentration of 50 mmol/L immediately after collection and incubated at room temperature for 15 min. In some experiments, cell viability was determined at the end of the experiment using the CellTiter-Blue assay (Promega) according to the manufacturer’s instructions.
Culture of THP-1 Cells
THP-1 monocytic cells were routinely cultured in RPMI (Sigma-Aldrich) supplemented with 10% fetal bovine serum and penicillin and streptomycin. For stimulation of THP-1 cells with LPS, cells were treated with 100 ng/mL of LPS in serum-free RPMI and cultured for 24 h before collecting cells and supernatants by centrifugation. NEM was again added to all supernatants and cell lysates to a final concentration of 50 mmol/L immediately after collection.
Western Blotting
293T cell culture supernatants or cleared cell lysates were applied without prior dilution to 12% acrylamide sodium dodecyl sulphate (SDS) gels and transferred to nitrocellulose membranes (Millipore) by electroblotting. After blocking in 5% bovine serum albumin (BSA) in PBS containing 0.05% Tween 20, membranes were probed with one of the following antibodies: polyclonal rabbit anti-human Prdx2 or Prdx1 antibody, mouse anti-His (Life Technologies [Thermo Fisher Scientific]), mouse anti-V5 (Life Technologies [Thermo Fisher Scientific]), mouse anti-Prdx4 (Santa Cruz Biotechnology Inc., Dallas, TX, USA), mouse anti-Prdx5 (Santa Cruz Biotechnology), rabbit anti-HSP70 (Sigma-Aldrich), mouse anti-actin (Sigma-Aldrich) followed by detection with anti-rabbit-IgG-horse-radish peroxidase conjugate (Sigma-Aldrich) or anti-mouse IgG horseradish peroxidase conjugate (Stratagene, Agilent Technologies). In some experiments, membranes were stripped using Restore stripping buffer (Pierce Scientific [Thermo Fisher Scientific]) before blocking and reprobing with an antibody as described above. Western blots were developed using advanced chemiluminescence (ECL) reagents (GE Healthcare, Amersham, UK) and exposed to autoradiography using Hyper-film (GE Healthcare). Films were developed using an AGFA Curix 60 developer (Agfa HealthCare, Brentford, Middle-sex, UK).
Cell Viability Assays
Cell viability was measured using the CellTiter-Glo assay (Promega) according to the manufacturer’s instructions. To confirm that this assay was sensitive enough to determine even relatively small levels of cellular toxicity, varying numbers of 293T cells were plated, incubated at 37°C for 24 h to allow cells to adhere, then cell viability measured.
IL-8 ELISA
The concentration of IL-8 secreted by HEK 293T cells was determined using as commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA).
Purification of Recombinant hPrdx2 from HEK293T Cells
HEK 293T cells transiently transfected with pcDNA6-hPrdx2 were lysed in ice-cold lysis buffer (50 mmol/L sodium phosphate pH 8.0, 300 mmol/L NaCl, 0.01% Tween20, Triton 100), centrifuged to remove cell debris and applied to a His Gravi-Trap Talon column (GE Healthcare). Columns were washed extensively with washing buffer (50 mmol/L sodium phosphate pH 8.0, 300 mmol/L NaCl, 0.01% Tween20, 50 mmol/L imidazole) and eluted with elution buffer (50 mmol/L sodium phosphate pH 8.0, 300 mmol/L NaCl, 0.01% Tween20, 300 mmol/L imidazole). The eluted Prdx2 was buffer exchanged into PBS (pH 7.4) using a PD10 desalting column (GE Healthcare).
Preparation of Exosomes
Serum-free medium (1 ml) from cultured cells was collected, treated with NEM to a final concentration of 50 mmol/L and incubated at room temperature for 15 min, then centrifuged at 2,000g for 10 min to remove cells and cell debris. Supernatants were then centrifuged at 100,000g at 4°C for 2 h, washed with PBS, and centrifuged again for 1 h and the resulting supernatants collected. Pellets (exosomes) were resuspended in SDS-PAGE sample buffer (50 mmol/L Tris-Cl [pH 6.8], 100 mmol/L dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) and subjected to 12% SDS-PAGE under nonreducing conditions for Western blot analysis as described above.
