

Abstract
Neonatal hypoxia-ischemia (H-I) is the leading cause of brain damage resulting from birth complications. Studies in neonatal rats have shown that H-I acutely expands the numbers of neural precursors (NPs) within the subventricular zone (SVZ). The aim of these studies was to establish which NPs expand after H-I and to determine how leukemia inhibitory factor (LIF) insufficiency affects their response. During recovery from H-I, the number of Ki67+ cells in the medial SVZ of the injured hemisphere increased. Similarly, the number and size of primary neurospheres produced from the injured SVZ increased approximately twofold versus controls, and, upon differentiation, more than twice as many neurospheres from the damaged brain were tripotential, suggesting an increase in neural stem cells (NSCs). However, multimarker flow cytometry for CD133/LeX/NG2/CD140a combined with EdU incorporation revealed that NSC frequency diminished after H-I, whereas that of two multipotential progenitors and three unique glial-restricted precursors expanded, attributable to changes in their proliferation. By quantitative PCR, interleukin-6, LIF, and CNTF mRNA increased but with significantly different time courses, with LIF expression correlating best with NP expansion. Therefore, we evaluated the NP response to H-I in LIF-haplodeficient mice. Flow cytometry revealed that one subset of multipotential and bipotential intermediate progenitors did not increase after H-I, whereas another subset was amplified. Altogether, our studies demonstrate that neonatal H-I alters the composition of the SVZ and that LIF is a key regulator for a subset of intermediate progenitors that expand during acute recovery from neonatal H-I.
Keywords: cerebral palsy, cytokines, glial progenitors, gliogenesis, inflammation, stem cells
Introduction
Neonatal hypoxia-ischemia (H-I) remains the major cause of mortality and permanent neurological disabilities in term infants. The prevalence of neonatal H-I in the United States remains at 2–4 per 1000 full-term births despite major advancements in obstetric monitoring and perinatal care (Wyatt et al., 2007). Permanent neurological handicaps, including cerebral palsy and epilepsy, as well as cognitive, motor, and behavioral disorders develop in many infants who survive H-I (Nelson and Lynch, 2004). Given that the quality of life for the child is often permanently affected by this insult, there is a need for regenerative medicine to repair the immature brain. To aid in the development of treatments, a rodent animal model known as the Vannucci model was developed, in which blood and oxygen delivery to the brain is disrupted (Rice et al., 1981). The Vannucci model was developed using neonatal rats; however, increasingly researchers have used adaptations of this model on neonatal mice, enabling studies to use genetically modified animals (Sheldon et al., 1998).
The subventricular zone (SVZ) is a heterogeneous reservoir of neural stem cells (NSCs) and progenitor cells that contribute new cells to the brain throughout life. At birth, the SVZ is at its maximal size in rodents, which is when the immature brain is at a dynamic state of development. Neonatal cerebral H-I occurs coincident with this critical stage. Studies have reported extensive loss of progenitors after H-I; however, previous data have suggested that the stem cells are resistant to the injury (Brazel et al., 2004; Romanko et al., 2004). Decades of research have provided insights into the mechanisms underlying neonatal H-I brain injury, and more recent studies have generated optimism that regeneration is occurring, at least modestly, after this injury (Parent et al., 2002; Plane et al., 2004; Yang et al., 2007, 2008). Our laboratory has generated strong in vivo and ex vivo neonatal rat data that reveal a transient compensatory increase in neural precursors (NPs) within the SVZ 48 h after recovery (Felling et al., 2006; Yang and Levison, 2006; Alagappan et al., 2009). However, an essential unanswered question is whether this injury amplifies the numbers of bona fide NSCs or whether it is increasing the numbers of multipotential progenitors (MPs).
The signals that regulate NP expansion after H-I also remain incompletely understood. NPs rely on a variety of extracellular signals that are produced by the niches that control their self-renewal, proliferation, and differentiation. With injury, the balance of growth factors and cytokines in the SVZ changes (Felling et al., 2006). In particular, there is a robust increase in leukemia inhibitory factor (LIF) mRNA within the injured neonatal rat SVZ that precedes NP expansion (Covey and Levison, 2007). Astrocytes are activated by CNS injury, and they are a major source of LIF (Ishibashi et al., 2006). Thus, we hypothesized that LIF secretion by astrocytes may be essential for NP expansion after neonatal H-I.
