

Abstract
Insulin sensitivity is critically dependent on the activity of PI3K (phosphoinositide 3-kinase) and generation of the PtdIns(3,4,5)P3 second messenger. PtdIns(3,4,5)P3 can be broken down to PtdIns(3,4)P2 through the action of the SHIPs (Src-homology-2-domain-containing inositol phosphatases). As PtdIns(3,4)P2 levels peak after those of PtdIns(3,4,5)P3, it has been proposed that PtdIns(3,4)P2 controls a negative-feedback loop that down-regulates the insulin and PI3K network. Previously, we identified two related adaptor proteins termed TAPP [tandem PH (pleckstrin homology)-domain-containing protein] 1 and TAPP2 that specifically bind to PtdIns(3,4)P2 through their C-terminal PH domain. To determine whether TAPP1 and TAPP2 play a role in regulating insulin sensitivity, we generated knock-in mice that express normal endogenous levels of mutant TAPP1 and TAPP2 that are incapable of binding PtdIns(3,4)P2. These homozygous TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice are viable and exhibit significantly enhanced activation of Akt, a key downstream mediator of insulin signalling. Consistent with increased PI3K and Akt activity, the double knock-in mice display enhanced whole body insulin sensitivity and disposal of glucose uptake into muscle tissues. We also generated wild-type and double TAPP1R211L/R211LTAPP2R218L/R218L knock-in embryonic fibroblasts and found that insulin triggered enhanced production of PtdIns(3,4,5)P3 and Akt activity in the double knock-in fibroblasts. These observations provide the first genetic evidence to support the notion that binding of TAPP1 and TAPP2 adaptors to PtdIns(3,4)P2 function as negative regulators of the insulin and PI3K signalling pathways.
Keywords: insulin signalling, phosphoinositide 3-kinase (PI3K), pleckstrin homology domain (PH domain), protein tyrosine phosphatase, tandem pleckstrin homology-domain-containing protein (TAPP)
INTRODUCTION
The PI3K (phosphoinositide 3-kinase) pathway plays a central role in regulating most cellular responses to insulin [1]. This is emphasized by the findings that marked insulin resistance is invariably induced by inhibiting the PI3K pathway in genetically modified animal models, or by administering inhibitors or by employing high-fat diets that suppress this signalling network [2,3]. There is also ample evidence that insufficient PI3K pathway activation is a hallmark of Type 2 diabetes in humans [2]. We now have a good understanding of how activation of the PI3K pathway controls insulin signalling responses. Once activated by insulin, PI3K phosphorylates the D3 hydroxy group of PtdIns(4,5)P2 to generate PtdIns(3,4,5)P3, the key second messenger of the insulin signalling pathway. PtdIns(3,4,5)P3 triggers cellular responses to insulin by recruiting the Akt protein kinases that possess a PtdIns(3,4,5)P3-binding PH (pleckstrin homology) domain to the plasma membrane. This induces a conformational change in Akt, permitting its activation by the upstream PDK1 (phosphoinositide-dependent kinase 1) and mTORC (mammalian target of rapamycin complex) 2 protein kinases [4]. Once activated, Akt phosphorylates numerous substrates to regulate responses to insulin [5], including phosphorylating AS160 (Akt substrate of 160 kDa) to stimulate glucose uptake [6] and GSK3 (glycogen synthase kinase 3) to promote glycogen synthesis [7].
The mechanisms that regulate the inactivation of the insulin signalling pathway also play a vital role in regulating overall insulin actions. For example, overactivation of the mTORC1 pathway as a result of obesity induces insulin resistance by stimulating phosphorylation and degradation of IRS (insulin receptor substrate) adaptor proteins required for activation of PI3K by insulin [8]. Furthermore, knock out of PTP (protein tyrosine phosphatase) 1B, which inactivates the insulin receptor, markedly sensitizes mice to insulin and protects animals to developing insulin resistance when fed on a high-fat diet [9]. The lipid phosphatases that break down PtdIns(3,4,5)P3 also play vital roles in controlling sensitivity to insulin. For example, a 50% reduction in the expression of the PTEN (phosphatase and tensin homologue deleted on chromosome 10), which converts PtdIns(3,4,5)P3 into PtdIns(4,5)P2, induces insulin sensitization [10] and is sufficient to reverse marked insulin resistance in a mouse knock-in model in which the ability of PDK1 to interact with PtdIns(3,4,5)P3 has been ablated [11]. PtdIns(3,4,5)P3 can also be converted into PtdIns(3,4)P2 by SHIP (Src-homology-2-domain-containing inositol phosphatase) 1 and SHIP2 [12,13]. Mice lacking SHIP2 display enhanced activation of Akt following insulin administration and are highly resistant to weight gain when placed on a high-fat diet [14]. There has been much discussion of whether PtdIns(3,4)P2 functions as a signalling molecule in its own right, since insulin and other agonists that activate the PI3K pathway significantly increase its levels [15]. Detailed analysis confirms that the majority of PtdIns(3,4)P2 originates from PtdIns(3,4,5)P3 and, consistent with this, levels of PtdIns(3,4)P2 peak after those of PtdIns(3,4,5)P3 [16].
To date, numerous PH-domain-containing proteins have been identified that interact with PtdIns(3,4,5)P3, specifically GRP1 (guanine-nucleotide-releasing protein 1), Btk and ARNO [ARF (ADP-ribosylation factor) nucleotide-binding-site opener], or bind both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 with similar affinity, such as Akt, PDK1 and DAPP1 (dual adaptor for phosphotyrosine and 3-phosphoinositides 1) [15,17]. However, to our knowledge, the only proteins to have been identified that specifically interact with PtdIns(3,4)P2 with high affinity are TAPP (tandem PH-domain-containing protein) 1 and TAPP2, related adaptor proteins consisting of two sequential PH domains in which the C-terminal PH domain binds PtdIns(3,4)P2. The N-terminal PH domain does not interact with any lipid tested [18]. The structure of the C-terminal PH domain of TAPP1 suggests that several conserved basic residues interacted with the 3- and the 4-phosphate groups of PtdIns(3,4)P2. Mutation of one of these residues, Arg211 on TAPP1 or the equivalent Arg218 residue in TAPP2, completely prevented interaction of these proteins with PtdIns(3,4)P2 [18]. Binding of TAPP1 to PtdIns(3,4,5)P3 is inhibited by steric hindrance of an alanine residue located close to the position in which the 5′-phosphate would be expected to reside [19]. Interestingly, the equivalent residue in PtdIns(3,4,5)P3-binding PH domains is frequently glycine. Mutation of the alanine residue to glycine in TAPP1 resulted in it being capable of interacting with PtdIns(3,4,5)P3 and PtdIns(3,4)P2 with similar affinity [19]. Evidence also suggests that TAPP1 binds PtdIns(3,4)P2 selectively in vivo, as TAPP1 relocated from the cytosol to the plasma membrane of cells following stimulation with agonists that induced PtdIns(3,4)P2, but not with those that induced mainly PtdIns(3,4,5)P3 [20]. Similarly, in B-cells, both TAPP1 and TAPP2 translocated to the plasma membrane in response to antigen stimulation and this correlated with the formation of PtdIns(3,4)P2 rather than production of PtdIns(3,4,5)P3 [21,22].