Collection of Human Plasma
Human peripheral blood was collected from healthy donors, all of whom gave written informed consent, under the approval of the local Research Governance and Ethics Committee. Blood was collected in vacutainers containing EDTA. Immediately after collection, blood was centrifuged at 1,000g to pellet cells. The plasma was removed and NEM added to a final concentration of 50 mmol/L. For preparation of exosomes from plasma, samples were centrifuged at 10,000g for 30 min at 4°C. The supernatants were then centrifuged at 100,000g at 4°C for 2 h, and the resulting pellets washed with PBS, before centrifuging at 100,000g at 4°C for 1 h. Pellets were lysed in SDS sample buffer for subsequent analysis by Western blotting.
All supplementary materials are available online at www.molmed.org.
RESULTS
TNF-α- and Oxidant-Induced Release of Prdx2 Homodimer
Our previous work showed that Prdx1 and 2 were released from cultured cells (RAW 267 and cultured human PBMCs) in response to inflammatory stimuli and, intriguingly, all of the released Prdx was in the dimeric form (10). To investigate the mechanisms of this release, we used a HEK 293 cell model system, with TNF-α as the proinflammatory stimulus. We first investigated the time course of release of Prdx2 from HEK 293 cells in response to stimulation with TNF-α (Figure 1A). The release of Prdx2 was maximal 24 h after stimulation with TNF-α. Intracellular Prdx2 was present both as monomer and dimer, while the released protein was all in the dimeric form (Figure 1B). Reduction with dithiothreitol (DTT) confirmed the presence of disulfide-linked homodimers. It is important to note that the concentration of TNF-α used was not significantly toxic, as assessed by CellTiter-Glo cell viability assay (Figure 2A), an assay which can detect even a 5% decrease in cell viability (Figure 2B). Furthermore, there was no actin detected in the supernatants of these cells by Western blotting (Figure 2C). We could also show that Prdx2 is released in response to the oxidant menadione and the vast majority of Prdx2 was present in the supernatant as a dimer (Figure 1C). The monomer was only observed in minute amounts, except with the highest concentrations of menadione, which resulted in 83% and 28% toxicity respectively (see Figure 2A), and substantial amounts of monomer released. Thus, the Prdx2 homodimer can be released not only upon cell death but also by a specific process of secretion induced by oxidants or TNF-α. Interestingly, while both menadione and TNF-α induced release of Prdx2, only TNF-α, but not menadione, induced release of IL-8 (IL-8 pg/mL: control, 4 ± 0.1; menadione, 0.1 ± 0.1; TNF-α, 2500 ± 140; n = 6).
Figure 1.

Endogenous (E) and recombinant (R) Prdx2 are released from HEK 293T cells in response to treatment with TNF-α. (A) Time course of Prdx2 release in response to treatment with TNF-α analyzed by Western blotting following nonreducing SDS-PAGE of conditioned media from 293T cells expressing recombinant Prdx2 using anti-Prdx2 antibody. The two arrows indicate rPrdx2 (upper band), which is larger due to V5, and His tags and endogenous Prdx2 (lower band). (B) Intracellular and extracellular levels of endogenous (E) and recombinant (R) Prdx2 in HEK 293T cells ± TNF-α treatment for 24 h (left, gel run under nonreducing conditions; right, reducing). The dimeric form disappears under reducing conditions. (C) Release of endogenous and recombinant Prdx2 from HEK 293T cells 24 h after menadione treatment.
Figure 2.

Cell toxicity does not account for the Prdx2 observed in HEK 293T cell culture supernatants. (A) Cell viability measured by CellTiter-Glo assay 24 h after treatment with 50 ng/mL TNF-α or varying concentrations of menadione. ** p < 0.0001; *p < 0.02, versus control by Dunnett test with one-way analysis of variance (ANOVA). (B) Sensitivity of the CellTiter-Glo assay demonstrated by plating varying numbers of 293T cells. *p < 0.005 versus control. (C) Western blot analysis of actin in the supernatants or (D) cell lysates from 293T cells.
TNF-α Induces Release of Prdx1 and 2 but Not Prdx4 or 5
Release of Prdx1 from HEK 293 cells upon stimulation with TNF-α is identical to that of Prdx2. As shown in Figure 3A, only the dimer of Prdx1 was released in response to TNF-α. By contrast, neither Prdx4 nor Prdx5 were released at detectable levels. Of note, while intracellular Prdx1, like Prdx2, also was present as a disulfide-linked dimer, Prdx4 and 5 were not. In fact, Prdx4 was detected (Figure 3B) as a 31-kDa membrane form and a 27-kDa processed form, consistent with previous reports (18). Despite the fact that Prdx4 has a signal peptide and reportedly can be secreted, none of it was detectable in the supernatant of HEK 293 cells upon TNF-α stimulation. Prdx5 was detected as a single 17-kDa monomer and did not form a dimer (Figure 3C), consistent with the knowledge that Prdx5, an atypical Prdx, forms an intramolecular disulfide rather than a dimer, during its catalytic cycle (19). Furthermore, no Prdx4 or 5 were detectable in the supernatants from LPS-stimulated RAW cells (data not shown), in contrast to Prdx1 and 2, indicating that these differences are not peculiar to HEK 293 cells or to the human system.