Materials and Methods
All experiments were performed in accordance with research guidelines of the institutional animal care and use committee of New Jersey Medical School. A breeding colony of C57BL/6 mice was established and maintained in the Cancer Center animal facility from mice initially purchased from Charles River Laboratories. C57BL/6 neonates of either sex were used in the experiments presented in Figures 1, 2, 3, 4, 5, and 6, while LIF heterozygous (Hets) of either sex on a CD-1 background were used for Figure 7. The LIF mouse line was provided by Dr. Douglas Fields (National Institutes of Health, Bethesda, MD) with permission from Dr. Colin Stewart (Institute of Medical Biology, Singapore) (Stewart et al., 1992).
Figure 1.

H-I increases the number of proliferating cells within mouse ipsilateral SVZ. A, B, Immunostaining for Ki67 (red) and DAPI (blue) of CL (A) and IL (B) SVZs at 4 d of recovery. C–F, Higher-magnification photomicrography of the bracketed areas in A (C, E) and B (D, F), respectively. Scale bars, 50 μm. G, Schematic diagram illustrates the three distinct regions within the dorsolateral SVZ. H, Ki67 indices within medial (M), mediolateral (ML), and lateral (L) regions of the dorsolateral SVZ, and ventral (V) SVZ. Bars represent the mean ± SEM from three animals per group. *p < 0.05, versus CL, by paired t test.
Figure 2.

Mouse neonatal H-I stimulates NP expansion. A–F, Representative 1° (A–C) and 2° spheres (D–F) at 7 DIV from control (CTL; A, D), CL (B, E), and IL (C, F) SVZs. G, Quantification of 1° and 2° sphere formation. H, Measurements of sphere volumes. Average sphere volume was measured from 60–100 spheres/group. N = 6 SVZs/group. Scale bars, 50 μm. Data represent the mean ± SEM of three independent experiments. +p < 0.05, ++p < 0.005, +++p < 0.0005, vs CTL; *p < 0.05, **p < 0.005, ***p < 0.0005, vs CL, by Student's t test.
Figure 3.
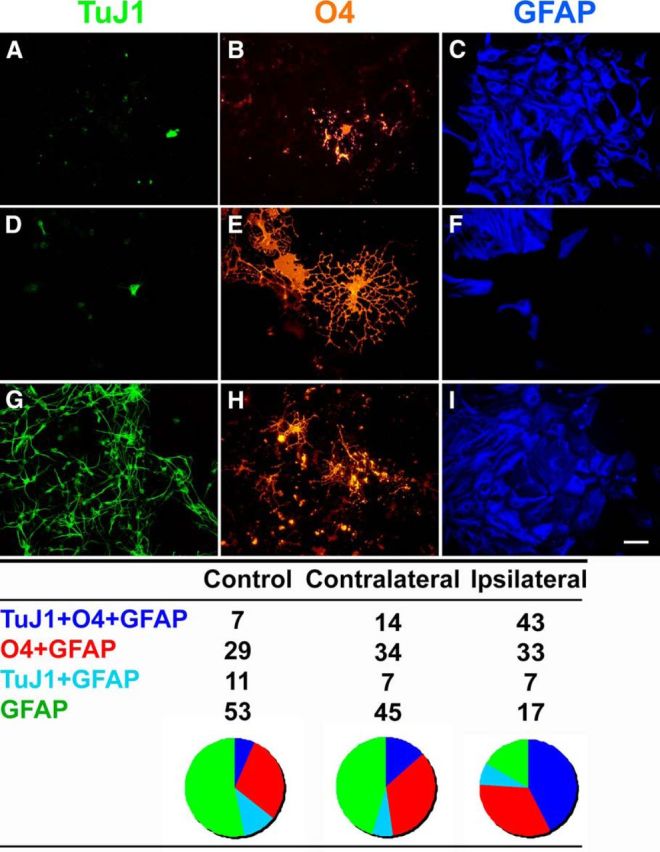
Hypoxia/ischemia increases the proportion of multipotential 1° neurospheres. A–I, Spheres generated from control (A–C) CL (D–F), and IL (G–I) SVZs were plated onto poly-d-lysine/laminin-coated chamber slides and differentiated in defined medium without growth factors for 5 d and then immunostained for neuronal (TuJ1, green), oligodendrocytic (O4, red), and astrocytic (GFAP, blue) markers. The table depicts the percentage of neurospheres that were unipotential, bipotential, or tripotential upon differentiation. At least 100 spheres were analyzed for each SVZ group. Scale bar, 25 μm.
