

Abstract
Motor neuron diseases are neurological disorders characterized primarily by the degeneration of spinal motor neurons, skeletal muscle atrophy, and debilitating and often fatal motor dysfunction. Spinal muscular atrophy (SMA) is an autosomal-recessive motor neuron disease of high incidence and severity and the most common genetic cause of infant mortality. SMA is caused by homozygous mutations in the survival motor neuron 1 (SMN1) gene and retention of at least one copy of the hypomorphic gene paralog SMN2. Early studies established a loss-of-function disease mechanism involving ubiquitous SMN deficiency and suggested SMN upregulation as a possible therapeutic approach. In recent years, greater knowledge of the central role of SMN in RNA processing combined with deep characterization of animal models of SMA has significantly advanced our understanding of the cellular and molecular basis of the disease. SMA is emerging as an RNA disease not limited to motor neurons, but one that involves dysfunction of motor circuits that comprise multiple neuronal subpopulations and possibly other cell types. Advances in SMA research have also led to the development of several potential therapeutics shown to be effective in animal models of SMA that are now in clinical trials. These agents offer unprecedented promise for the treatment of this still incurable neurodegenerative disease.
Introduction
Essential human behaviors such as swallowing, breathing, and locomotion depend on precise motor control. The coordinated activity of motor neurons within complex neuronal networks is critical for motor system function. Disruption of this network leads to fatal human disorders such as spinal muscular atrophy (SMA) and amyotrophic lateral sclerosis (ALS), the two most common motor neuron diseases of infancy and adulthood, respectively. Increasing evidence links these diseases to mutations in ubiquitously expressed genes implicated in RNA processing, but this has not yet led to an effective treatment for SMA or ALS. The multidisciplinary efforts of both academic and industry researchers supported by public and private foundations and patient advocacy groups have led to tremendous scientific progress in the SMA field. Here, we discuss advances in our understanding of the molecular and cellular basis of SMA and the development of candidate therapeutics that promise to mitigate the burden of this devastating motor disorder in the near future.
Clinical features of SMA
SMA is an inherited neuromuscular disease primarily affecting children. It is the leading genetic cause of infant death and the second most common autosomal-recessive genetic disorder after cystic fibrosis (Lunn and Wang, 2008). The disease affects 1 of every 6000 to 10,000 newborns and ∼1 in 50 people are carriers. SMA causes degeneration and loss of α motor neurons in the anterior horn of the spinal cord, leading to progressive muscle weakness and, in severe cases, respiratory failure and death (Crawford and Pardo, 1996). Proximal muscles are preferentially affected in the disease, as are lower more than upper extremities. The differential vulnerability of distinct motor neuron pools in SMA is also demonstrated by the marked involvement of intercostal and axial muscles and the relative sparing of the diaphragm, which results in the characteristic bell-shaped chest and paradoxical breathing.
SMA presents across a broad clinical spectrum in terms of age of onset and severity of disease and is classified into several different types based on onset and highest motor function milestones achieved (Munsat and Davies, 1992). Patients with the severe type I form of SMA, which is also the most common form, show signs of the disease soon after birth (<6 months of age), never gain the ability to sit, and typically do not survive past 2 years of age. Patients with the intermediate form of the disease (type II) are affected before 18 months of age and are able to sit upright, but never gain the ability to stand without support. A milder form of the disease (type III) begins after 2 years of age and patients maintain the ability to stand on their own and live a normal lifespan. Additional forms of SMA have been identified at both ends of the severity spectrum: type 0 SMA is a prenatal form of the disease that is uniformly fatal in utero or a few months after birth (MacLeod et al., 1999) and type IV SMA presents as an adult-onset form of the disease with mild proximal muscle weakness and normal life expectancy (Lunn and Wang, 2008). In all SMA patients, clinical and electrophysiological findings are consistent with a neurogenic disorder in which loss of motor units and rapid functional decline occur soon after the onset of symptoms, usually followed by a long plateau phase during which muscle strength remains relatively stable (Crawford and Pardo, 1996; Iannaccone et al., 2000; Swoboda et al., 2005). In milder, but not severe, forms of SMA, some preservation of muscle power is associated with collateral axonal sprouting and myofiber reinnervation, evidenced by the presence of large motor units and fiber type grouping (Crawford and Pardo, 1996). Treatment of SMA remains limited to symptomatic and supportive care.
Molecular genetics of SMA
Although no effective therapies are yet available to SMA patients, the molecular genetics underlying the disease has been well characterized in the two decades since it was discovered that SMA is caused by homozygous deletion or mutation of the survival motor neuron 1 (SMN1), a gene that encodes the SMN protein (Lefebvre et al., 1995). Due to a large inverted duplication located at chromosome 5q, the human genome contains two SMN-coding genes, SMN1 and SMN2. Although the two genes are nearly identical, a single C to T transition in exon 7 of SMN2 disrupts an exon splicing enhancer and concomitantly creates an exon splicing silencer element (Cartegni and Krainer, 2002; Kashima and Manley, 2003). As a result, this single base change potently redirects alternative splicing so that exon 7 is excluded from the majority of SMN2-derived mRNA transcripts (Lorson et al., 1999; Monani et al., 1999). The exon 7-excluded mRNA (SMNΔ7) encodes a truncated SMN protein that is rapidly degraded (Lorson and Androphy, 2000; Cho and Dreyfuss, 2010). However, a small proportion of transcripts derived from SMN2 includes exon 7 and encodes full-length, functional SMN that—in the context of SMA, in which SMN1 is mutated or deleted—provides a sufficient amount of SMN to prevent lethality yet not enough to fully compensate for the loss of SMN1, resulting in motor neuron disease. Importantly, due to the intrinsic instability of the 5q chromosomal duplication, multiple copies of SMN2 can be present in the human genome and a higher SMN2 copy number correlates inversely with disease severity (McAndrew et al., 1997; Wirth et al., 2006). In an extreme example of the disease-modifying capacity of SMN2, individuals with homozygous SMN1 deletions have been identified who lack SMA symptoms due to the presence of five copies of SMN2 (Prior et al., 2004). Furthermore, variants of SMN2 containing single nucleotide changes that increase the efficiency of exon 7 inclusion have been identified in SMA patients with a milder phenotype despite carrying only two copies of SMN2 (Prior et al., 2009; Vezain et al., 2010). Therefore, SMA is the result of a deficiency—but not a complete absence—of functional SMN protein and SMN2 is the most potent genetic modifier of the disease.