Figure 3.

Release of other Prdx proteins from HEK 293T cells. (A) Release of endogenous (E) and recombinant (R) Prdx1 in response to treatment with TNF-α analyzed by Western blotting following nonreducing SDS-PAGE. Intracellular expression of Prdx1 consists of both monomers and dimers and DTT treatment results in the disappearance of the dimeric form. (B) and (C). Absence of Prdx4 (B) and Prdx5 (C) in the conditioned media of HEK 293T cells ± TNF-α. Both proteins (indicated by arrows) were detected in cell lysates by Western blotting following nonreducing SDS-PAGE with anti-Prdx4 and anti-Prdx5 respectively.
Stability of the Monomer in Cell Culture Conditions
The observation that only dimeric Prdx1 and 2 were ever detected in the supernatants of cultured cells raised the question of whether this reflected the fact that only dimers were released or whether a mixture of monomers and dimer were released with subsequent oxidation of monomer to dimer. To distinguish between these two possibilities, we purified recombinant Prdx2 from HEK 293T cells. The resulting preparation consisted of a mixture of monomers and dimers (Figure 4A). The purified Prdx2 was incubated with HEK 293T cultures at 37°C for 24 h and analyzed by Western blotting. Both monomer and dimer were still present in the cell-conditioned medium, with an even higher ratio of monomer to dimer than the input material (Figure 4B).
Figure 4.
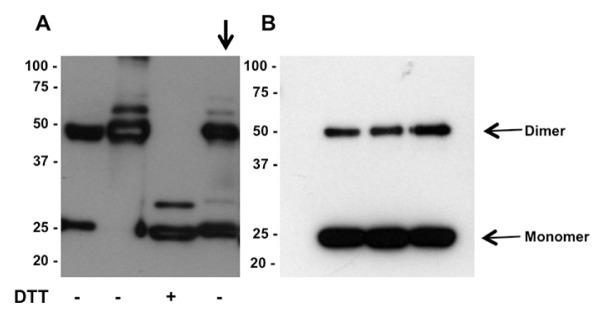
Purified recombinant Prdx2 does not form dimers in HEK 293T cell culture. (A) Recombinant Prdx2 consists of monomer and dimer, which can be reduced to monomer by treatment with DTT. Removal of DTT results in reversion to a mixture of monomers and dimers. (B) Incubation of recombinant Prdx2 (shown by arrow in panel A) with HEK 293T cell for 24 h does not result in oxidation of monomer to dimer.
Disulfide-Mediated Dimerization Is Essential for Release of Prdx1 and 2
The data shown above led us to hypothesize that cysteine oxidation, by formation of a disulfide-linked dimer, is a prerequisite for the release of Prdx1 and 2. We thus overexpressed single-cysteine mutants of Prdx1 and Prdx2 in HEK 293 cells and studied their secretion in response to TNF-α. As shown in Figure 5D, intracellular wild-type recombinant Prdx1 (detected with an anti-V5 tag antibody) is present as both monomer and dimer. Mutating either Cys52 or Cys173 prevented dimerization of Prdx1, while mutations of Cys71 or Cys83 had no effect. Prdx1 was secreted in response to TNF-α solely in the dimeric form, and blocking dimerization by mutating Cys52 or Cys173 prevented secretion (Figure 5F). We performed a similar set of experiments for Prdx2, this time with three cysteine mutations, as Prdx2 has one cysteine fewer than Prdx1. The Western blot for intra-cellular recombinant Prdx2 (E) and secreted Prdx2 (G) is shown in Figure 5. Intracellular recombinant wild-type Prdx2 (detected using an anti-His tag antibody) was present both as monomer and dimer. Mutation of Cys51 or Cys172 prevented the formation of the dimer, while mutation of Cys70 had no effect on dimerization. Secretion of the recombinant protein also was stimulated by TNF-α, in a manner similar to the endogenous protein, although it should be noted that this is likely also due to the increased expression of the recombinant proteins (Figure 5G, arrow). Mutation of either Cys51 or Cys172 inhibited the release of the proteins, while mutation of noncatalytic cysteines did not inhibit secretion. In fact, mutation of Cys71 or Cys83 in Prdx1 (see Figure 5F) or Cys70 in Prdx2 (Figure 5G) slightly increased their release.