The biological functions of TAPP1 and TAPP2 are not well characterized. Apart from the PH domains, the only other known functional region is a C-terminal PDZ-binding motif that interacts with several PDZ-binding proteins, including PTPN13 (known previously as PTPL1 or FAP-1) [23] as well as the scaffolding proteins MUPP1 (multiple PDZ-domain-containing protein 1) [20], syntrophin [24] and utrophin [25]. One hypothesis was that TAPP1 and TAPP2, by specifically recognizing PtdIns(3,4)P2, could function to recruit signalling molecules or complexes to the plasma membrane that down-regulate the PI3K signalling pathways [23]. As a first step to exploring this idea, we generated knock-in mice expressing point mutants of TAPP1 and TAPP2 unable to interact with PtdIns(3,4)P2. The resulting TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice are viable and displayed significantly enhanced whole body insulin sensitivity in a hyperinsulinaemic–euglycaemic clamp study. We have also demonstrated that TAPP1R211L/R211L TAPP2R218L/R218L double knock-in fibroblast cells display enhanced PtdIns(3,4,5)P3 levels and Akt activation in response to insulin. Our results suggest that enhanced insulin sensitivity is mediated by increased Akt activation, thereby stimulating glucose uptake in heart and skeletal muscle. TAPP1R211L/R211LTAPP2R218L/R218L knock-in mice represent the first mouse model for these proteins and support the notion that TAPP1 and TAPP2 operate as negative regulators of the PI3K signalling pathway.
MATERIAL AND METHODS
Materials and general buffers
Protein G–Sepharose and [γ-32P]ATP were purchased from GE Healthcare. Human insulin (Actrapid) from Novo-Nordisk was obtained from Ninewells Pharmacy, Dundee. PI-103 PI3K inhibitor [26] was synthesized by Natalia Shpiro (MRC Protein Phosphorylation Unit, Dundee). Lysis buffer consisted of 50 mM Tris/HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 0.3% CHAPS, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 0.27 Msucrose, 0.1%2-mercaptoethanol and Complete™ protease inhibitor cocktail (Roche).
Generation and genotyping of TAPP1R211L/R211L and TAPP2R218L/R218L knock-in mice
TaconicArtemis generated the TAPP1R211L/R211L and TAPP2R218L/R218L knock-in mice described in Figure 1. The knock-in mice were generated and maintained on an inbred C57BL/6J background. Genotyping was performed by PCR using genomic DNA isolated from ear biopsies. For TAPP1 knock-in mice, Primer 1 (5′-CCTCATTCAGAGTATGCAGC-3′) and Primer 2 (5′-CTACCTTCAAGGAAGTGTTCC-3′), and for TAPP2 knock-in mice, Primer 1 (5′-GGATGTTCTCAAGACTCACG-3′) and Primer 2 (5′-CATCATGGGAGTAAGAGTAGG-3′) were used to detect the wild-type and knock-in alleles. The PCR programme consisted of 5 min at 95°C, then 35 cycles of 30 s at 95°C, 30 s at 60°C and 30 s at 72°C, and 5min at 72°C. DNA sequencing was performed by DNA Sequencing & Services (MRC Protein Phosphorylation Unit, College of Life Sciences, University of Dundee; http://www.dnaseq.co.uk) using Applied Biosystems Big-Dye version 3.1 chemistry on an Applied Biosystems model 3730 automated capillary DNA sequencer.
Figure 1. Generation and analysis of TAPP1R211L/R211L and TAPP2R218L/R218L knock-in mice.

(A) The knock-in construct, the endogenous TAPP1 allele containing exons 4–9 and the targeted allele with the neomycin cassette (NEO) removed by Flp recombinase are depicted. The black/grey rectangles represent exons, the grey triangles represent FRT sites and the black triangles represent loxP sites. TK, thymidine kinase. (B) To confirm correct vector insertion, genomic DNA purified from the targeted ES cells was digested with either HpaI or KpnI and subjected to Southern blot analysis. Sizes are indicated in kb. (C) To genotype mice, genomic DNA was PCR-amplified with primers P1 and P2. Sizes are indicated in bp. (D) Genomic DNA (gDNA) obtained from mice was subjected to PCR to generate a product encompassing the knock-in mutation region. To verify the presence of the knock-in mutation, the PCR products were sequenced. (E) The knock-in construct, the endogenous TAPP2 allele containing exons 4–10 and the targeted allele with the neomycin cassette (NEO) removed by Flp recombinase are depicted. Symbols are as in (A). (F) To confirm correct vector insertion, genomic DNA purified from the targeted embryonic stem cells was digested with either XhoI or EcoRV and subjected to Southern blot analysis. Sizes are indicated in kb. (G) To genotype mice genomic DNA was PCR-amplified with primers P1 and P2. Sizes are indicated in bp. (H) Genomic DNA (gDNA) obtained from mice was amplified by PCR to generate a product encompassing the knock-in mutation region. To verify the presence of the knock-in mutation, PCR products were sequenced.
Animals
Mice were maintained under specific pathogen-free conditions and all procedures were carried out in accordance with the regulations set by the University of Dundee and the U.K. Home Office.
Blood glucose and plasma insulin measurement
Blood glucose levels were determined using the Ascensia Breeze 2 blood glucose monitoring system (Bayer) following tail incision. For plasma insulin measurement, blood was collected from mice following tail incision and using sodium-heparinized capillary tubes (Hawksley). The blood was centrifuged at 3000 g for 15 min, and the supernatant was collected. Plasma insulin levels were determined using a rat/mouse insulin ELISA kit from Millipore (No EZRMI-13K) according to the instructions of the manufacturer. Rat insulin ranging from 0.2 to 10 ng/ml was used as a standard.
PtdIns(3,4)P2–agarose pull-down
Mouse tissues were homogenized in 10 mM Hepes (pH 7.4), 150 mM NaCl, 0.25% Nonidet P40, 1 mM benzamidine, 0.1 mM PMSF and 2 mM sodium orthovanadate. Tissue lysates were centrifuged at 18000 g for 15 min at 4°C, and the supernatant was snap-frozen and stored at −80°C. Then, 1 mg of tissue lysate was incubated with 25 μl of PtdIns(3,4)P2 PIP beads (Echelon) for 1.5 h at 4°C. Beads were washed three times with 10 mM Hepes (pH 7.4), 150 mM NaCl and 0.25% Nonidet P40 and proteins were eluted with SDS/PAGE sample buffer (250 mM Tris/HCl, pH 6.8, 5% SDS, 5% 2-mercaptoethanol and 32.5% glycerol) at 95°C for 5 min. Eluted proteins were analysed by immunoblotting.