Cellular basis of motor system dysfunction in SMA
Motor dysfunction associated with motor neuron loss and skeletal muscle atrophy is a defining clinical feature of SMA across the disease spectrum. The development of various animal models of SMA has been key to defining the cellular and molecular basis of the disease (Schmid and DiDonato, 2007; Burghes and Beattie, 2009). Complete absence of SMN in model organisms, all of which have only a single Smn gene that is equivalent to SMN1, is lethal as it likely is in humans, in whom the absence of both SMN1 and SMN2 has never been found. In contrast, ubiquitous SMN reduction to levels found in severe SMA patients appears to be relatively well tolerated by most tissues, but not in the nervous system (Burghes and Beattie, 2009). Thus far, the genetic and phenotypic hallmarks of the human disease have been most faithfully reproduced in the mouse, which has proven to be the most powerful platform for elucidating pathological mechanisms in SMA (Burghes and Beattie, 2009; Sleigh et al., 2011). Through similar design, the first mouse models of SMA were engineered to express low levels of SMN by harboring two copies of the human SMN2 transgene with concomitant knock-out of the single mouse Smn gene (Hsieh-Li et al., 2000; Monani et al., 2000). These SMA mice are born phenotypically normal but begin to display severe motor deficits, muscle weakness, and reduced weight gain early after birth and die at ∼1 week of age (Hsieh-Li et al., 2000; Monani et al., 2000). Addition of the SMNΔ7 cDNA transgene to this genetic background was found to extend survival of SMNΔ7 SMA mice to ∼2 weeks of age (Le et al., 2005), yielding one of the most widely studied SMA models to date. Confirming the disease-modifying role of SMN2, Smn knock-out mice with eight or more copies of SMN2 are phenotypically normal (Monani et al., 2000).
Examination of cellular pathology reveals selective loss of motor neurons in the spinal cord of SMA mice (Hsieh-Li et al., 2000; Monani et al., 2000; Le et al., 2005; Mentis et al., 2011; Fig. 1). Interestingly, SMA motor neurons display differential, segment-specific vulnerabilities along the rostrocaudal axis (Mentis et al., 2011): those residing in lumbar level L1 that innervate axial and proximal muscles are affected earlier and more profoundly than those in the L5 region, which innervate distal hindlimb muscles. Moreover, motor neurons in the medial motor column that innervate axial muscles are more affected than those in the lateral motor column that innervate distal muscles, consistent with findings in SMA patients (Mentis et al., 2011). The realization that some motor neuron pools are spared both in the human disease and in mouse models offers unique opportunities to identify candidate vulnerability or resistance factors through differential screening approaches similar to those used successfully in ALS (Kaplan et al., 2014).
Figure 1.

Schematic representation of the key morphological and functional abnormalities induced by SMN deficiency in the motor system of SMA mouse models. Multiple aspects of the motor system are disrupted in SMA. For simplicity, only the excitatory premotor neurons of the motor circuit affected by the disease are depicted. Motor neurons (dark blue) in the ventral horn of the spinal cord receive excitatory synaptic inputs from proprioceptive neurons residing in the dorsal root ganglion (green) and local interneurons (light blue). Upon sufficient excitatory drive to generate action potentials, motor neurons innervating skeletal muscle induce muscle contraction through cholinergic neurotransmission at the NMJ. The specific deficits within the SMA motor system are indicated and are described in detail in the text.
Beyond death of spinal motor neurons, SMA pathology is characterized by additional defects that occur both centrally, at synapses impinging on somata and dendrites of motor neurons, and distally, at the neuromuscular junction (NMJ) (Fig. 1). The NMJ defects include presynaptic neurofilament accumulation, reduced vesicle content, and impaired synaptic transmission, as well as defective postsynaptic acetylcholine receptor clustering and motor endplate development (Kariya et al., 2008; Murray et al., 2008; Kong et al., 2009; Ling et al., 2010; Ruiz et al., 2010; Lee et al., 2011). Furthermore, alterations in intracellular calcium homeostasis and defective clustering of Cav2.2 channels have been found in SMA motor neuron terminals both in vivo and in vitro (Jablonka et al., 2007; Ruiz et al., 2010). These morphological and functional abnormalities of the NMJ are associated with delayed development, decreased myofiber size, and muscle denervation, all of which are well documented and particularly pronounced for select vulnerable muscles (Kariya et al., 2008; Murray et al., 2008; Kong et al., 2009; Ling et al., 2012).
In addition to proper connectivity between motor neuron and muscle, accurate wiring of motor neurons with other neurons into functional circuits is essential for motor behavior, and abnormalities in neural networks likely contribute to SMA. In support of this, SMA motor neurons receive fewer glutamatergic synaptic inputs from proprioceptive sensory neurons and local spinal interneurons (Ling et al., 2010; Mentis et al., 2011; Fig. 1). Furthermore, severe impairment of sensory-motor neurotransmission associated with strong dysfunction of proprioceptive synapses has been demonstrated in SMA mice (Mentis et al., 2011). Intriguingly, SMA motor neurons have increased intrinsic excitability and altered firing properties (Mentis et al., 2011), possibly reflecting a homeostatic response to reduced excitatory drive from SMN-deficient premotor neurons. Altogether, these data suggest that the severe motor dysfunction associated with SMA pathology is in part due to synaptic abnormalities induced by SMN deficiency in the sensory-motor system. The observation that structural and functional alterations of sensory-motor connectivity precede motor neuron loss further supports the idea that these early events contribute to motor neuron dysfunction and death in SMA. It will be important to determine whether hyperexcitability and death of SMA motor neurons are linked events.
In sum, SMA is emerging as a disease characterized by dysfunction of multiple components of the motor system and not motor neurons alone. In addition, defects in tissues outside of the nervous system, such as heart, pancreas, and liver, have been documented in severe SMA mice (Bevan et al., 2010; Heier et al., 2010; Shababi et al., 2010; Hua et al., 2011; Bowerman et al., 2012b), although the role of peripheral tissue dysfunction in human SMA pathology remains to be established. These defects have been covered in several excellent reviews (Hamilton and Gillingwater, 2013; Shababi et al., 2014) and thus will not be discussed further here.
Temporal and spatial requirement of SMN
The preferential motor system pathology caused by deficiency of a ubiquitously expressed protein raises key questions about the temporal and spatial requirement of SMN in vivo. This has begun to be addressed using animal models in which SMN is conditionally restored or depleted in a tissue- and time-dependent manner. These studies showed that a ubiquitous postnatal increase of SMN rescues the phenotype in severe SMA mice even after disease onset (Le et al., 2011; Lutz et al., 2011). Importantly, SMN restoration after the first postnatal week is ineffective, demonstrating that this increase must occur early during postnatal development (Le et al., 2011; Lutz et al., 2011). It was also found that early postnatal SMN depletion rapidly elicited a motor phenotype and death, whereas SMN reduction after the second postnatal week did not lead to an overt SMA phenotype in mice (Le et al., 2011; Kariya et al., 2014). Therefore, a high level of SMN is required during the embryonic and early postnatal phase of mouse development, which coincides with maturation of the motor system.