Figure 5.

Mutation of cysteines involved in disulfide bond formation prevents release of Prdx1 and Prdx2 from HEK 293T cells. (A) Catalytic mechanism of Prdx2. The peroxidatic cysteine C51 (C52 in Prdx1) reacts with peroxide then with the resolving cysteine C172 (C173 in Prdx1) on the second subunit of the dimer to form disulfide bonds. (B) Prdx1 and (C) Prdx2 cysteine mutants. Intracellular expression of recombinant wild-type and mutated Prdx1 (D) and Prdx2 (E) in HEK 293T cell lysates analyzed by Western blotting with anti-V5 or anti-His antibody, respectively, following nonreducing SDS-PAGE to allow identification of monomers and dimers. Release of Prdx1 (F) and Prdx2 (G) also were analyzed by Western blotting of conditioned media from HEK 293T cells ± TNF-α using specific antibodies following nonreducing SDS-PAGE.
An appreciable amount of monomer was observed in the cell supernatants for the first time, but only when disulfide bond formation was prevented by mutation of Cys51 or Cys172. One possibility was that overexpression of these mutant proteins was causing protein aggregation and consequent cellular toxicity. To determine whether this was the case and whether cellular location was affected by the mutation, we expressed wild-type and C51S Prdx2 as GFP-fusion proteins for analysis by confocal microscopy. wild-type GFP-Prdx2 was visible as punctate cytoplasmic staining, in contrast to the GFP control, which showed diffuse staining throughout the cytoplasm and nucleus (Supplementary Figures 1A, B). The C51S mutant GFP-fusion was present in much larger aggregates within the cytoplasm (Supplementary Figure 1C). Staining of endogenous Prdx2 by immunofluorescent labeling confirmed that the punctate cytoplasmic staining observed for the wild-type recombinant Prdx2 accurately reflected the localization of the endogenous protein (Supplementary Figure 1D).
An important question was whether this phenomenon of cysteine oxidation and resultant dimerization as a requirement for release from stimulated cells applied to the release of other proteins in response to inflammatory stimuli. The obvious candidate to investigate was the well-characterized inflammatory danger signal, HMGB1. This protein previously has been shown to switch between redox states in response to inflammation (20) and, importantly, the nucleocytoplasmic localization is impaired by mutation of one of the three cysteine residues. We generated four forms of recombinant HMGB1, wild-type, C23S, C106S and triple cysteine-to-serine mutations to investigate whether cysteine mutation could impair secretion in the manner observed for Prdx1 and 2. Recombinant HMGB-1 was released from HEK 293 cells in response to TNF-α but none of the cysteine mutations affected the release of HMGB1 from HEK 293 cells in response to TNF-α (Figure 6).
Figure 6.

Mutation of cysteine residues does not prevent release of HMGB-1 from HEK 293T cells. (A) Intracellular expression of recombinant wild-type and mutated HMGB-1 in HEK 293T cell lysates analyzed by Western blotting with anti-V5 antibody following reducing SDS-PAGE. Cell lysates were diluted 50-fold before applying to gel. (B) Release of HMGB-1 from HEK 293T cells analyzed by Western blotting of conditioned media applied to gel without dilution using anti-V5 antibody following reducing SDS-PAGE. (C) Densitometric analysis of released HMGB-1 expressed as a percentage of intracellular HMGB-1.