Antibodies
The following antibodies were raised in sheep and affinity-purified on the appropriate antigen. The total antibody used for immunoprecipitation and immunoblotting of Akt1 (S742B, third bleed) was raised against full-length His–Akt1. The anti-TAPP1 antibody (S022C, second bleed) was raised against residues 252–356 of mouse GST (glutathione transferase)–TAPP1. The anti-TAPP2 antibody (S392A, first bleed) was raised against residues 396–415 of mouse GST–TAPP2. The anti-PRAS40 (proline-rich Akt substrate of 40 kDa) antibody (S115B, first bleed) was raised against the sequence DLPRPRLNTSDFQKLKRKY corresponding to residues 238–256 of human PRAS40. An antibody that recognizes PRAS40 phosphorylated at Thr246 (S114B, second bleed) was raised against the peptide CRPRLNpTSDFQK. The anti-FOXO1 (forkhead box O1) antibody (S457, third bleed) was raised against GST–FOXO1 comprising residues 2–655 of human FOXO1. The antibodies against Akt pThr308 (#9275), Akt pSer473 (#9271), FOXO1 pThr24 (#9464) and GSK3α/β pSer21/pSer9 (#9331) were purchased from Cell Signaling Technology. The anti-GSK3α/β antibody (#44–610) was purchased from Biosource. The anti-IRS1 antibody (06–248) was from Millipore and the antibody against IRS1 pTyr612 (44–816G) was purchased from Invitrogen. The anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (ab8245) was purchased from Abcam. Detection of immune complexes was performed using either fluorophore-conjugated secondary antibodies (Molecular Probes) followed by visualization using an Odyssey® LI-COR imaging system or by HRP (horseradish peroxidase)-conjugated secondary antibodies (Pierce) and an enhanced chemiluminescence reagent.
Preparation of tissue lysates, immunoblotting and Akt kinase assay
Following a 5 h fast, a bolus of insulin (1 m-unit/g of body weight) was intravenously injected through the inferior vena cava to mice that had been anaesthetized by pentobarbital (86 μg/g of body weight, intraperitoneally injected). After 20 min, tissues (heart, liver, gastrocnemius muscle) were extracted, frozen in liquid nitrogen and stored at −80°C. Tissues were homogenized on ice in a 10-fold mass excess of ice-cold lysis buffer using a Kinematica Polytron. Tissue lysates were centrifuged at 18 000 g for 15 min at 4°C, and the supernatant was snap-frozen and stored at −80°C. Lysates (20 μg) were analysed by immunoblotting using the antibodies, as indicated in the Figures. The activity of Akt1 was assessed either by immunoblotting of tissue lysate (20 μg) using phosphospecific antibodies or by kinase activity assays. Briefly, Akt1 was immunoprecipitated from 1 mg of tissue lysate, and kinase activity was measured using the Crosstide peptide (GRPRTSSFAEG) as described previously [27]. Values of Akt activity are given as means±S.E.M. for the number of mice indicated in the Figure legends, and significance was determined by unpaired two-tailed Student’s t tests.
Generation and stimulation of MEFs (mouse embryonic fibroblasts)
MEFs isolated from mouse embryos at E13.5 (embryonic day 13.5) were generated as described previously [28] and immortalized by continuous passaging. Cells were cultured in DMEM (Dulbecco’s modified Eagle’s medium) containing 10% serum (Sigma), 2 mM L-glutamine, 50 units/ml penicillin G and 50 μg/ml streptomycin (Life Technologies). Cells were serum-starved in DMEM with L-glutamine, penicillin and streptomycin for 16 h before stimulation. Cells were stimulated with insulin (10 nM) for 15 or 30 min. Cells were subsequently lysed in lysis buffer and the lysates were centrifuged at 18 000 g for 15 min at 4°C. The supernatant was snap-frozen and stored at −80°C. Lysates (10 μg) were analysed by immunoblotting using the antibodies, as indicated in the Figures.
Hyperinsulinaemic–euglycaemic clamp studies
Male mice at 6 months of age were used for a hyperinsulinaemic–euglycaemic clamp studies which were performed at Vanderbilt-NIH Mouse Metabolic Phenotyping Center, Nashville, TN, U.S.A. (http://www.mc.vanderbilt.edu/MMPC/), as described previously in detail [29]. Catheters were implanted in a carotid artery and a jugular vein of mice for sampling and infusions respectively 5 days before study [30]. Insulin clamps were performed on 5 h fasted mice [29]. [3–3H]glucose (2.4 μCi) was primed and continuously infused for a 90-min equilibration period (0.04 μCi/min) and a 2 h clamp period (0.12 μCi/min). Baseline blood or plasma parameters were determined as the mean of values obtained in blood samples collected at −15 and −5 min. At zero time, insulin infusion (4 m-units·kg−1·min−1) was started and continued for 165 min. Blood glucose was clamped at 150–160 mg/dl using a variable GIR (glucose infusion rate). Mice received heparinized saline-washed erythrocytes from donors at 5 μl/min to prevent a fall of haematocrit. Insulin clamps were validated by assessment of blood glucose over time. Blood glucose was monitored every 10 min, and the GIR was adjusted as needed. Blood was taken at 80–120 min for the determination of [3–3H]glucose. Clamp insulin was determined at 100 and 120 min. At 120 min, 13 μCi of [14C]2DG (2-[14C]deoxyglucose) was administered as an intravenous bolus. Blood was taken at 122, 125, 130 and 135 min for the determination of [14C]2DG. After the last sample, mice were anaesthetized and tissues were collected.
Plasma insulin was determined by ELISA (Millipore). Non-esterified fatty acids were assayed enzymatically using the Wako Diagnostics. Radioactivity of [3–3H]glucose, [14C]2DG, and [14C]2DG 6-phosphate in plasma and tissue samples were determined by liquid-scintillation counting [30]. Whole body glucose appearance (Ra) and disappearance (Rd) rates were determined using non-steady-state equations [31]; EndoRa was determined by subtracting the GIR from total Ra. Glucose uptake was calculated as described previously [32].
The area under the curve was calculated using GraphPad Prism software. Values are given as means±S.E.M. for the number of mice indicated in the Figure legends. Significance was determined by unpaired two-tailed Student’s t tests.
Measurement of PtdIns(3,4,5)P3 levels
PtdIns(3,4,5)P3 levels were measured employing a TR-FRET (time-resolved fluorescence resonance energy transfer) displacement assay that was described previously [33]. Briefly, wild-type or knock-in MEFs cultured on 10-cm-diameter dishes were deprived of serum overnight and then left untreated in the absence or presence of 1 μM PI-103 or stimulated with insulin (10 nM) for the indicated times. Cells were lysed by incubation with 1.5 ml of ice-cold 0.5 M trichloroacetic acid. Cells were then pelleted by centrifugation at 13 000 g for 1 min, and non-charged lipids were extracted by washing cell pellets with 1 ml of a neutral solvent (methanol/chloroform, 2:1 v/v) for 20 min. PtdIns(3,4,5)P3 was extracted by washing with 0.5 ml of an acidic solvent (methanol/chloroform/12 M HCl, 80:40:1, by vol.) for 20 min. The acidic extract containing PtdIns(3,4,5)P3 was phase-split by the addition of 0.18 ml of chloroform and 0.3 ml of 0.1M HCl. After centrifugation at 12 000 g for 1 min, the lower organic phase containing PtdIns(3,4,5)P3, was collected and dried under vacuum. The lipid pellet was resuspended by sonication (15 s at 40 W in a cup sonicator bath) in 0.06 ml of TR-FRET assay buffer consisting of 50 mM Tris/HCl (pH 7.4), 0.15 M NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 2mM dithiothreitol and 1.2% (w/v) sodium cholate. Duplicate 25 μl samples were assayed for PtdIns(3,4,5)P3 content using the TR-FRET displacement assay described previously [33]. The estimated PtdIns(3,4,5)P3 levels were normalized to cell density by estimating protein levels in the extracted cell pellets. This was achieved by recovering cells from the upper phase of the phase-split material by the addition of 1 ml of acetone (to remove residual chloroform) and centrifugation. The air-dried pellets were then dissolved overnight by gentle shaking in 0.2 M NaOH and 1% (w/v) SDS at 50°C. Protein in the dissolved pellets was estimated by Micro BCA (bicinchoninic acid) protein assay kit (Thermo Scientific) according to the manufacturer’s instructions.