In addition to the temporal requirement for SMN, several observations suggest that SMA is a disease affecting motor system development. Although embryonic specification and patterning of motor neurons appear normal (McGovern et al., 2008; Murray et al., 2010), several synaptic abnormalities appear early during postnatal development and increase over the disease course in SMA mice. First, NMJs display signs of delayed maturation, as indicated by a failure to develop properly into the highly perforated, rosette-like, complex structures characteristic of mature neuromuscular synapses (Kariya et al., 2008; Kong et al., 2009). Second, the developmental increase in the number of proprioceptive synapses on motor neuron dendrites is halted (Mentis et al., 2011). Last, the physiological switch in gene expression from embryonic to adult forms of myosin heavy chains and acetylcholine receptors is impaired in SMA skeletal muscle (Avila et al., 2007; Kong et al., 2009).
Is motor system pathology in SMA the result of cell autonomous defects solely in motor neurons? Establishing the spatial requirement of SMN is key, not only to distinguishing primary cellular deficits from secondary effects, but also to identifying the cellular targets for therapy. Early work showed that pan-neuronal SMN expression was able to fully rescue the disease phenotype in SMA mice, whereas SMN restoration in muscle had no effect (Gavrilina et al., 2008). Although intrinsic muscle deficits were later found to contribute to muscle hypotrophy (Martinez et al., 2012), these findings indicated a key role for neuronal dysfunction in SMA pathology. Surprisingly, selective depletion of SMN in motor neuron progenitors caused motor neuron death, but only a mild SMA phenotype (Park et al., 2010). Furthermore, motor-neuron-specific SMN restoration improved motor neuron loss, but had little benefit in terms of survival of SMA mice (Gogliotti et al., 2012; Martinez et al., 2012). Therefore, although motor neuron death appears to be a cell autonomous event, SMN deficiency must induce defects elsewhere that also contribute to motor system pathology in animal models of SMA. This scenario is supported by a recent study using a Drosophila model of SMA in which reduced muscle growth and defective locomotion induced by SMN deficiency were accompanied by aberrant motor neuron output and increased NMJ neurotransmission (Imlach et al., 2012). Remarkably, whereas restoration of SMN in either muscles or motor neurons did not alter these phenotypes, selective expression of SMN in proprioceptive neurons and interneurons of the motor circuit was sufficient to correct defects in motor neurons and muscles (Imlach et al., 2012), revealing the non-cell-autonomous origin of these abnormalities in this model system. To date, evidence for dysfunction of proprioceptive neurons and excitatory spinal interneurons has accumulated in mouse models of SMA (Ling et al., 2010; Mentis et al., 2011), but the effect of selective SMN restoration in these premotor neurons on motor neuron function and the disease phenotype has yet to be evaluated.
Collectively, these findings highlight motor system disruption as a prominent feature of SMA pathology in which dysfunction of neuronal networks has a key role. In the future, identification of the specific neurons within the motor circuit in which SMN is essential and of the molecular pathways and cellular activities that are disrupted by SMN deficiency will be critical for understanding the underlying mechanisms of motor system dysfunction in SMA and the development of effective therapeutics.
Molecular mechanisms of SMA: dysregulation of RNA processing
After the identification of SMN as the disease-causing gene product of SMA, several studies established the basic biological function of SMN and began to provide insight into how a deficiency in SMN could elicit motor neuron disease. SMN is an evolutionarily conserved and ubiquitously expressed protein that localizes to both the cytoplasm and the nucleus of all eukaryotic cells (Liu and Dreyfuss, 1996; Burghes and Beattie, 2009). A series of biochemical studies firmly established that SMN, along with eight core protein components (Gemins2–8 and unrip), constitutes a large, multiprotein complex (Pellizzoni, 2007). The best-characterized function of the SMN complex is in the assembly of small nuclear ribonucleoproteins (snRNPs) of the major (U2-dependent) and minor (U12-dependent) spliceosomes (Fischer et al., 2011; Li et al., 2014). Specifically, SMN is essential for the assembly of a heteroheptameric ring of Sm proteins on snRNAs (Meister et al., 2001; Pellizzoni et al., 2002; Fig. 2A), which is an obligate step in the biogenesis pathway of these essential RNP components of the RNA splicing machinery. In addition to spliceosomal snRNPs, the SMN complex also mediates the assembly of a variant core comprising Sm and Sm-like (LSm10 and LSm11) proteins on the U7 snRNA (Pillai et al., 2003; Tisdale et al., 2013; Fig. 2A). U7 snRNP functions, not in splicing, but in the unique 3′-end processing of replication-dependent histone mRNAs that comprise the most abundant class of intronless and non-polyadenylated transcripts in metazoans (Marzluff et al., 2008). Accordingly, SMN deficiency impairs U7 biogenesis, leading to histone mRNA processing deficits in SMA (Tisdale et al., 2013). Therefore, generation of both intron-containing mRNAs and replication-dependent histone transcripts depend on SMN activity for their proper processing and expression. SMN has also been implicated in the formation of many other cellular RNPs containing both coding and noncoding RNAs (Fallini et al., 2012; Li et al., 2014), with the most prominent of these putative functions relating to the axonal transport and local translation of mRNAs at the distal end of developing neurons (Rossoll et al., 2003; Fallini et al., 2011). Although these additional activities have not yet been well defined mechanistically, SMN undoubtedly plays multiple key roles in the posttranscriptional regulation of gene expression.
Figure 2.