Figure 4.

LIF, interleukin-6 (IL6), and CNTF mRNA. Time course of LIF, interleukin-6, and CNTF mRNA expression in the mouse SVZ after neonatal H-I. Cytokine mRNAs in the ipsilateral SVZ were measured by quantitative PCR and normalized to control SVZ. n = 3–5 SVZs for each group at all time points. Values represent the mean ± SEM.
Figure 5.

Flow cytometric isotype control gating and experimental profiles of C57BL/6 SVZs injected with EdU after H-I. A, Gate 1: live cell scatter, side scattering area (SSC-A) versus forward scatter area (FSC-A). B, Gate 2: live cell gating with DAPI exclusion, FSC-A versus DAPI. The gate is of live cells, while debris and dead cells are to the right of gate, respectively. C, Gate 3: single-cell gating to exclude doublets, FSC-A versus FSC-W. D, Histogram of EdU in unlabeled control. EdU− cells are to the left of the marker gate, and EdU+ cells are to the right. E, Isotype controls of CD140a-PE versus NG2-Alexa Fluor 700 and (F) CD133-APC versus LeX-FITC. G, Representative histogram from EdU-injected and Click-iT-treated animals showing two distinct EdU− and EdU+ populations after H-I. H, I, Representative sample of EdU+/CD140a-PE versus EdU+/NG2− Alexa Fluor 700 precursors (H) and depicts the relatively rare EdU+/LeX-v450 versus EdU+/CD133-APC from the NG2−/CD140 population (I). FSC-A, forward scatter area; FSC-W, forward scatter width.
Figure 6.

Neonatal H-I diminishes NSCs to generate multipotential and glial-restricted progenitors. SVZs from WT C57BL/6 mice were analyzed by flow cytometry at 48 h of recovery using fluorescently labeled probes to the following cell surface antigens: CD133/LeX/NG2/CD140a/EGFR. A total of 50,000 cell events were analyzed per group after excluding debris and dead cells by LIVE/DEAD Violet. Two injury-matched SVZs were pooled per sample, and three samples were analyzed per condition. Data are presented as the mean ± SEM and are representative of two independent experiments. Control (CTL) represents the combined mean of control and sham-operated animals. *p < 0.05; **p < 0.01; ***p < 0.001, vs control by Tukey's post hoc test. CONTRA, Contralateral; IPSI, ipsilateral.
Figure 7.

LIF haplodeficiency diminishes NP expansion after H-I. SVZs from LIF HET mice were analyzed by flow cytometry at 48 h using the following: CD133/LeX/NG2/CD140a/EGFR. A total of 70,000 cell events were analyzed per group after excluding debris and dead cells using LIVE/DEAD Violet. Data represent mean values. Two injury-matched SVZs were pooled per sample, and three samples were analyzed per condition. Data are presented as the mean ± SEM and are representative of two independent experiments. Control (CTL) represents the combined mean of control and sham-operated animals. *p < 0.05; **p < 0.01; ***p < 0.001, vs control by Tukey's post hoc test. CONTRA, Contralateral; IPSI, ipsilateral.
Mouse neonatal hypoxia-ischemia.