RESULTS
Generation and analysis of TAPP1R211L/R211L and TAPP2R218L/R218L knock-in mice
To study the physiological role of TAPP1 and TAPP2, we generated knock-in mice in which the critical arginine residues required for PtdIns(3,4)P2 binding (Arg211 of TAPP1 and Arg218 of TAPP2 [18,19]) were mutated to leucine in order to abolish the ability of these adaptors to interact with phosphoinositides. The strategy to generate and genotype the TAPP1R211L/R211L and TAPP2R218L/R218L knock-in mice is summarized in Figure 1. The knock-in mice were generated and maintained on an inbred C57BL/6J background.
Single TAPP1R211L/R211L or TAPP2R218/R218L or double TAPP1R211L/R211LTAPP2R218L/R218L were viable and born at the expected Mendelian frequency (Table 1). The strategy used to breed wild-type TAPP1+/+TAPP2+/+ control and experimental TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice is outlined in Table 1. The TAPP1 and TAPP2 proteins were expressed at the same levels in all tissues examined of wild-type TAPP1+/+TAPP2+/+ and homozygous TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice, establishing that mutation does not affect protein stability (Figure 2A). TAPP1 (Figure 2B) and TAPP2 (Figure 2C) proteins derived from wild-type TAPP1+/+TAPP2+/+ tissues, but not those from TAPP1R211L/R211LTAPP2R218L/R218L double knock-in tissues, interacted with agarose resin conjugated to PtdIns(3,4)P2, confirming that the knock-in mutation ablated the ability of TAPP1 and TAPP2 to interact with PtdIns(3,4)P2. The TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice compared with wild-type TAPP1+/+TAPP2+/+ mice displayed normal body weight (Figure 2D) as well as fed and fasted levels of blood glucose (Figure 2E) and plasma insulin (Figure 2F). We have maintained the double knock-in mice over a 14-month period and no discernable overt phenotype was noted.
Table 1. Breeding strategy used to generate TAPP1R211L/R211L, TAPP2R218L/R218L and TAPP1R211L/R211LTAPP2R218L/R218L knock-in mice.
Single homozygous TAPP1R211L/R211L and TAPP2R218L/R218L, as well as double TAPP1R211L/R211LTAPP2R218L/R218L, knock-in mice were bred using the strategy depicted in the Table and the progeny was genotyped as described in the Materials and methods section. The number and the percentage of each genotype are indicated followed by its expected Mendelian frequency. In the case of the double TAPP1R211L/R211LTAPP2R218L/R218L knock-in mice, as wild-type littermates and double knock-in mice can only be obtained at a ratio of 1:16 in the same cross, we interbred TAPP1R211L/R211LTAPP2R218L/R218L and TAPP1R211L/R211LTAPP2R218L/R218L as well as TAPP1+/+TAPP2+/+ and TAPP1+/+TAPP2+/+ littermates derived from a 1:16 ratio mating.
| Cross | Genotype | Number born | Expected Mendelian frequency (%) |
|---|---|---|---|
| TAPP1+/R211L and TAPP1+/R211L | TAPP1+/+ | 11 (24.45 %) | 25 |
| TAPP1+/R2111L | 23 (51.1 %) | 50 | |
| TAPP1R211L/R211L | 11 (24.45 %) | 25 | |
| TAPP2+/R218L and TAPP2+/R218L | TAPP2+/+ | 21 (31.8 %) | 25 |
| TAPP2+/R218L | 28 (42.4 %) | 50 | |
| TAPP2R218L/R218L | 17 (25.8 %) | 25 | |
| TAPP1+/R211LTAPP2+/R218L and TAPP1+/R211LTAPP2+/R218L | TAPP1+/+TAPP2+/+ | 6 (5.7 %) | 6.25 |
| TAPP1R211L/R211LTAPP2R218L/R218L | 5 (4.8 %) | 6.25 | |
| TAPP1+/R211LTAPP2+/R218L | 28 (26.7 %) | 25 | |
| TAPP1+/R211LTAPP2+/+ | 19 (18.1 %) | 12.5 | |
| TAPP1+/+TAPP2+/R218L | 14 (13.3 %) | 12.5 | |
| TAPP1R211L/R211LTAPP2+/+ | 4 (3.8 %) | 6.25 | |
| TAPP1+/+TAPP2R218L/R218L | 5 (4.8 %) | 6.25 | |
| TAPP1R211L/R211LTAPP2+/R218L | 14 (13.3 %) | 12.5 | |
| TAPP1+/R211LTAPP2R218L/R218L | 10 (9.5 %) | 12.5 | |
| TAPP1+/+TAPP2+/+ and TAPP1+/+TAPP2+/+ | TAPP1+/+TAPP2+/+ | 100% | |
| TAPP1R211L/R211LTAPP2R218L/R218L and TAPP1R211L/R211LTAPP2R218L/R218L | TAPP1R211L/R211LTAPP2R218L/R218L | 100% |
Figure 2. Analysis of TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice.

(A) Tissue lysates from wild-type TAPP1+/+TAPP2+/+ (WT) and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in (KI) mice were subjected to immunoblot analysis using anti-TAPP1, anti-TAPP2 and anti-GAPDH antibodies. (B) PtdIns(3,4)P2–agarose pull-downs of lung extracts from wild-type TAPP1+/+TAPP2+/+ (WT) and TAPP1R211L/R211LTAPP2R218L/R218L (R211L) mice and total lysates were subjected to immunoblot analysis using anti-TAPP1 antibody. (C)PtdIns(3,4)P2–agarose pull-downs of spleen extracts from wild-type TAPP1+/+TAPP2+/+ (WT) and TAPP1R211L/R211LTAPP2R218L/R218L (R218L) mice and total lysates were subjected to immunoblot analysis using anti-TAPP2 antibody. (D) The body weight of wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice was monitored over a period of 16 weeks. Results are mean ± S.E.M. body weights for male (left-hand panel) and female (right-hand panel) animals (TAPP1+/+TAPP2+/+, n =12; TAPP1R211L/R211LTAPP2R218L/R218L, n =10). (E) Blood glucose and (F) plasma insulin levels of fed and overnight fasted wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L knock-in mice. Results are presented as means ± S.E.M. for male mice, 3 months of age (TAPP1+/+TAPP2+/+, n =10; TAPP1R211L/R211LTAPP2R218L/R218L, n =10).