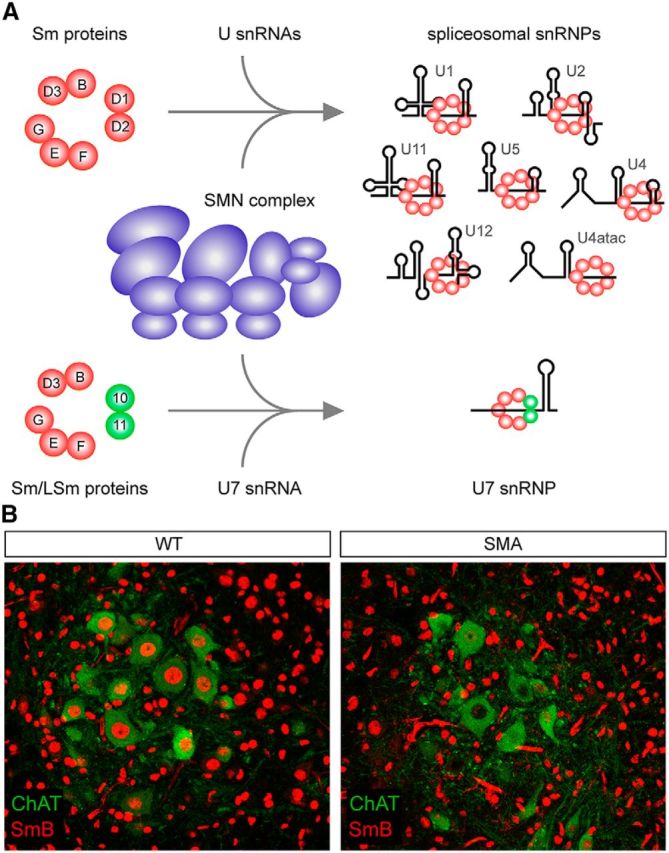
The biogenesis of snRNPs requires SMN and is disrupted in SMA motor neurons. A, Schematic representation of the only molecularly established RNP assembly functions of SMN. The multisubunit SMN complex (Pellizzoni, 2007) binds to seven Sm proteins (B, D1, D2, D3, E, F, G) or a subset of Sm proteins and LSm10/11 to mediate their respective assembly onto specific snRNAs, yielding spliceosomal snRNPs that function in pre-mRNA splicing or U7 snRNP that functions in histone mRNA processing. B, SMN deficiency causes a more prominent reduction of snRNPs in the nucleus of SMA motor neurons than other spinal ventral horn cells. Double-label immunohistochemistry and confocal microscopy analysis of lumbar L1 spinal cord sections from wild-type (WT) or SMNΔ7 SMA mice (Le et al., 2005) at postnatal day 10 was performed using antibodies against SmB (Carissimi et al., 2006) to label snRNPs and the motor-neuron-specific marker ChAT, as described previously (Ruggiu et al., 2012).
Despite increased knowledge of SMN′s functions in RNA regulation, how a deficiency in a ubiquitous protein with housekeeping roles in gene expression elicits selective motor dysfunction in SMA is poorly understood. Because pathology is not restricted to motor neurons, but also involves impairment of the entire sensory-motor system, SMA is likely not caused by disruption of a single SMN-dependent RNA pathway in motor neurons, but rather by disruption of multiple pathways across the entire motor circuit. Defining the specific pathways and associated downstream events disrupted by SMN deficiency that elicit disease relevant effects is one of the most challenging tasks in understanding molecular mechanisms of SMA.
To date, multiple lines of evidence point to a role for spliceosomal snRNP dysfunction in SMA. Defects in snRNP assembly correlate with disease severity and lead to a reduction in the steady-state levels of snRNPs in mouse models of SMA (Gabanella et al., 2007; Zhang et al., 2008). These changes are both tissue specific and not uniform across all snRNPs: those of the minor spliceosome that carry out U12-dependent splicing are more strongly affected than those of the major spliceosome. Moreover, SMN deficiency induces a particularly prominent snRNP reduction in motor neurons compared with other neurons and non-neuronal cells in the spinal cord of SMA mice (Ruggiu et al., 2012; Fig. 2B). This has been attributed to both the especially low degree of exon 7 inclusion in SMN2 transcripts within normal motor neurons and the activation of a negative feedback loop in which reduced snRNPs further decrease exon 7 splicing in SMA (Jodelka et al., 2010; Ruggiu et al., 2012), exacerbating dysfunction of SMN-dependent pathways and contributing to the selective vulnerability of SMA motor neurons. Therefore, ubiquitous SMN deficiency has remarkably selective consequences on the snRNP content of SMA cells. Differential changes—both qualitative and quantitative—in snRNP content across tissues induced by SMN deficiency could provide an explanation for how SMN-dependent snRNP reduction selectively affects the motor system. Consistent with snRNP involvement in the disease process, restoration of snRNP levels coincides with phenotypic correction in animal models (Winkler et al., 2005; Workman et al., 2009).
One of the greatest challenges in SMA research is to identify specific SMN-dependent RNA-processing events that contribute to neuronal dysfunction and ultimately to the disease. Toward this end, a recent study identified Stasimon as a gene essential for the normal synaptic transmission in Drosophila motor neurons whose function is disrupted by SMN deficiency due to defective splicing of a U12 intron within the gene (Lotti et al., 2012). Importantly, Stasimon restoration rescued select aspects of motor neuron pathology in animal models of SMA, including NMJ neurotransmission and muscle size defects in Drosophila and motor axon outgrowth in zebrafish (Lotti et al., 2012). Therefore, this study linked neuronal circuit perturbations that are important determinants of SMA pathogenesis directly to disruption of an SMN-dependent RNA splicing event in animal models. Dysregulation of additional RNA processing events downstream of SMN function in splicing (See et al., 2014; Wishart et al., 2014) and other less-defined SMN activities (Akten et al., 2011; Kye et al., 2014; Sleigh et al., 2014) have been shown to contribute to SMA phenotypes in animal models. Collectively, these findings suggest that SMA pathology may result from a combination of specific functional deficits in distinct cell types, each triggered by select gene expression changes among many transcriptome alterations. This scenario is not unprecedented in human disease because it bears conceptual and mechanistic similarities with other RNA disorders of the motor system such as myotonic dystrophy (Udd and Krahe, 2012) and a paraneoplastic neurologic syndrome linked to the neuronal splicing factor Nova (Darnell and Posner, 2006; Ruggiu et al., 2009).
Future studies aiming to identify the full complement of RNA-processing defects that contribute to SMA pathology will help to build a comprehensive molecular view of SMN-dependent RNA regulation in motor circuit function and disease. The individual contribution of each RNA pathway regulated by SMN to the disease phenotype in mouse models will require the development of multipronged approaches for selectively enhancing RNP biogenesis, restoring downstream targets with altered expression, or reducing RNP turnover to compensate for the imbalance induced by defects in biogenesis.
Insights into SMA pathogenesis from disease modifiers
Identifying specific cellular activities disrupted in SMA is a key, logical complement to the molecular dissection of SMN-dependent RNA pathways and downstream targets. Studies of chemical and genetic modifiers of SMA phenotypes have helped to identify cellular events disrupted in the disease (Wirth et al., 2013), allowing for a better understanding of the disease process.