Prdx1 and 2 Dimers Are Secreted via a Nonclassical Pathway and Are Associated with the Exosomes
The lack of signal peptides in Prdx1 and 2 suggests a nonclassical pathway for secretion. We wondered, however, whether the dimer could be secreted through the Golgi. Therefore, we tested the effect of a known inhibitor of classical secretion, BFA, on the secretion of Prdx2 from TNF-α-stimulated HEK 293 cells. As shown in Supplementary Figure 2A, BFA did not inhibit TNF-α-induced Prdx2 secretion. In fact, BFA alone induced some release of Prdx2, possibly due to nonspecific toxicity. To confirm the inhibitory effect of BFA on the classical secretary pathway, we measured the concentration of a classically secreted cytokine, IL-8. As shown in Supplementary Figure 2B, BFA completely blocked release of IL-8. We also tested the effects of glyburide and nocodazole, known inhibitors of the release of a nonclassically secreted cytokine, IL-1β. As shown in Supplementary Figures 2C and D, glyburide and nocodazole actually increased the amount of Prdx2 dimer released. Because several inflammatory proteins can be secreted though the exosomal pathway (21,22), we investigated whether the Prdx1 and 2 released from HEK 293 cells were associated with exosomes. For this purpose, we isolated exosomes from the supernatants of control or TNF-α-stimulated HEK 293 cells using ultracentrifugation. Figure 7 shows the levels of Prdx1 and 2 present in the exosomes and in the supernatant left after removal of exosomes (Figures 7A, B). It can be seen that there is a strong association of Prdx1 and 2 with the exosomal fraction. To assess the validity of the fractionation method used here, we also measured an exosomal marker, HSP70 (23), and the results shown in Figures 7A and B confirm its enrichment in the exosomal fraction. Identical results were obtained using a second method of exosome isolation, further confirming the association of Prdx2 with the exosomes (Supplementary Figure 3). Similar results were obtained in THP-1 cells (a human monocytic cell line) when stimulated with LPS. Dimeric Prdx1 and 2 were released from THP-1 cells in response to LPS, and are also associated with the exosomal fraction as shown in Figures 7C and D.
Figure 7.

Both Prdx1 and Prdx2 are associated with exosomes in cell culture supernatants and in healthy human serum. Western blot analysis of (A,C) Prdx1 or (B,D) Prdx2 present in exosomal or cytosolic fractions of supernatants from (A,B) HEK 293T cells or from (C,D) THP-1 cells. Blots were stripped and reprobed with anti-HSP70 antibody to confirm the presence of exosomes. (E) Western blotting of human serum from four healthy donors using anti-Prdx2 following nonreducing SDS-PAGE. Addition of DTT reduces the dimers to monomers. (F) Exosomes were prepared from serum samples by ultracentrifugation and analyzed by Western blotting with anti-Prdx2 in nonreducing and reducing conditions.
Prdx2 Is Present as Dimers in Human Plasma
To determine whether this mechanism also occurs in vivo, we analyzed Prdx2 in human plasma. Prdx2 was detected in human plasma by Western blotting using specific antibodies as shown in Figures 7E and F. The majority of Prdx2 was present as oligomers, with some dimer present. Upon treatment of the samples with DTT, the oligomeric form disappeared and most of the protein was reduced to monomers (Figure 7E). We could also show that, similar to the conditioned medium from cultured cells, the Prdx2 present in plasma was associated with exosomes (see Figure 7F).
DISCUSSION
Here we show a novel redox-regulated mechanism for the extracellular release of Prdx1 and 2 in response to inflammatory stimuli. The lack of a signal peptide in these proteins meant that classical secretion was unlikely and this was confirmed by the fact that release of these proteins was unaffected by BFA. The aim of the study was to test the hypothesis that cysteine oxidation is implicated in the release of Prdx1 and 2. The observation that only the dimer is present in the supernatant raised the possibility that secreted Prdx, whether in monomeric or dimeric form, would be oxidized after its release in our culture conditions. However, when recombinant Prdx2 originally present both as monomer and dimer was added to cultured cells, we could still detect both monomer and dimer. Thus, if a mixture of monomer and dimer was secreted, this would be detected in the supernatant.
Prdx1 and 2 are typical 2-Cys Prdxs, which rely on two specific cysteine residues for their catalytic activity, with a peroxidatic cysteine that is oxidized to a sulfenic acid by H2O2 and a resolving one which then reduces it by forming a disulfide bridge (Figure 5A). The peroxidatic cysteines are Cys52 or 51, while the resolving ones are Cys173 or 172, in Prdx1 and 2 respectively. Prdx2 has one (Cys70) and Prdx1 has two (Cys71 and Cys83) additional cysteines. Consistent with the mechanism shown in Figure 5A, we found that mutation of the peroxidatic or resolving cysteines prevented dimerization, while mutation of the additional cysteine had no effect on dimerization. Intriguingly, mutation of either the peroxidatic or the resolving cysteines in both Prdx1 and 2 prevented secretion from cells
The one exception to this was the C51S mutation of Prdx2, which was observed in the supernatant in the monomeric form. It is possible, however, that the appearance of the Prdx2 monomer in the supernatants of cells expressing the C51S mutant is an artifact of recombinant expression and aggregation of this protein. This is supported by experiments showing that, when expressed as a GFP-fusion protein, the C51S mutant is present in large aggregates in the cytoplasm, while the wild-type protein is present as uniformly distributed punctate staining in the cytoplasm. Secretion of misfolded or overexpressed proteins is a phenomenon previously described for various proteins and is thought to represent a means by which the cytoplasm can be cleared of aggregated or accumulated protein (24,25). As there does not appear to be any difference in the extent of expression for the wild-type and C51S mutant, it seems more likely that the release of the monomer in the case of the C51S mutant is due to the lack of dimerization which seems to cause aggregation within the cells. Of note, the corresponding C52S mutant in Prdx1 did not behave in this manner, with the overall evidence suggesting that mutations which prevented dimerization also prevented secretion. It is clear from the preceding discussion that, although mutation of individual cysteines is the only way of determining their importance, there are some limitations to this approach. However, with the exception of the experiments with the C51S mutant, the evidence points to a role for cysteine oxidation in the secretion of Prdx1 and 2.