Increased activation of Akt in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice
To investigate how inhibiting the ability of TAPP1 and TAPP2 to interact with PtdIns(3,4)P2 affects the insulin signalling pathway, we injected mice deprived of food for 5 h with insulin (1 m-unit/g of body weight) and analysed phosphorylation and activation of Akt in various insulin-responsive tissues, including liver (Figure 3A), heart (Figure 3B) and gastrocnemius skeletal muscle (Figure 3C). We observed that there was no difference in the basal Akt activity between wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice, whereas insulin-stimulated Akt activation was significantly enhanced in tissues derived from insulin-injected TAPP1R211L/R211LTAPP2R218L/R218L double knock-in compared with wild-type TAPP1+/+TAPP2+/+ animals (Figure 3). Increased activation of Akt in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice was accompanied by enhanced phosphorylation of Akt at Thr308 (PDK1 site) and Ser473 (mTORC2 site). Quantitative immunoblotting analysis confirmed that insulin stimulation of double knock-in mice induced a statistically significant increase in Thr308 and Ser473 phosphorylation in heart and gastrocnemius tissues (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/434/bj4340265add.htm). Despite enhancement of Akt activity, this was not translated into a significant increase in the phosphorylation of Akt substrates examined (PRAS40, GSK3α/β and FOXO1). Moreover, no marked changes were observed in IRS1 phosphorylation at Tyr612 (Figures 4A–4C), a key-binding site for PI3K, which is phosphorylated by the insulin receptor [34]. This is considered further in the Discussion.
Figure 3. Increased Akt activation in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice upon insulin stimulation.
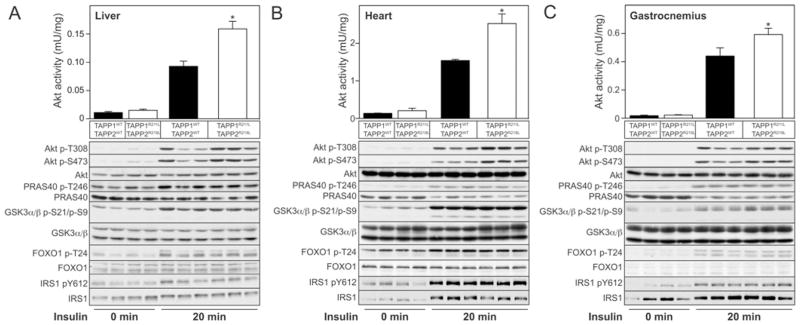
Wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in male mice, 3 months of age, were starved for 5 h and injected (intravenously) with insulin (1 m-unit/g of body weight) for 20 min. Tissue extracts of liver (A), heart (B) and gastrocnemius muscle (C) were generated and analysed by immunoblotting using the indicated antibodies. Samples from every lane in each blot shown are derived from a separate mouse. Akt was also immunoprecipitated and its activity in the different tissues was measured by in vitro kinase assay (*P < 0.05, n =3–4). Results area means ± S.E.M.
Figure 4. Enhanced insulin sensitivity and glucose metabolic index in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice.

(A)GIRs in wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in male mice, 6months of age, during the hyerinsulinaemic–euglycaemic clamp study. The area under the curve for GIRs at steady state (80–120 min) was determined (**P< 0.004). (B)Whole body glucose disappearance rates (Rd) of wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice under basal and clamp conditions (*P < 0.02). (C) Endogenous glucose appearance rate (EndoRa) of wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP-2R218L/R218L double knock-in mice under basal and clamp conditions. (D) [14C]2DG uptake during the clamp was determined in adipose, gastrocnemius muscle, diaphragm and heart tissues of wild-type TAPP1+/+TAPP2+/+ (TAPP1/2 Wt) and TAPP1R211L/R211LTAPP2R218L/R218L (TAPP1/2 Ki) double knock-in mice (**P < 0.005 gastrocnemiusmuscle, *P < 0.01 heart). TAPP1+/+TAPP2+/+, n =7; TAPP1R211L/R211LTAPP2R218L/R218L, n =8 for all panels. Results are means ± S.E.M.
Enhanced whole body insulin sensitivity in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice
To investigate whether increased insulin-induced activation of Akt in the TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice enhanced insulin sensitivity in the most physiological context, we performed a hyperinsulinaemic–euglycaemic clamp, a method that is considered the gold standard for assessing insulin actions on whole body glucose homeostasis in vivo [35], on TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice and wild-type TAPP1+/+TAPP2+/+ control mice. These studies were undertaken at the Vanderbilt NIH Mouse Metabolic Phenotyping Center and we employed the clamp study in conscious and unrestrained animals [29]. Blood glucose levels were maintained by a variable glucose infusion with a constant insulin infusion rate of 4m-units·kg−1·min−1 (see Supplementary Figure S2A at http://www.BiochemJ.org/bj/434/bj4340265add.htm). Interestingly, the key index of whole body insulin sensitivity, the GIR required to maintain euglycaemia, was increased at all time points in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice compared with wild-type TAPP1+/+TAPP2+/+ control mice (Figure 4A). Calculation of the area under the curve during establishment of the clamp between 80 and 120 min revealed significantly higher (25%) GIR in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice compared with wild-type TAPP1+/+TAPP2+/+ control mice. The steady-state whole body glucose disappearance rate (Rd) during the clamp was also significantly higher (23%) in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice in comparison with wild-type TAPP1+/+TAPP2+/+ mice (Figure 4B).
These results demonstrate that TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice display significantly enhanced whole body insulin sensitivity measured in conscious unrestrained animals in vivo. Endogenous glucose appearance (EndoRa) is the sum of hepatic glycogenolysis and gluconeogenesis in the postabsorptive state. In the basal state, EndoRa was comparable in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice and wild-type TAPP1+/+TAPP2+/+ mice. EndoRa was suppressed equally in both genotypes during insulin clamps (Figure 4C). EndoRa therefore did not account for the enhanced insulin sensitivity observed in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice. The optimal insulin dose used to examine liver is much lower than the optimal insulin dose used to examine muscle. The insulin dose used was chosen specifically to amplify an insulin effect on glucose flux in cardiac and skeletal muscle. EndoRa was maximally suppressed and therefore an enhanced effect at the liver could not be elucidated. Considering the elevated Akt activation in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice, it is very possible that an enhanced insulin suppression of EndoRa would have been observed at a lower insulin dose (<2.0 m-units·kg−1·min−1). In addition, non-esterified fatty acid concentrations were comparable in wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice, indicating that insulin’s effect on lipolysis was similar between the two genotypes (see Supplementary Figure S2B). Glucose uptake measured by infusion of [14C]2DG during the steady-state phase of the clamp revealed comparable glucose uptake into adipose and diaphragm of wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice (Figure 4D). Interestingly, significant increases in rates of glucose uptake in gastrocnemius muscle (77% increase) and heart (75% increase) were detected in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice in comparison with wild-type TAPP1+/+TAPP2+/+ mice in which we also found increased activation of Akt (Figure 3). As is often the case in insulin-sensitive mice [30], the basal and clamp insulin levels were reduced. This is important to consider, since the increased insulin action was observed in the TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice despite reduced insulin concentrations (see Supplementary Figure S2C). These results indicate that the enhanced whole body insulin sensitivity observed in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice is likely to be mediated through increased glucose uptake into skeletal muscle and heart tissues.