A prominent example is the emerging role of disrupted actin regulation and cytoskeletal dynamics at SMA synapses. A study in human patients identified increased expression of the F-actin-bundling protein Plastin 3 as a protective modifier in asymptomatic patients who carried SMN1 deletions with identical SMN2 copy number as their SMA-affected siblings (Oprea et al., 2008). Although higher Plastin 3 levels can also be observed in female SMA patients relative to unaffected siblings (Bernal et al., 2011), Plastin 3 has been found to be a modifier of neuromuscular phenotypes induced by SMN deficiency in both invertebrate and vertebrate models of SMA (Dimitriadi et al., 2010; Hao le et al., 2012; Ackermann et al., 2013). In SMA mice, overexpression of human Plastin 3 increased proprioceptive inputs onto motor neurons and both acetylcholine receptor clustering and endplate size at the NMJ (Ackermann et al., 2013). The stabilizing effect of Plastin 3 on synapses was linked to higher presynaptic levels of F-actin and resulted in partial improvement of NMJ neurotransmission and muscle fiber size, yet had little effect on motor function and survival of SMA mice (Ackermann et al., 2013). Consistent with these observations, another study revealed that SMN deficiency induces activation of RhoA, a regulator of actin dynamics, in an intermediate SMA mouse model (Bowerman et al., 2010). Interestingly, pharmacological inhibition of the RhoA/ROCK pathway was found to improve NMJ development and myofiber size, but not motor neuron loss, and to extend survival in SMA mice (Bowerman et al., 2010; Bowerman et al., 2012a). Collectively, these findings implicate dysregulation of presynaptic actin dynamics in SMA pathology, although the mechanisms underlying this defect remain elusive. Reduced synthesis of β-actin at motor neuron terminals resulting from disruption of the proposed function of SMN in axonal mRNA transport could affect the actin cytoskeleton in SMA (Rossoll et al., 2003); however, genetic ablation of β-actin in mouse motor neurons has no phenotypic effects on the neuromuscular system (Cheever et al., 2011).
Efforts aimed at the discovery and characterization of modifiers of SMN biology and SMA pathology will provide invaluable insights to help link disruption of SMN-dependent RNA processing events and the downstream functional deficits induced by SMN deficiency in the motor system. They may also point to novel therapeutic approaches that are complementary to SMN upregulation.
Advances toward SMA therapy
Based on our understanding of SMA etiology, restoration of SMN expression is the most promising strategy to treat the disease. Because all SMA patients carry at least one copy of SMN2, the most attractive approach to treat SMA is to manipulate SMN2 to produce higher amounts of functional SMN protein. This can be accomplished in several ways, including enhancing SMN2 promoter activation, increasing exon 7 inclusion in SMN2 transcripts, and promoting stabilization of SMN mRNA and protein. In addition, SMN expression could be increased by gene therapy. Basic research and preclinical studies in animal models have demonstrated the validity of these approaches, leading to several ongoing clinical trials (Fig. 3). For brevity, only the most advanced candidate treatments that have been tested successfully in animal models and have moved to clinical development are discussed below. For additional information and updates on the clinical trials, we refer the reader to www.clinicaltrials.gov and websites of the individual biotechnology companies involved in these drug development programs.
Figure 3.

Candidate SMA therapeutics and their progress through the clinical development pipeline. The chart summarizes the current status of the most advanced programs in SMA therapeutic development based on publicly available information. Small molecules and biologics listed with blue bars aim to increase the functional levels of SMN, and those denoted with red bars act by improving motor system function through SMN-independent mechanisms.
Gene therapy
Viral-mediated SMN gene delivery has been remarkably successful in preclinical studies. Both systemic and intracerebroventricular injection of self-complementary adeno-associated viral vectors (scAAV) expressing SMN showed efficient transduction of motor neurons in both mice and non-human primates, as well as nearly complete correction of the SMA phenotype in mice (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010; Dominguez et al., 2011). Recently, proof of principle for phenotypic correction of SMA-like phenotypes induced by SMN knock-down in a large animal model has also been obtained using AAV-SMN gene delivery (Duque et al., 2014). AveXis is conducting the first gene therapy phase 1 clinical trial to assess the safety of multidose intravenous delivery of scAAV9-SMN in type I SMA infants and is also planning an additional trial of scAAV9-SMN by intrathecal delivery in milder SMA patients. In addition to safety and efficacy, a potential hurdle of SMN gene therapy may be the production of enough AAV vector for large-scale administration to SMA patients.
Antisense oligonucleotides
Alternative splicing of exon 7 is subject to an intricate regulatory network comprising multiple cis-acting elements and trans-acting factors (Singh and Singh, 2011). Antisense oligonucleotides (ASOs) that specifically target and inactivate a splicing silencer motif within intron 7 of SMN2 bound by hnRNPA1 (Singh et al., 2006; Hua et al., 2008) have been characterized extensively. These ASOs are highly effective at promoting inclusion of exon 7 in SMN2 transcripts and at increasing SMN protein levels both in vitro and in vivo. Importantly, in addition to being highly stable for months within the CNS and showing little to no toxicity, a single administration of ASO splicing modifiers either systemically or in the CNS of severe SMA mice rescues the disease phenotype (Hua et al., 2011; Porensky et al., 2012). To date, ISIS-SMNRx, a 2′-O-methoxyethyl modified ASO developed by Isis Pharmaceuticals/Biogen to correct SMN2 splicing, has shown good safety and tolerability profiles in open-label phase 2 clinical trials after intrathecal delivery in type I SMA infants and types II and III SMA children. The drug displayed wide CNS distribution, target engagement as indicated by higher SMN levels, and concentrations in a range associated with phenotypic correction in mouse models. Preliminary but encouraging evidence for efficacy is further suggested by dose-dependent increases in the scores of motor function scales observed in treated patients relative to measures from natural history studies. ISIS-SMNRx is currently undergoing phase 3 clinical trials.
Small molecules
The development of orally bioavailable, brain-penetrable small molecules that upregulate SMN expression is particularly advantageous in treating SMA due to the ease of delivery and ability to target both peripheral and CNS-specific aspects of the disease. Early studies investigated the therapeutic potential of histone deacetylase inhibitors and demonstrated their ability to increase SMN2 transcription through modification of chromatin structure and to generate some phenotypic benefit in SMA mice; however, these agents were mostly ineffective in the clinical setting (Mohseni et al., 2013).
Cell-based high-throughput screens were performed to identify small molecules that increase SMN expression and their therapeutic potential was investigated in SMA mice (Jarecki et al., 2005; Cherry et al., 2013; Naryshkin et al., 2014). Quinazoline derivatives, a group of compounds that inhibit the RNA decapping scavenger enzyme DcpS (Singh et al., 2008), were the first compounds to be identified in high-throughput drug screens as candidate activators of SMN2 expression (Jarecki et al., 2005). After extensive chemical improvement, treatment of SMA mice with orally bioavailable and CNS-penetrant DcpS inhibitors—including the most optimized RG3039 compound—showed only moderate effects on SMN upregulation and motor function and survival (Butchbach et al., 2010; Gogliotti et al., 2013; Van Meerbeke et al., 2013). Interestingly, the drug exerts effects beyond motor neurons because the degree of phenotypic improvement in SMA mice of combining RG3039 treatment with selective, genetic restoration of SMN in motor neurons exceeds that observed for each condition separately (Van Meerbeke et al., 2013). RG3039 (PF-06687859) was found to be safe and well tolerated in a phase 1 study in healthy volunteers, but Pfizer recently terminated further clinical development of this compound in partnership with Repligen for undisclosed reasons.