There is a growing list of proteins that are secreted by nonclassical pathways (26), and our finding might define a class of them whose secretion is mediated by the redox state of the protein. Interestingly, dimerization via the formation of covalent dimers is also essential for the secretion of fibroblast growth factor (FGF) 1 (27), as point mutation of Cys30 inhibits export of this protein (28). FGF1 is also one of a group of nonclassically secreted proteins that are not released spontaneously, but that require cell stress to be secreted (26). However, the similarities between release of FGF1 and Prdx 1 and 2 end there. In fact, although FGF1 homodimers are absolutely necessary for secretion, they form only one part of a much larger multiprotein complex that is required for release (29,30). Export from the cell then occurs via translocation through the cell membrane (31) and not via exosomal release, as shown here for Prdx1 and 2.
The release of the dimer in response to proinflammatory stimulation or oxidant treatment must reflect a change in intra-cellular trafficking of the dimer to associate with exosomes. It is interesting to note that oxidation and subsequent dimerization of Prdx2 does seem to promote membrane-association of the protein as shown in human erythrocytes (32) and rat myocardium (33). As both monomer and dimer are present in un-stimulated cells, but with little or no release in the absence of stimulation (10) (this study), it may be the ratio of monomer to dimer that determines the association of the dimer with the exosomes and subsequent export from the cell as postulated in Figure 8.
Figure 8.

Schematic model of redox-regulated release of Prdx 1 and 2. Oxidized Prdx 1 and 2 are targeted to exosomes for release following exposure of the cells to inflammatory agents or oxidants.
It is difficult to say whether glutathionylation is important for this process. Our previous work has shown that both Prdx1 and 2 are released from mouse macrophage cells in response to LPS as glutathionylated dimers (10). One possibility is that the glutathionylation releases monomers and dimers from larger multimeric structures that, as such, would normally be retained in the cell. In fact, Prdx1 dimers can organize in decamers (34) and it was shown that, while normally 97% of the protein is in this form, glutathionylation completely eliminates these decamers, releasing monomers and dimers (35). Although the decameric form was not investigated specifically in this study, it is likely that the dimers that are secreted are released from these higher-order oligomers.
The mechanism of release of Prdx1 and 2 is distinct from that for Il-1β, which can be inhibited by glyburide or nocodazole. It is unsurprising that glyburide does not inhibit the release of Prdxs, as this compound inhibits activation of the NLRP3 inflammasome, hence inhibiting the processing of pro-IL1 to mature IL-1β for secretion, while Prdx1 and 2 do not undergo proteolytic processing during secretion. However, secretion of IL-1β also can be inhibited by cytoskeletal inhibitors such as nocodazole (36), demonstrating the involvement of microtubules in the exocytosis of the vesicles containing IL-1β (36). It appears from the results shown here that microtubules are unimportant for the release of Prdx1 and 2 and the major determining factor in their release is dimerization via cysteine oxidation. The fact that the oxidant menadione also induces the release of Prdx1 and 2 suggest that oxidation per se is sufficient to cause their release.
The redox regulation of these Prdxs is of particular importance as the proinflammatory activity of both Prdx1 (11,12) and Prdx2 (10,13) has been demonstrated previously. The fact that these Prdxs are released in a redox-regulated manner in response to inflammatory stimuli as well as oxidant treatment is the first evidence for the potential generation of inflammatory mediators by cellular redox changes. The redox enzyme TXN also is released from RAW cells in response to LPS in an oxidized (glutathionylated) form (10) and also is released during infection and inflammation and has immunoregulatory effects (37). Similarly to the Prdxs shown here, secretion of TXN occurs by a nonclassical pathway (38), but intriguingly, secretion is independent of redox state (39).