Increased levels of PtdIns(3,4,5)P3 and Akt activation in TAPP1R211L/R211LTAPP2R218L/R218L knock-in fibroblasts
We generated immortalized MEFs derived from wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in E13.5 stage embryos. These cells were stimulated with 10 nM insulin for various time points and PtdIns(3,4,5)P3 levels were measured employing a TR-FRET assay described previously [33]. This revealed similar levels of PtdIns(3,4,5)P3 in non-stimulated wild-type and or double knock-in cells (Figure 5A). There was also no marked difference in PtdIns(3,4,5)P3 levels between cells treated with the PI3K inhibitor PI-103. However, following insulin stimulation, at all time points examined, we observed a 1.5–2-fold increase in PtdIns(3,4,5)P3 levels in the TAPP1R211L/R211LTAPP2R218L/R218L cells compared with TAPP1+/+TAPP2+/+ fibroblasts (Figure 5A). We also observed that Akt was phosphorylated and activated to a significantly greater extent in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in fibroblasts compared with wild-type TAPP1+/+TAPP2+/+ cells (Figure 5B). Moreover, unlike in mouse tissues, we observed that the TAPP1R211L/R211LTAPP2R218L/R218L double knock-in fibroblasts stimulated with insulin displayed a modest increase in the phosphorylation of PRAS40 and GSK3α/β compared with wild-type TAPP1+/+TAPP2+/+ cells (Figure 5B).
Figure 5. Increased PtdIns(3,4,5)P3 production and Akt activation in TAPP1R211L/R211LTAPP2R218L/R218L double knock-in MEFs upon insulin stimulation.
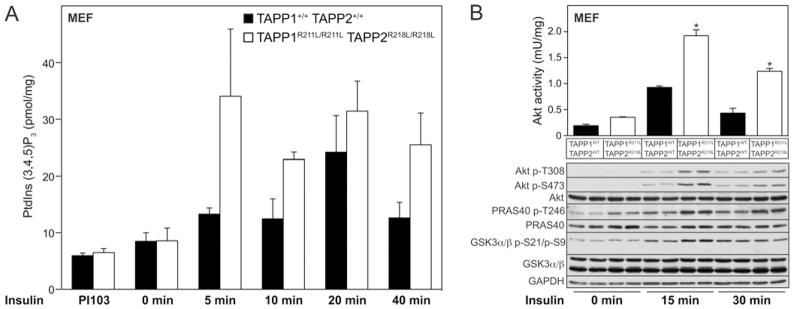
MEFs derived from wild-type TAPP1+/+TAPP2+/+ and TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice were serum-starved overnight. Cells were left untreated or treated with 1 μM PI-103 or 10 nM insulin for the indicated times. (A) PtdIns(3,4,5)P3 levels were quantified and are expressed as pmol/mg of cellular protein. Results are means ± S.E.M. for a triplicate experiment. Similar results were obtained in two separate experiments. (B) Akt was immunoprecipitated and activity was measured by in vitro kinase assay (*P < 0.05, n =3–4). Results are mean ± S.E.M. activity (m-unit/mg). Cell extracts were analysed by immunoblotting using the indicated antibodies. Samples from every lane in each blot shown are derived from a separate cell dish.
DISCUSSION
The key finding of the present study is that the TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice displayed significantly enhanced whole body insulin sensitivity. This provides the first genetic evidence establishing that the TAPP1 and TAPP2 adaptor proteins regulate insulin sensitivity by binding to PtdIns(3,4)P2. Our data from in vivo clamp studies suggest that insulin sensitivity results from increased glucose disposal into heart and skeletal muscle. This is supported by the 1.5–2- fold increase in activity of Akt which is well known to control glucose transport in response to insulin [36]. The increased activation of Akt observed in TAPP1R211L/R211LTAPP2R218L/R218L knock-in mice is likely to result from enhanced activation of PI3K that would account for the increased phosphorylation of Akt at Thr308 (PDK1 T-loop site) and Ser473 (mTORC2 site, hydrophobic motif) (Figure 3). This increased Akt phosphorylation and activation is also observed in fibroblasts derived from TAPP1R211L/R211LTAPP2R218L/R218L double knock-in embryos (Figure 5B). Overall, these findings support the notion that binding of TAPP1 and TAPP2 to PtdIns(3,4)P2 results in down-regulation of PI3K and the insulin signalling pathway.
It should be noted that the increased activation of Akt observed in TAPP1 and TAPP2 double knock-in mice did not lead to a marked increase in the phosphorylation of Akt substrates that we have analysed (PRAS40, GSK3 and FOXO1) in various insulin-responsive tissues (Figure 3). Similar results have been obtained in other studies where marked alterations in insulin sensitivity were correlated with changes in Akt activity, which were not reflected by monitoring phosphorylation of PRAS40, GSK3 or FOXO1. For example, knock-in mutation of the PDK1 PH domain to prevent interaction with PtdIns(3,4,5)P3 in mice results in a ~2-fold inhibition of Akt, resulting in marked insulin resistance, without significantly affecting the phosphorylation of Akt substrates [11,37]. This lack of effect on phosphorylation of Akt substrates is likely to be a result of the inherent spare capacity and amplification of signalling pathways. These data suggest that in vivo insulin sensitivity can be intricately correlated with Akt activation and even 1.5–2-fold changes in Akt activity, which are relatively modest, are sufficient to induce profound changes in insulin sensitivity. More work needs to be undertaken to identify the key substrates that Akt phosphorylates to regulate insulin sensitivity and how modest changes in Akt activity influence this process. It is also likely that PtdIns(3,4,5)P3 will stimulate other Akt-independent pathways that modulate overall insulin sensitivity.
It makes sense to employ PtdIns(3,4)P2 as a signal to down-regulate the PI3K pathway, as the levels of this 3-phosphoinositide peak later than those of PtdIns(3,4,5)P3. PtdIns(3,4)P2 would serve to down-regulate PI3K and signal the need for decreased formation of PtdIns(3,4,5)P3 production. Functionally, this would accelerate the return of insulin action to the pre-fed state. Further evidence supporting the notion that PtdIns(3,4)P2 acts as a negative regulator of the PI3K pathway is provided by analysis of SHIP2-knockout mice. Mice lacking SHIP2 that converts PtdIns(3,4,5)P3 into PtdIns(3,4)P2 displayed similar increased Akt activation in response to insulin in liver and muscle, as we have found in the TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice [14]. These results have been interpreted to result from increased levels of PtdIns(3,4,5)P3 in the SHIP2-knockout mice leading to increased activation of Akt. However, it would be interesting to explore whether the increased activation of Akt results instead from diminished recruitment of the TAPP1 and TAPP2 adaptor proteins to the plasma membrane as a result of decreased levels of PtdIns(3,4)P2 being produced in SHIP2-knockout mice. SHIP1 and SHIP2 have also been implicated in having roles in the negative regulation of the PI3K pathway in immune cells [38]. The activity of SHIP1 and SHIP2 is exquisitely controlled by interaction with growth factor receptors and by phosphorylation of tyrosine residues [39]. Presumably, being able to intricately regulate the activity of the SHIPs is vital, as they play dual roles in directly controlling absolute levels of PtdIns(3,4,5)P3, as well as acting as gatekeepers of PtdIns(3,4)P2 production that could in turn influence PI3K signalling networks via recruitment of the TAPP1 and TAPP2 adaptors.