After normal delivery, cerebral H-I was induced on postnatal day 9 (P9) mouse pups (day of birth was designated as P0) by a right common carotid artery (RCCA) cauterization followed by systemic hypoxia. Pups were lightly anesthetized with isoflurane (5% induction, 3% maintenance) mixed with medical oxygen. Once fully anesthetized, a midline neck incision was made, and the RCCA was identified. The RCCA was separated from the vagus nerve and then cauterized at two distinct locations with a bipolar cauterizer at power setting of 10 (Model 80-1140, Codman & Shurtleff, Inc.). The neck incision was then sutured, and the pups were returned to the dam for 1.5 h. The pups were prewarmed for 10 min in jars submerged in a 36°C water bath. They were then exposed to 65 min of 10% O2 balanced N2. After this hypoxic interval, the pups were returned to their dam for recovery periods of 2, 3, or 4 d, at which time they were either decapitated for the neurosphere assay (NSA) or anesthetized and intracardially perfused with cold 4% PFA in PBS. Controls included unoperated animals as well as sham-operated animals that were anesthetized and had the RCCA isolated but not cauterized. Controls were separated from the dam for the same amount of time as experimental animals. Controls were not subjected to a hypoxic interval, whereas sham-operated neonates were exposed to 65 min of 10% O2 balanced N2.
Immunohistochemistry.
Free-floating 35 μm cryostat sections were stained using standard immunofluorescence procedures. Proliferating cells were detected using a rabbit polyclonal anti-Ki67 (1:500; Vector Laboratories), detected using Rhodamine Red-x-conjugated donkey anti-rabbit IgG (1:200; Jackson ImmunoResearch). Sections were counterstained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; 1 μg/ml; Sigma-Aldrich). Fluorescently immunolabeled sections were analyzed on an Olympus Optical AX-70 microscope interfaced with IPLab Scanalytics scientific imaging software. To obtain a Ki67 index, three to five sections were sampled at 150 μm intervals with five animals per group. The dorsolateral SVZ was divided into three distinct anatomical subregions, as previously described (Rothstein and Levison, 2002). Briefly, the medial region consists of the densely packed SVZ cells within 20 μm of the ependymal layer. The mediolateral region is ∼200 μm lateral from the medial region and is much less dense than the medial region. Finally, the lateral SVZ is 200 μm lateral to the mediolateral region and consists of densely packed cells. The ventral SVZ was defined as a subregion of the SVZ ∼2 mm ventral to the corpus callosum. Two fields per SVZ subregion were assessed in the ipsilateral (IL) and contralateral (CL) hemisphere per section by an observer blinded to the experimental and age groups. Each counting field occupied an area of 50 × 50 μm. The numbers of Ki67+ and total DAPI+ cells in each region of the dorsolateral SVZ were quantified, converted into a percentage of Ki67+/DAPI+ cells, and averaged. The data are presented as the percentage of labeled cells per field after using the Abercrombie correction, as previously described (Romanko et al., 2004).
Neurosphere propagation.
C57BLK/6 pups at 3 d of recovery after H-I (P12) were decapitated. SVZs from coronal sections of control and H-I brains were isolated and placed in dishes containing cold PGM (PBS with 1 mm MgCl2 and 0.6% dextrose). The SVZs were isolated by microdissection, mechanically minced (six SVZs for each group), and then incubated for 15 min at 37°C in 3 ml containing 0.25% trypsin-mixture trypsin-EDTA, 10 U/ml papain (Worthington Biochemical), 0.01% collagenase (Roche Applied Science), 40 μg/ml DNase 1 (Sigma-Aldrich), and 2.8 mm MgSO4 in MEM-HEPES buffer (Invitrogen). Subsequently, an equal volume of 0.02% trypsin inhibitor (Sigma) in Pro-N media (DMEM/F-12 media containing 10 ng/ml d-biotin, 25 μg/ml insulin, 20 nm progesterone, 100 μm putrescine, 5 ng/ml selenium, 50 μg/ml apo-transferrin, and 50 μg/ml gentamycin) was added. After centrifuging for 5 min at 200 × g, the supernatant was removed. The tissue was mechanically dissociated by trituration in Pro-N media. The cell suspension was then passed through 100 and 40 μm cell strainers (BD Bioscience) to ensure an even single-cell suspension and collected by centrifugation at 200 × g for 5 min. The number of viable cells was determined with a hemocytometer by exclusion of Trypan Blue dye. Cells were then plated into 12-well plates at a density of 2.5 × 104 cells in 0.5 ml of Pro-N media supplemented with 20 ng/ml EGF (PeproTech) and 10 ng/ml FGF2 (referred to as EF; PeproTech). Cell cultures were fed every 2 d by removing approximately half of the media and replacing it with an equal volume of fresh media. For 2° sphere propagation, 1° spheres were collected from 12-well plates at 9 d in vitro (DIV) and pelleted by centrifugation at 200 × g for 5 min. The same dissociation procedures were followed to obtain single suspension cells from 1° spheres. Cells were plated into a 12-well plate at a density of 5 × 103 cells in 0.5 ml of Pro-N media with EF. Cells were grown for 7 d before analysis. A sphere was defined as a free-floating, cohesive cluster that was at least 30 μm in diameter, although the majority of spheres were larger than this. Plates were gently shaken to distribute the spheres before counting. Five random 10× fields were counted per well and six wells were evaluated per group. The frequency of sphere-forming cells was calculated by the average number of spheres per field, the area of the field, and the area of the well using a Zeiss Axio Observer.Z1 microscope. Sphere volume was measured using IPLab Scanalytics Software.