Small-molecule compounds that are orally available and potently correct SMN2 splicing have been developed by a consortium including Roche, PTC Therapeutics, and the SMA Foundation. These compounds have shown remarkable results in preclinical studies in SMA mice, where they induced strong splicing correction and SMN upregulation, resulting in a very robust rescue of both motor function and survival (Naryshkin et al., 2014). Despite the unclear nature of the mechanism of action of the compounds, whole-genome analysis has generated considerable enthusiasm for their therapeutic relevance by suggesting that they are highly specific to SMN with negligible effects on other mRNA targets. Investigation of the mechanism of action of these compounds is bound to yield interesting insight into the basic biology of splicing regulation. In human trials, a clinical candidate compound developed by Roche (RO06885247) was well tolerated in healthy individuals and the profile of safety and tolerability in SMA patients is currently being evaluated, together with efficacy measures on motor function as secondary endpoints. A phase 1 study of an orally available SMN2-splicing modulator developed by Novartis (LMI070) is also about to begin in type I SMA infants.
SMN-independent therapeutics
Restoration of SMN levels through any mechanism will likely only be effective if intervention occurs early in the disease. This is supported by work in mouse models that has demonstrated a critical window early in disease progression in which SMN upregulation has meaningful phenotypic benefit (Foust et al., 2010; Le et al., 2011; Lutz et al., 2011). Beyond this point, it will be especially important to target the molecular pathways and cellular activities that are disrupted downstream of SMN. Therefore, precise characterization of SMN-dependent pathways that are both affected and relevant to the disease remains a critical aspect of therapeutic development for SMA. In addition to SMN upregulation, other possibly viable approaches to SMA treatment are emerging from studies using agents that improve motor circuit function in an SMN-independent manner.
Olesoxime is a cholesterol-like neuroprotective compound developed by the pharmaceutical company Trophos that may act by preserving the integrity of mitochondria in neurons under conditions of cellular stress. Originally identified as a protective agent against motor neuron death induced by withdrawal of neurotrophic factors, olesoxime was found to be beneficial in an ALS mouse model (Bordet et al., 2007), but not in ALS clinical trials (Lenglet et al., 2014). In a phase 2 clinical trial in type II and non-ambulatory type III SMA patients, however, olesoxime was shown to maintain motor function and to reduce disease-related adverse events. Based on these results, the compound has been granted orphan drug designation for SMA treatment by US and EU regulatory authorities.
Knowledge of disease mechanisms will undoubtedly yield alternative therapeutic targets and approaches in SMA. One notable example comes from a recent study in which the motor circuit dysfunction induced by SMN deficiency in a Drosophila model of SMA was partially corrected by pharmacological treatment with 4-aminopyridine (4-AP) (Imlach et al., 2012). Inhibiting K+ channels by treatment with 4-AP likely provides phenotypic benefit in the fly model of SMA by increasing the excitability of SMA motor circuits. 4-AP (Ampyra) is a FDA-approved drug developed by Acorda Therapeutics for the treatment of fatigue in multiple sclerosis. Although not yet tested in SMA mice, the clinical efficacy of the drug in type III SMA patients is being assessed in an ongoing trial led by investigators at Columbia University.
As a complement to SMN-restoring strategies, agents that promote motor neuron survival and neural circuit function—and possibly muscle-enhancing approaches as well—deserve consideration as important therapeutic avenues for SMA. In the future, the combination of SMN-dependent and SMN-independent treatment approaches hold the greatest promise in the fight against SMA.
Conclusions
SMA is mechanistically well defined compared with many other neurodegenerative diseases and has an advanced therapeutic pipeline in place. However, SMA continues to be a fertile research area with many important questions yet to be addressed for both basic and clinical studies. Of particular importance will be to elucidate the full spectrum of SMN functions in posttranscriptional gene regulation and to further define the SMN-dependent RNA-processing events and downstream cellular activities that contribute to the motor system dysfunction seen in SMA. As these efforts increase our knowledge of disease mechanisms, they might also help to optimize current treatments and lead to the development of novel therapeutic approaches to treat SMA.
On the clinical front, SMA is one of the few neurological diseases for which an effective treatment seems possible in the foreseeable future. Clinical development of current candidate SMA therapeutics still has to face a variety of challenges related to safety, efficacy, and route of delivery in patients before a treatment is established conclusively. Further efforts are also needed in the area of outcome measures to identify reliable disease biomarkers and improved functional tests for scoring motor performance milestones in SMA patients. Nevertheless, results from ongoing clinical trials are eagerly awaited and evidence of therapeutic benefit would favor the implementation of universal newborn screening, in turn allowing both earlier diagnosis and therapeutic intervention and possibly improved clinical outcome. It is anticipated that continuing progress in SMA research will strongly affect, not only this devastating disease of childhood, but also other neurodegenerative conditions of the motor system.
Footnotes
Editor's Note: Disease Focus articles provide brief overviews of a neural disease or syndrome, emphasizing potential links to basic neural mechanisms. They are presented in the hope of helping researchers identify clinical implications of their research. For more information, see http://www.jneurosci.org/misc/ifa_minireviews.dtl.
The authors declare no competing financial interests.