An important question for future research is to define the mechanism by which recruitment of TAPP1 and TAPP2 to plasma membrane down-regulates the PI3K pathway. To date, TAPP1 and TAPP2 have been reported to interact via their C-terminal residues with at least three PDZ-domain-binding proteins, including PTPN13 (known previously as PTPL1 or FAP-1) [23], as well as the scaffolding proteins MUPP1 [20], syntrophin [24] and utrophin [25]. PTPN13 [40], MUPP1 [41], syntrophin and utrophin [42] are known to interact with a large number of binding partners involved in regulating numerous signalling pathways, so it may not be straightforward to deconvolute which of these is involved in regulating insulin sensitivity. It is also possible that TAPP1 and TAPP2 bind to other regulators of the PI3K pathway that have not been identified. PTPN13 is an attractive candidate to be a mediator of PI3K pathway signalling, as the three-dimensional structure of PTPN13 closely resembles the structure of PTP1B, one of the physiological PTPs that acts on the insulin receptor [43]. PTPN13 contains a positively charged pocket located near the catalytic site, reminiscent of the second phosphotyrosine-binding site in PTP1B, which is required to dephosphorylate peptides containing two adjacent phosphotyrosine residues as occurs, for example, in the activated insulin receptor [44]. Consistent with this, PTPN13, like PTP1B, interacted with and dephosphorylates a diphosphorylated insulin receptor peptide much more efficiently than monophosphorylated peptides. This indicates that PTPN13 may down-regulate the PI3K pathway by dephosphorylating the insulin or potentially other growth factor receptors that contain tandem phosphotyrosine residues [43].
To address the role of PTPN13 in regulating insulin sensitivity, we have generated catalytically inactive PTPN13C2374A/C2374A knock-in mice (S. Wullschleger and D.R. Alessi, unpublished work). These mice are viable, display no obvious phenotype and did not display marked insulin sensitization in the initial studies that we have undertaken (S. Wullschleger and D.R. Alessi, unpublished work). This suggests that PTPN13 may not be rate-limiting in the mechanism by which TAPP1 and TAPP2 control insulin sensitivity. In future work, it will be important to study which proteins are associated with TAPP1 and TAPP2 in insulin-responsive tissues and evaluate whether these are involved in down-regulating insulin signalling when recruited to the plasma membrane.
In conclusion, the TAPP1R211L/R211LTAPP2R218L/R218L double knock-in mice represent the first mouse model for these adaptor proteins and support the notion that TAPP1 and TAPP2 operate as negative regulators of the PI3K signalling pathway. As TAPP1 and TAPP2 are expressed in all tissues examined, these adaptors may have roles to play in modulating PI3K activity and PtdIns(3,4,5)P3 levels in systems beyond controlling insulin sensitivity. In future work, it will be important to define the mechanism by which TAPP1 and TAPP2 induce down-regulation of PI3K by binding to PtdIns(3,4)P2 and establish whether this system plays more general roles in other biological systems, such as in B-cells and T-cells where TAPP1 and TAPP2 are highly expressed. These results also indicate that if compounds could be developed that inhibit binding of TAPP1 and TAPP2 to PtdIns(3,4)P2, these could be deployed to improve insulin sensitivity in insulin-resistant diabetic patients. Interestingly, a recent study has shown that it is possible to design small molecules that bind to the 3-phosphoinositide-binding sites of PH domains [45] and it would be interesting to employ a similar approach to develop compounds that prevent TAPP1/TAPP2 binding to PtdIns(3,4)P2. It will also be important to identify the proteins that interact with TAPP1 and TAPP2 to control insulin sensitivity, as these might also represent new therapeutic targets for the treatment of insulin resistance.
Supplementary Material
Acknowledgments
We acknowledge Gail Fraser for genotyping of mice, the Sequencing Service (College of Life Sciences, University of Dundee) for DNA sequencing co-ordinated by Nicholas Helps, and the protein production and antibody purification teams [Division of Signal Transduction Therapy (DSTT), University of Dundee] co-ordinated by Hilary McLauchlan and James Hastie for generation of antibodies. Surgery and clamps were performed by Bingle Bracy and Dr Li Kang of Vanderbilt University School of Medicine.
FUNDING
We thank the Medical Research Council and the pharmaceutical companies supporting the Division of Signal Transduction Therapy Unit (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck KGaA and Pfizer) for financial support. The research was also supported by the National Institutes of Health [grant numbers U24 DK059637 and R01 DK054902].
Abbreviations used
- [14C]2DG
2-[14C]deoxyglucose
- DMEM
Dulbecco’s modified Eagle’s medium
- E13.5
embryonic day 13.5
- FOXO1
forkhead box O1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GIR
glucose infusion rate
- GSK3
glycogen synthase kinase 3
- GST
glutathione transferase
- IRS
insulin receptor substrate
- MEF
mouse embryonic fibroblast
- mTORC
mammalian target of rapamycin complex
- MUPP1
multiple PDZ-domain-containing protein 1
- PDK1
phosphoinositide-dependent kinase 1
- PH
pleckstrin homology
- PI3K
phosphoinositide 3-kinase
- PRAS40
proline-rich Akt substrate of 40 kDa
- PTP
protein tyrosine phosphatase
- SHIP
Src-homology-2-domain-containing inositol phosphatase
- TAPP
tandem PH-domain-containing protein
- TR-FRET
time-resolved fluorescence resonance energy transfer
Footnotes
AUTHOR CONTRIBUTION
Stephan Wullschleger undertook most of the experimentation, Kei Sakamoto provided key expertise with mice studies and helped with insulin injections, David Wasserman undertook the clamp studies and Alex Gray undertook the PtdIns(3,4,5)P3 measurements. All authors were involved in planning and analysing the experimental data. Stephan Wullschleger and Dario Alessi wrote the paper.