Neurosphere differentiation.
Spheres were plated onto chamber slides previously coated with 1% poly-d-lysine (w/v) and 10 μg/ml laminin, in ProN with 0.5% FBS without growth factors. After 5–7 d, chamber slides were stained using O4 culture supernatant (1:10) in 10% lamb serum in DMEM/F12 with 15 mm HEPES at room temperature (RT) for 45 min, washed in BCH (10% newborn bovine serum in DMEM/F12 with HEPES), and then incubated for 30 min in goat anti-mouse (GAM) IgM Lissamine rhodamine sulfonyl chloride (1:200; Jackson ImmunoResearch). Cells were fixed in 3% PFA, quenched with 100 mm glycine, rinsed with PBS, and then permeabilized with methanol for 20 min at −20°C. Cells were blocked in TGB superblock (0.3% Triton X-100, 10% goat serum, and 10% bovine serum albumin in Tris buffer) for 45 min and immunostained using mouse monoclonal anti-neuronal class III α-tubulin (Tuj1 clone; 1:300; Covance) and rabbit anti-GFAP (1:500; Dako) in diluent O/N at 4°C. After washing thoroughly, cells were incubated for 30 min at RT in GAM DyLight 488 IgG2a for TuJ1 and goat anti-rabbit DyLight 649 IgG for GFAP (both at 1:300; Jackson ImmunoResearch). Cells were counterstained using DAPI and then coverslipped with ProLong Gold with DAPI (Invitrogen). Images of stained cells were collected using a Sensys cooled-coupled device camera interfaced with IPLab scientific imaging software on an Olympus AX-70 microscope.
Quantitative real-time PCR.
Microdissected dorsolateral SVZs were processed for mRNA extraction using Qiagen RNeasy Mini Kit. RNA samples were then reverse transcribed to cDNA using Superscript III (Invitrogen). We used a 7300 real-time PCR system (Applied Biosystems) and MicroAmp optical 96-well reaction plates (Applied Biosystems) for quantitative real-time PCR analysis. As an internal standard, we used QuantumRNA 18S (Applied Biosystems) in a 4:6 primer/Competimer reaction mix with QuantiTect SYBR Green PCR kit (Qiagen). Sample size consisted of 100 ng of cDNA per well of each reaction plate. We used the QuantiTect PCR protocol and the following primers: Mm_IL6_1_SG (IL6), Mm_Lif_1_SG (LIF), and Mm_Cntf_2_SG [ciliary neurotrophic factor (CNTF); Qiagen]. The ΔΔCt method of the 7300 Real-Time PCR software was used for relative quantification (Applied Biosystems).
Flow cytometry.