References
- Ackermann B, Kröber S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, Wunderlich TF, Riessland M, Tabares L, Wirth B. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. 2013;22:1328–1347. doi: 10.1093/hmg/dds540. [DOI] [PubMed] [Google Scholar]
- Akten B, Kye MJ, Hao le T, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, Steen JA, Sahin M. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci U S A. 2011;108:10337–10342. doi: 10.1073/pnas.1104928108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal S, Also-Rallo E, Martínez-Hernández R, Alías L, Rodríguez-Alvarez FJ, Millán JM, Hernández-Chico C, Baiget M, Tizzano EF. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord. 2011;21:413–419. doi: 10.1016/j.nmd.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, Ward JG, Petruska JC, Lucchesi PA, Burghes AH, Kaspar BK. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordet T, Buisson B, Michaud M, Drouot C, Galéa P, Delaage P, Akentieva NP, Evers AS, Covey DF, Ostuni MA, Lacapere JJ, Massaad C, Schumacher M, Steidl EM, Maux D, Delaage M, Henderson CE, Pruss RM. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J Pharmacol Exp Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- Bowerman M, Beauvais A, Anderson CL, Kothary R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet. 2010;19:1468–1478. doi: 10.1093/hmg/ddq021. [DOI] [PubMed] [Google Scholar]
- Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 2012a;10:24. doi: 10.1186/1741-7015-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman M, Swoboda KJ, Michalski JP, Wang GS, Reeks C, Beauvais A, Murphy K, Woulfe J, Screaton RA, Scott FW, Kothary R. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann Neurol. 2012b;72:256–268. doi: 10.1002/ana.23582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchbach ME, Singh J, Thorsteinsdóttir M, Saieva L, Slominski E, Thurmond J, Andrésson T, Zhang J, Edwards JD, Simard LR, Pellizzoni L, Jarecki J, Burghes AH, Gurney ME. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carissimi C, Saieva L, Gabanella F, Pellizzoni L. Gemin8 is required for the architecture and function of the survival motor neuron complex. J Biol Chem. 2006;281:37009–37016. doi: 10.1074/jbc.M607505200. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- Cheever TR, Olson EA, Ervasti JM. Axonal regeneration and neuronal function are preserved in motor neurons lacking ss-actin in vivo. PLoS One. 2011;6:e17768. doi: 10.1371/journal.pone.0017768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JJ, Osman EY, Evans MC, Choi S, Xing X, Cuny GD, Glicksman MA, Lorson CL, Androphy EJ. Enhancement of SMN protein levels in a mouse model of spinal muscular atrophy using novel drug-like compounds. EMBO Mol Med. 2013;5:1035–1050. doi: 10.1002/emmm.201201864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Dreyfuss G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010;24:438–442. doi: 10.1101/gad.1884910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Darnell RB, Posner JB. Paraneoplastic syndromes affecting the nervous system. Semin Oncol. 2006;33:270–298. doi: 10.1053/j.seminoncol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Dimitriadi M, Sleigh JN, Walker A, Chang HC, Sen A, Kalloo G, Harris J, Barsby T, Walsh MB, Satterlee JS, Li C, Van Vactor D, Artavanis-Tsakonas S, Hart AC. Conserved genes act as modifiers of invertebrate SMN loss of function defects. PLoS Genet. 2010;6:e1001172. doi: 10.1371/journal.pgen.1001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez E, Marais T, Chatauret N, Benkhelifa-Ziyyat S, Duque S, Ravassard P, Carcenac R, Astord S, Pereira de Moura A, Voit T, Barkats M. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- Duque SI, Arnold WD, Odermatt P, Li X, Porensky PN, Schmelzer L, Meyer K, Kolb SJ, Schümperli D, Kaspar BK, Burghes AH. A large animal model of spinal muscular atrophy and correction of phenotype. Ann Neurol. 2014;77:399–414. doi: 10.1002/ana.24332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Zhang H, Su Y, Silani V, Singer RH, Rossoll W, Bassell GJ. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci. 2011;31:3914–3925. doi: 10.1523/JNEUROSCI.3631-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res. 2012;1462:81–92. doi: 10.1016/j.brainres.2012.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Englbrecht C, Chari A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip Rev RNA. 2011;2:718–731. doi: 10.1002/wrna.87. [DOI] [PubMed] [Google Scholar]
- Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, Kaspar BK. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilina TO, McGovern VL, Workman E, Crawford TO, Gogliotti RG, DiDonato CJ, Monani UR, Morris GE, Burghes AH. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Quinlan KA, Barlow CB, Heier CR, Heckman CJ, Didonato CJ. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci. 2012;32:3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, Jorgensen M, Durens M, Xia B, Barlow C, Heier CR, Plasterer HL, Jacques V, Kiledjian M, Jarecki J, Rusche J, DiDonato CJ. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum Mol Genet. 2013;22:4084–4101. doi: 10.1093/hmg/ddt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med. 2013;19:40–50. doi: 10.1016/j.molmed.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Hao le T, Wolman M, Granato M, Beattie CE. Survival motor neuron affects plastin 3 protein levels leading to motor defects. J Neurosci. 2012;32:5074–5084. doi: 10.1523/JNEUROSCI.5808-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannaccone ST, Russman BS, Browne RH, Buncher CR, White M, Samaha FJ. Prospective analysis of strength in spinal muscular atrophy. DCN/Spinal Muscular Atrophy Group. J Child Neurol. 2000;15:97–101. doi: 10.1177/088307380001500207. [DOI] [PubMed] [Google Scholar]
- Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–439. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol. 2007;179:139–149. doi: 10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005;14:2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- Jodelka FM, Ebert AD, Duelli DM, Hastings ML. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum Mol Genet. 2010;19:4906–4917. doi: 10.1093/hmg/ddq425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81:333–348. doi: 10.1016/j.neuron.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Obis T, Garone C, Akay T, Sera F, Iwata S, Homma S, Monani UR. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest. 2014;124:785–800. doi: 10.1172/JCI72017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kye MJ, Niederst ED, Wertz MH, Gonçalves Ido C, Akten B, Dover KZ, Peters M, Riessland M, Neveu P, Wirth B, Kosik KS, Sardi SP, Monani UR, Passini MA, Sahin M. SMN regulates axonal local translation via miR-183/mTOR pathway. Hum Mol Genet. 2014;23:6318–6331. doi: 10.1093/hmg/ddu350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Le TT, McGovern VL, Alwine IE, Wang X, Massoni-Laporte A, Rich MM, Burghes AH. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet. 2011;20:3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YI, Mikesh M, Smith I, Rimer M, Thompson W. Muscles in a mouse model of spinal muscular atrophy show profound defects in neuromuscular development even in the absence of failure in neuromuscular transmission or loss of motor neurons. Dev Biol. 2011;356:432–444. doi: 10.1016/j.ydbio.2011.05.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy- determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lenglet T, Lacomblez L, Abitbol JL, Ludolph A, Mora JS, Robberecht W, Shaw PJ, Pruss RM, Cuvier V, Meininger V, Mitotarget study group A phase II-III trial of olesoxime in subjects with amyotrophic lateral sclerosis. Eur J Neurol. 2014;21:529–536. doi: 10.1111/ene.12344. [DOI] [PubMed] [Google Scholar]