Other proinflammatory molecules secreted in exosomes or vesicles include HSP70 (21) and HMGB-1 (40). HSP70 is released from cells in exosomes (23), but there is no evidence that its association with exosomes is associated with changes in its redox status. Although redox changes are important in controlling the intracellular localization of HMGB-1 in terms of nuclear versus cytoplasmic localization (41), extracellular secretion of HMGB-1 is independent of redox state.
CONCLUSION
We have uncovered a novel mechanism for nonclassical secretion of inflammatory mediators that relies on cysteine oxidation and might potentially define a “redox secretome.” The presence of Prdx1 and 2 in healthy human plasma suggests that redox changes, by controlling their secretion, regulate their role in inflammation. This may be particularly important in pathologies such as ischemia-reperfusion injury, where an inflammatory response is observed in the absence of infection or autoimmunity, and where a redox imbalance may act as the trigger of inflammation. A comprehensive definition of the redox secretome in pathological conditions might open new perspectives in terms of pharmacological targets or biomarkers for several diseases.
Supplemental Data
ACKNOWLEDGMENTS
Funded by the EU, European Regional Development Fund, France (Channel) England (PeReNE Project), and by the Brighton and Sussex Medical School.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Mullen L, Hanschmann E-M, Lillig CH, Herzenberg LA, Ghezzi P. (2015) Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 21:98–108.
REFERENCES
- 1.Rhee SG, et al. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–9. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 2.Kang SW, Rhee SG, Chang TS, Jeong W, Choi MH. 2-Cys peroxiredoxin function in intracellular signal transduction: therapeutic implications. Trends Mol Med. 2005;11:571–8. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang JW, et al. Peroxiredoxin-I is an autoimmunogenic tumor antigen in non-small cell lung cancer. FEBS Lett. 2005;579:2873–7. doi: 10.1016/j.febslet.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 4.Gharib SA, et al. Of mice and men: comparative proteomics of bronchoalveolar fluid. Eur Respir J. 2010;35:1388–95. doi: 10.1183/09031936.00089409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerrits EG, et al. Serum peroxiredoxin 4: a marker of oxidative stress associated with mortality in type 2 diabetes (ZODIAC-28) PLoS One. 2014;9:e89719. doi: 10.1371/journal.pone.0089719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szabo-Taylor KE, et al. Lymphocytes from rheumatoid arthritis patients have elevated levels of intracellular peroxiredoxin 2, and a greater frequency of cells with exofacial peroxiredoxin 2, compared with healthy human lymphocytes. Int J Biochem Cell Biol. 2012;44:1223–31. doi: 10.1016/j.biocel.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shau H, Gupta RK, Golub SH. Identification of a natural killer enhancing factor (NKEF) from human erythroid cells. Cell Immunol. 1993;147:1–11. doi: 10.1006/cimm.1993.1043. [DOI] [PubMed] [Google Scholar]
- 8.Shau H, Kim A. Identification of natural killer enhancing factor as a major antioxidant in human red blood cells. Biochem Biophys Res Commun. 1994;199:83–8. doi: 10.1006/bbrc.1994.1197. [DOI] [PubMed] [Google Scholar]
- 9.Fratelli M, et al. Identification of proteins undergoing glutathionylation in oxidatively stressed hepatocytes and hepatoma cells. Proteomics. 2003;3:1154–61. doi: 10.1002/pmic.200300436. [DOI] [PubMed] [Google Scholar]
- 10.Salzano S, et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc Natl Acad Sci U S A. 2014;111:12157–62. doi: 10.1073/pnas.1401712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 12.Riddell JR, Wang XY, Minderman H, Gollnick SO. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J Immunol. 2010;184:1022–30. doi: 10.4049/jimmunol.0901945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shichita T, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18:911–7. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 14.Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for inter-leukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990;9:1503–10. doi: 10.1002/j.1460-2075.1990.tb08268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardella S, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razonable RR, Henault M, Watson HL, Paya CV. Nystatin induces secretion of interleukin (IL)-1beta, IL-8, and tumor necrosis factor alpha by a toll-like receptor-dependent mechanism. Antimicrob Agents Chemother. 2005;49:3546–9. doi: 10.1128/AAC.49.8.3546-3549.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derouazi M, et al. Serum-free large-scale transient transfection of CHO cells. Biotechnol Bioeng. 2004;87:537–45. doi: 10.1002/bit.20161. [DOI] [PubMed] [Google Scholar]