SVZs were dissociated with 0.45 Wünsch unit/ml Liberase DH (Roche) and 250 μg of DNase1 (Sigma-Aldrich) in PGM and were shaken at 220 rpms (Innova 2300, New Brunswick Scientific) at 37°C for 30 min. Enzymatic digestions were quenched with 10 ml of PGB (PBS without Mg2+ and Ca2+ with 0.6% dextrose and 2 mg/ml fraction V of BSA; Thermo Fisher Scientific), and cells were centrifuged for 5 min at 200 × g. Cells were dissociated by repeated trituration, collected by centrifugation, counted using a ViCell (Beckman Coulter), and diluted to at least 106 cells per 50 μl of PGB. All staining was performed in 96 V-bottom plates using 150 μl volume. For surface marker analysis, cell were incubated in PGB for 25 min with antibodies against Alexa Fluor 647 EGF complex (Invitrogen), CD24-PE/Cy7 (1:300, M1/69; BioLegend), Lewis-X (1:20, LeX/CD15, MMA; BD Bioscience), CD133-APC (1:50, 13A4; eBioscience), CD140a (1:400, APA5; BioLegend), and NG2 Chondroitin Sulfate Proteoglycan (1:50, AB5320; Millipore). Cells were washed with PGB by centrifugation at 278 × g. Anti-mouse IgM PerCP-eFluor 710 (1:200; eBioscience) was used to detect LeX and goat anti-rabbit IgG Alexa Fluor 700 (1:100; Invitrogen) was used to detect NG2. Cells were incubated with secondary antibodies and LIVE/DEAD fixable Violet Dead Cell Stain Kit (Invitrogen) in PGB for 20 min and then washed by centrifugation in PGB at 278 × g. Cells from SVZ were fixed with 1% ultrapure formaldehyde (50,000; Polysciences, Inc.) for 20 min, collected by centrifugation for 9 min at 609 × g, resuspended in PGB, and stored at 4°C overnight. All sample data were collected on a BD LSR II flow cytometer (BD Biosciences Immunocytometry Systems). Matching isotype controls for all antibodies were used, and gates were set based on these isotype controls. Data were analyzed using FlowJo software (Tree Star, Inc.).
5-Ethynyl-2′-deoxyuridine injections and Click-iT 5-ethynyl-2′-deoxyuridine flow cytometry.
At 20 and 22 h after irradiation, C57BL/6 mice were injected intraperitoneally with 100 mg/kg 5-ethynyl-2′-deoxyuridine (EdU) dissolved in saline (Life Technologies). Two hours later (i.e., 24 h after H-I), the mice were killed by carbon dioxide inhalation, and the SVZs were isolated by microdissection and dissociated as described above with the following modifications. Cells were incubated in DAPI (1:5000) with secondary antibodies. Following overnight fixation in 1% ultrapure PFA, cells were resuspended in PG and washed by centrifugation at 400 × g. Cells were resuspended and permeabilized with saponin, and EdU was visualized with Click-iT reagent (Life Technologies) following the manufacturer instructions. Cells were washed by centrifugation in PG at 400 × g, resuspended in PG, and acquired on a BD LSR II flow cytometer.
Statistical analysis.
Results from immunofluorescence assays replicates were analyzed for statistical significance using paired Student's t test. Comparisons were interpreted as significant with p < 0.05. All data are expressed as the mean ± SEM. Flow cytometric data were analyzed using one-way ANOVA followed by Tukey's post hoc analysis (Prism 4 software, Graphpad).
Results
H-I injury response in neonatal mice is similar to neonatal rats
In our previous studies (Felling et al., 2006; Yang and Levison, 2006), we demonstrated that within 2–3 d of recovery from neonatal H-I there is a transient compensatory increase in the number of NPs within the rat SVZ. However, the mechanisms regulating this expansion are incompletely understood. Mice at P9 sustained injuries after H-I that were similar to those seen in P6 rats (Brazel et al., 2004). To begin to determine which precursors in the neonatal mouse SVZ proliferate in response to H-I, Ki67 staining was performed on cryosections at 4 d of recovery, and a Ki67 index was calculated. As different precursors are known to reside within specific niches, with the NSCs in particular, residing in the most medial aspect, we subdivided the SVZ into four subregions, as described previously for studies of both H-I and traumatic brain injury (Rothstein and Levison, 2002; Felling et al., 2006; Goodus et al., 2015). Within the injured IL medial subarea of the dorsolateral SVZ (SVZdl), which we defined as being within 20 μm of the ependymal cell lining, there was an increase in cell proliferation above that seen in the same region of the uninjured CL hemisphere (Fig. 1A,C,E). There also were more Ki67+ NPs in the lateral SVZ of the IL hemisphere; however, no differences were seen in the mediolateral region of the SVZdl or in SVZ cells located more ventrally along the wall of the lateral ventricle. Quantifying the Ki67 index, revealed a 33% and 54% increase in the proportion of Ki67+ cells located in the medial and the lateral regions of the SVZ, respectively (Fig. 1).