- Li DK, Tisdale S, Lotti F, Pellizzoni L. SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol. 2014;32:22–29. doi: 10.1016/j.semcdb.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KK, Lin MY, Zingg B, Feng Z, Ko CP. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One. 2010;5:e15457. doi: 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KK, Gibbs RM, Feng Z, Ko CP. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15:3555–3565. [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, Pellizzoni L. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE, Li DK, Pellizzoni L, Rojas J, Valenzuela DM, Murphy AJ, Winberg ML, Monani UR. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest. 2011;121:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod MJ, Taylor JE, Lunt PW, Mathew CG, Robb SA. Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. 1999;3:65–72. doi: 10.1053/ejpn.1999.0184. [DOI] [PubMed] [Google Scholar]
- Martinez TL, Kong L, Wang X, Osborne MA, Crowder ME, Van Meerbeke JP, Xu X, Davis C, Wooley J, Goldhamer DJ, Lutz CM, Rich MM, Sumner CJ. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci. 2012;32:8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet. 2008;9:843–854. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN, Mendell JR, Prior TW, Burghes AH. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Bühler D, Pillai R, Lottspeich F, Fischer U. A multiprotein complex mediates the ATP-dependent assembly of spliceosomal U snRNPs. Nat Cell Biol. 2001;3:945–949. doi: 10.1038/ncb1101-945. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohseni J, Zabidi-Hussin ZA, Sasongko TH. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet Mol Biol. 2013;36:299–307. doi: 10.1590/S1415-47572013000300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Munsat TL, Davies KE. International SMA consortium meeting (26–28 June 1992, Bonn, Germany) Neuromuscul Disord. 1992;2:423–428. doi: 10.1016/S0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Murray LM, Lee S, Bäumer D, Parson SH, Talbot K, Gillingwater TH. Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum Mol Genet. 2010;19:420–433. doi: 10.1093/hmg/ddp506. [DOI] [PubMed] [Google Scholar]
- Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KK, Karp GM, Qi H, Woll MG, Chen G, Zhang N, Gabbeta V, Vazirani P, Bhattacharyya A, Furia B, Risher N, Sheedy J, Kong R, Ma J, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693. doi: 10.1126/science.1250127. [DOI] [PubMed] [Google Scholar]
- Oprea GE, Kröber S, McWhorter ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park GH, Maeno-Hikichi Y, Awano T, Landmesser LT, Monani UR. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci. 2010;30:12005–12019. doi: 10.1523/JNEUROSCI.2208-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini MA, Bu J, Roskelley EM, Richards AM, Sardi SP, O'Riordan CR, Klinger KW, Shihabuddin LS, Cheng SH. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340–345. doi: 10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Grimmler M, Meister G, Will CL, Lührmann R, Fischer U, Schümperli D. Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev. 2003;17:2321–2333. doi: 10.1101/gad.274403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porensky PN, Mitrpant C, McGovern VL, Bevan AK, Foust KD, Kaspar BK, Wilton SD, Burghes AH. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. 2004;130a:307–310. doi: 10.1002/ajmg.a.30251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85:408–413. doi: 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiu M, Herbst R, Kim N, Jevsek M, Fak JJ, Mann MA, Fischbach G, Burden SJ, Darnell RB. Rescuing Z+ agrin splicing in Nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc Natl Acad Sci U S A. 2009;106:3513–3518. doi: 10.1073/pnas.0813112106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiu M, McGovern VL, Lotti F, Saieva L, Li DK, Kariya S, Monani UR, Burghes AH, Pellizzoni L. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol. 2012;32:126–138. doi: 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz R, Casañas JJ, Torres-Benito L, Cano R, Tabares L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci. 2010;30:849–857. doi: 10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid A, DiDonato CJ. Animal models of spinal muscular atrophy. J Child Neurol. 2007;22:1004–1012. doi: 10.1177/0883073807305667. [DOI] [PubMed] [Google Scholar]
- See K, Yadav P, Giegerich M, Cheong PS, Graf M, Vyas H, Lee SG, Mathavan S, Fischer U, Sendtner M, Winkler C. SMN deficiency alters Nrxn2 expression and splicing in zebrafish and mouse models of spinal muscular atrophy. Hum Mol Genet. 2014;23:1754–1770. doi: 10.1093/hmg/ddt567. [DOI] [PubMed] [Google Scholar]
- Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- Shababi M, Lorson CL, Rudnik-Schöneborn SS. Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J Anat. 2014;224:15–28. doi: 10.1111/joa.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Salcius M, Liu SW, Staker BL, Mishra R, Thurmond J, Michaud G, Mattoon DR, Printen J, Christensen J, Bjornsson JM, Pollok BA, Kiledjian M, Stewart L, Jarecki J, Gurney ME. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem Biol. 2008;3:711–722. doi: 10.1021/cb800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NN, Singh RN. Alternative splicing in spinal muscular atrophy underscores the role of an intron definition model. RNA Biol. 2011;8:600–606. doi: 10.4161/rna.8.4.16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Gillingwater TH, Talbot K. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis Model Mech. 2011;4:457–467. doi: 10.1242/dmm.007245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh JN, Barreiro-Iglesias A, Oliver PL, Biba A, Becker T, Davies KE, Becker CG, Talbot K. Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum Mol Genet. 2014;23:855–869. doi: 10.1093/hmg/ddt477. [DOI] [PubMed] [Google Scholar]
- Swoboda KJ, Prior TW, Scott CB, McNaught TP, Wride MC, Reyna SP, Bromberg MB. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57:704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale S, Lotti F, Saieva L, Van Meerbeke JP, Crawford TO, Sumner CJ, Mentis GZ, Pellizzoni L. SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell Rep. 2013;5:1187–1195. doi: 10.1016/j.celrep.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- Valori CF, Ning K, Wyles M, Mead RJ, Grierson AJ, Shaw PJ, Azzouz M. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- Van Meerbeke JP, Gibbs RM, Plasterer HL, Miao W, Feng Z, Lin MY, Rucki AA, Wee CD, Xia B, Sharma S, Jacques V, Li DK, Pellizzoni L, Rusche JR, Ko CP, Sumner CJ. The DcpS inhibitor RG3039 improves motor function in SMA mice. Hum Mol Genet. 2013;22:4074–4083. doi: 10.1093/hmg/ddt257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezain M, Saugier-Veber P, Goina E, Touraine R, Manel V, Toutain A, Fehrenbach S, Frébourg T, Pagani F, Tosi M, Martins A. A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat. 2010;31:E1110–E1125. doi: 10.1002/humu.21173. [DOI] [PubMed] [Google Scholar]
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 2005;19:2320–2330. doi: 10.1101/gad.342005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth B, Brichta L, Schrank B, Lochmüller H, Blick S, Baasner A, Heller R. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. 2006;119:422–428. doi: 10.1007/s00439-006-0156-7. [DOI] [PubMed] [Google Scholar]
- Wirth B, Garbes L, Riessland M. How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev. 2013;23:330–338. doi: 10.1016/j.gde.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, Eaton SL, Fuller HR, Roche SL, Somers E, Morse R, Young PJ, Lamont DJ, Hammerschmidt M, Joshi A, Hohenstein P, Morris GE, Parson SH, Skehel PA, Becker T, et al. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J Clin Invest. 2014;124:1821–1834. doi: 10.1172/JCI71318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman E, Saieva L, Carrel TL, Crawford TO, Liu D, Lutz C, Beattie CE, Pellizzoni L, Burghes AH. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet. 2009;18:2215–2229. doi: 10.1093/hmg/ddp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]