References
- 1.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 2.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- 3.Ihle NT, Paine-Murrieta G, Berggren MI, Baker A, Tate WR, Wipf P, Abraham RT, Kirkpatrick DL, Powis G. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther. 2005;4:1349–1357. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 5.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakamoto K, Holman GD. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab. 2008;295:E29–E37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, Alessi DR. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knock-in analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 9.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 10.Wong JT, Kim PT, Peacock JW, Yau TY, Mui AL, Chung SW, Sossi V, Doudet D, Green D, Ruth TJ, et al. Pten (phosphatase and tensin homologue gene) haploinsufficiency promotes insulin hypersensitivity. Diabetologia. 2007;50:395–403. doi: 10.1007/s00125-006-0531-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wullschleger S, Sakamoto K, Johnstone L, Duce S, Fleming S, Alessi DR. How moderate changes in Akt T-loop phosphorylation impact on tumorigenesis and insulin resistance. Dis Models Mech. 2010;4:95–103. doi: 10.1242/dmm.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase. Proc Natl Acad Sci USA. 1996;93:1689–1693. doi: 10.1073/pnas.93.4.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pesesse X, Deleu S, De Smedt F, Drayer L, Erneux C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem Biophys Res Commun. 1997;239:697–700. doi: 10.1006/bbrc.1997.7538. [DOI] [PubMed] [Google Scholar]
- 14.Sleeman MW, Wortley KE, Lai KM, Gowen LC, Kintner J, Kline WO, Garcia K, Stitt TN, Yancopoulos GD, Wiegand SJ, Glass DJ. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat Med. 2005;11:199–205. doi: 10.1038/nm1178. [DOI] [PubMed] [Google Scholar]
- 15.Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- 16.Stephens LR, Jackson TR, Hawkins PT. Agonist-stimulated synthesis of phosphatidylinositol(3,4,5)-trisphosphate: a new intracellular signalling system? Biochim Biophys Acta. 1993;1179:27–75. doi: 10.1016/0167-4889(93)90072-w. [DOI] [PubMed] [Google Scholar]
- 17.Manna D, Albanese A, Park WS, Cho W. Mechanistic basis of differential cellular responses of phosphatidylinositol 3,4-bisphosphate- and phosphatidylinositol 3,4,5-trisphosphate-binding pleckstrin homology domains. J Biol Chem. 2007;282:32093–32105. doi: 10.1074/jbc.M703517200. [DOI] [PubMed] [Google Scholar]
- 18.Dowler S, Currie RA, Campbell DG, Deak M, Kular G, Downes CP, Alessi DR. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J. 2000;351:19–31. doi: 10.1042/0264-6021:3510019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas CC, Dowler S, Deak M, Alessi DR, van Aalten DM. Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain-containing protein 1 (TAPP1): molecular basis of lipid specificity. Biochem J. 2001;358:287–294. doi: 10.1042/0264-6021:3580287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimber WA, Trinkle-Mulcahy L, Cheung PC, Deak M, Marsden LJ, Kieloch A, Watt S, Javier RT, Gray A, Downes CP, et al. Evidence that the tandem-pleckstrin-homology-domain-containing protein TAPP1 interacts with Ptd(3,4)P2 and the multi-PDZ-domain-containing protein MUPP1 in vivo. Biochem J. 2002;361:525–536. doi: 10.1042/0264-6021:3610525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung SM, Kornelson JC, Al-Alwan M, Marshall AJ. Regulation of phosphoinositide 3-kinase signaling by oxidants: hydrogen peroxide selectively enhances immunoreceptor-induced recruitment of phosphatidylinositol (3,4) bisphosphate-binding PH domain proteins. Cell Signalling. 2007;19:902–912. doi: 10.1016/j.cellsig.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 22.Marshall AJ, Krahn AK, Ma K, Duronio V, Hou S. TAPP1 and TAPP2 are targets of phosphatidylinositol 3-kinase signaling in B cells: sustained plasma membrane recruitment triggered by the B-cell antigen receptor. Mol Cell Biol. 2002;22:5479–5491. doi: 10.1128/MCB.22.15.5479-5491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimber WA, Deak M, Prescott AR, Alessi DR. Interaction of the protein tyrosine phosphatase PTPL1 with the PtdIns(3,4)P2-binding adaptor protein TAPP1. Biochem J. 2003;376:525–535. doi: 10.1042/BJ20031154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hogan A, Yakubchyk Y, Chabot J, Obagi C, Daher E, Maekawa K, Gee SH. The phosphoinositol 3,4-bisphosphate-binding protein TAPP1 interacts with syntrophins and regulates actin cytoskeletal organization. J Biol Chem. 2004;279:53717–53724. doi: 10.1074/jbc.M410654200. [DOI] [PubMed] [Google Scholar]
- 25.Costantini JL, Cheung SM, Hou S, Li H, Kung SK, Johnston JB, Wilkins JA, Gibson SB, Marshall AJ. TAPP2 links phosphoinositide 3-kinase signaling to B-cell adhesion through interaction with the cytoskeletal protein utrophin: expression of a novel cell adhesion-promoting complex in B-cell leukemia. Blood. 2009;114:4703–4712. doi: 10.1182/blood-2009-03-213058. [DOI] [PubMed] [Google Scholar]
- 26.Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, Raynaud FI, et al. Synthesis and biological evaluation of pyrido[3′,2′:4,5]furo[3,2-d]pyrimidine derivatives as novel PI3 kinase p110α inhibitors. Bioorg Med Chem Lett. 2007;17:2438–2442. doi: 10.1016/j.bmcl.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 27.Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol. 2000;10:439–448. doi: 10.1016/s0960-9822(00)00441-3. [DOI] [PubMed] [Google Scholar]
- 28.Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic–euglycemic clamps in the conscious mouse. Diabetes. 2006;55:390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- 30.Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes. 2007;56:1025–1033. doi: 10.2337/db06-0883. [DOI] [PubMed] [Google Scholar]
- 31.Steele R, Wall JS, De Bodo RC, Altszuler N. Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am J Physiol. 1956;187:15–24. doi: 10.1152/ajplegacy.1956.187.1.15. [DOI] [PubMed] [Google Scholar]
- 32.Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose–response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol. 1985;248:E353–E362. doi: 10.1152/ajpendo.1985.248.3.E353. [DOI] [PubMed] [Google Scholar]
- 33.Gray A, Olsson H, Batty IH, Priganica L, Downes CP. Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal Biochem. 2003;313:234–245. doi: 10.1016/s0003-2697(02)00607-3. [DOI] [PubMed] [Google Scholar]
- 34.Esposito DL, Li Y, Cama A, Quon MJ. Tyr612 and Tyr632 in human insulin receptor substrate-1 are important for full activation of insulin-stimulated phosphatidylinositol 3-kinase activity and translocation of GLUT4 in adipose cells. Endocrinology. 2001;142:2833–2840. doi: 10.1210/endo.142.7.8283. [DOI] [PubMed] [Google Scholar]
- 35.Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Models Mech. 2010;3:525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang S, Czech MP. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Bayascas JR, Wullschleger S, Sakamoto K, Garcia-Martinez JM, Clacher C, Komander D, van Aalten DM, Boini KM, Lang F, Lipina C, et al. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol Cell Biol. 2008;28:3258–3272. doi: 10.1128/MCB.02032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, Mitchell CA. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J. 2009;419:29–49. doi: 10.1042/BJ20081673. [DOI] [PubMed] [Google Scholar]
- 39.Batty IH, van der Kaay J, Gray A, Telfer JF, Dixon MJ, Downes CP. The control of phosphatidylinositol 3,4-bisphosphate concentrations by activation of the Src homology 2 domain containing inositol polyphosphate 5-phosphatase 2, SHIP2. Biochem J. 2007;407:255–266. doi: 10.1042/BJ20070558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erdmann KS. The protein tyrosine phosphatase PTP-basophil/basophil-like: interacting proteins and molecular functions. Eur J Biochem. 2003;270:4789–4798. doi: 10.1046/j.1432-1033.2003.03895.x. [DOI] [PubMed] [Google Scholar]
- 41.Adachi M, Hamazaki Y, Kobayashi Y, Itoh M, Tsukita S, Furuse M. Similar and distinct properties of MUPP1 and Patj, two homologous PDZ domain-containing tight-junction proteins. Mol Cell Biol. 2009;29:2372–2389. doi: 10.1128/MCB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haenggi T, Fritschy JM. Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell Mol Life Sci. 2006;63:1614–1631. doi: 10.1007/s00018-005-5461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villa F, Deak M, Bloomberg GB, Alessi DR, van Aalten DM. Crystal structure of the PTPL1/FAP-1 human tyrosine phosphatase mutated in colorectal cancer: evidence for a second phosphotyrosine substrate recognition pocket. J Biol Chem. 2005;280:8180–8187. doi: 10.1074/jbc.M412211200. [DOI] [PubMed] [Google Scholar]
- 44.Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol Cell. 2000;6:1401–1412. doi: 10.1016/s1097-2765(00)00137-4. [DOI] [PubMed] [Google Scholar]
- 45.Miao B, Skidan I, Yang J, Lugovskoy A, Reibarkh M, Long K, Brazell T, Durugkar KA, Maki J, Ramana CV, et al. Small molecule inhibition of phosphatidylinositol-3,4,5-trisphosphate (PIP3) binding to pleckstrin homology domains. Proc Natl Acad Sci USA. 2010;107:20126–20131. doi: 10.1073/pnas.1004522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.