- 18.Tavender TJ, Sheppard AM, Bulleid NJ. Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem J. 2008;411:191–9. doi: 10.1042/BJ20071428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seo MS, et al. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J Biol Chem. 2000;275:20346–54. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- 20.Venereau E, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anand PK, Anand E, Bleck CK, Anes E, Griffiths G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS One. 2010;5:e10136. doi: 10.1371/journal.pone.0010136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burke M, Choksawangkarn W, Edwards N, Ostrand-Rosenberg S, Fenselau C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J Proteome Res. 2014;13:836–43. doi: 10.1021/pr400879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem. 2005;280:23349–55. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- 24.Sloan IS, Horowitz PM, Chirgwin JM. Rapid secretion by a nonclassical pathway of overexpressed mammalian mitochondrial rhodanese. J Biol Chem. 1994;269:27625–30. [PubMed] [Google Scholar]
- 25.Tanudji M, Hevi S, Chuck SL. Improperly folded green fluorescent protein is secreted via a non-classical pathway. J Cell Sci. 2002;115:3849–57. doi: 10.1242/jcs.00047. [DOI] [PubMed] [Google Scholar]
- 26.Prudovsky I, et al. Secretion without Golgi. J Cell Biochem. 2008;103:1327–43. doi: 10.1002/jcb.21513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson A, Tarantini F, Gamble S, Friedman S, Maciag T. The release of fibroblast growth factor-1 from NIH 3T3 cells in response to temperature involves the function of cysteine residues. J Biol Chem. 1995;270:33–6. doi: 10.1074/jbc.270.1.33. [DOI] [PubMed] [Google Scholar]
- 28.Tarantini F, Gamble S, Jackson A, Maciag T. The cysteine residue responsible for the release of fibroblast growth factor-1 residues in a domain independent of the domain for phosphatidylserine binding. J Biol Chem. 1995;270:29039–42. doi: 10.1074/jbc.270.49.29039. [DOI] [PubMed] [Google Scholar]
- 29.LaVallee TM, et al. Synaptotagmin-1 is required for fibroblast growth factor-1 release. J Biol Chem. 1998;273:22217–23. doi: 10.1074/jbc.273.35.22217. [DOI] [PubMed] [Google Scholar]
- 30.Mouta Carreira C, et al. S100A13 is involved in the regulation of fibroblast growth factor-1 and p40 synaptotagmin-1 release in vitro. J Biol Chem. 1998;273:22224–31. doi: 10.1074/jbc.273.35.22224. [DOI] [PubMed] [Google Scholar]
- 31.Mohan SK, Rani SG, Yu C. The hetero-hexameric complex structure, a component in the non-classical pathway for fibroblast growth factor 1 (FGF1) secretion. J Biol Chem. 2010;285:15464–75. doi: 10.1074/jbc.M109.066357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matte A, et al. Membrane association of peroxiredoxin-2 in red cells is mediated by the N-terminal cytoplasmic domain of band 3. Free Radic Biol Med. 2013;55:27–35. doi: 10.1016/j.freeradbiomed.2012.10.543. [DOI] [PubMed] [Google Scholar]
- 33.Schroder E, Brennan JP, Eaton P. Cardiac peroxiredoxins undergo complex modifications during cardiac oxidant stress. Am J Physiol Heart Circ Physiol. 2008;295:H425–33. doi: 10.1152/ajpheart.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroder E, et al. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 A resolution. Structure. 2000;8:605–15. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- 35.Park JW, Piszczek G, Rhee SG, Chock PB. Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry. 2011;50:3204–10. doi: 10.1021/bi101373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carta S, et al. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: role of micro-tubules. Blood. 2006;108:1618–26. doi: 10.1182/blood-2006-03-014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertini R, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–9. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R. Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem. 1992;267:24161–4. [PubMed] [Google Scholar]
- 39.Tanudji M, Hevi S, Chuck SL. The nonclassic secretion of thioredoxin is not sensitive to redox state. Am J Physiol Cell Physiol. 2003;284:C1272–9. doi: 10.1152/ajpcell.00521.2002. [DOI] [PubMed] [Google Scholar]
- 40.Schiraldi M, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209:551–63. doi: 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoppe G, Talcott KE, Bhattacharya SK, Crabb JW, Sears JE. Molecular basis for the redox control of nuclear transport of the structural chromatin protein Hmgb1. Exp Cell Res. 2006;312:3526–38. doi: 10.1016/j.yexcr.2006.07.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.