Finding a similar increase in proliferation in the mice as observed in rats after neonatal H-I (Felling et al., 2006), we wanted to establish which NPs were proliferating and accumulating within the injured hemisphere. We used the NSA to quantify the numbers of self-renewing cells that formed spheres. At 4 d of recovery from H-I, SVZs were microdissected from control, CL, and IL hemispheres; dissociated; and plated at low density in the presence of EGF and FGF2 plus heparin (i.e., EF). By 7 DIV, the cells had formed 1° neurospheres (Fig. 2A–F). Quantifying the number of 1° spheres from the three groups revealed a doubling of sphere-forming cells from the injured hemisphere, while the CL hemisphere displayed a 36% increase in the generation of spheres; both were compared with control (Fig. 2G). Primary spheres isolated from ipsilateral hemispheres were visibly larger than spheres generated from CL and control SVZs (Fig. 2, compare A, C). More detailed analyses of these neurospheres showed that the increase in size was due to a larger number of cells and not to larger cells (Buono, 2011). To assess their potentiality, 1° spheres were differentiated for 5 d in the absence of mitogens, immunostained for lineage-specific neural cell markers, and classified as tripotential, bipotential, or unipotential. This assay revealed a sevenfold increase in the proportion of tripotential spheres from the injured hemisphere and a twofold increase in tripotential spheres from the CL hemisphere compared with control (Fig. 3). Interestingly, the injured hemisphere also displayed an approximately threefold decrease in spheres that only generated astrocytes compared with control. Thus, the sphere data suggest that H-I stimulates the expansion of proliferating, multipotential NPs.
To determine whether these NPs could sustain their self-renewal in vitro, we passaged the primary neurospheres. Under normal conditions, NSCs divide asymmetrically; however, if there is a change in the microenvironment, these cells can divide symmetrically to either expand or deplete the pool of stem cells. We hypothesized that if the NPs from the ipsilateral hemisphere were symmetrically expanding, there would be a higher 2° sphere number upon passaging. If these NPs were symmetrically depleting, then there would be a reduction in sphere number upon passage, indicating a decrease in their self-renewal capacity and an increase in lineage-restricted progeny. The third possibility was that if these cells required sustained exposure to injury-induced signals, then their in vitro mode of division would return to normal (asymmetric division), and there would be no change in sphere number compared with control. To test these possible modes of division, 1° spheres from the three groups were dissociated, plated at clonal density, and grown for 7 d with EF to form 2° spheres. There was no change in 2° sphere number compared with control (Fig. 2G), suggesting that NPs from the ipsilateral and the contralateral hemispheres returned to the asymmetric division mode under the growth factor conditions used to propagate them. However, 2° sphere size remained elevated, similar to 1° spheres from the ipsilateral and contralateral hemispheres, compared with control (5.5-fold and 2.5-fold increase, respectively; Fig. 2D–F,H). Thus, these experiments reveal species differences in the proliferative responses of mouse SVZ NPs versus rat NPs after neonatal H-I. This variance between the two species could have several causes, as follows: (1) cell-plating density differences; (2) alterations in the secreted factors after H-I; (3) intrinsic differences in rat versus mouse NPs; and (4) differences in the extent of or quality of H-I injury.
Interleukin-6 and LIF rapidly but transiently increase after neonatal H-I
Several studies, including ours, have shown that the LIF receptor (LIFR) is essential for maintaining the normal complement of NPs (Shimazaki et al., 2001; Pitman et al., 2004; Bonaguidi et al., 2005; Gregg and Weiss, 2005). LIF, CNTF, and cardiotrophin-1 all signal through the LIFR/gp130 complex, but only LIF and CNTF have been reported to maintain NPs (Shimazaki et al., 2001; Bauer and Patterson, 2006). Recently, we demonstrated that interleukin-6, which signals through a gp130 homodimer and is known to be significantly elevated after brain injury, also promotes rat NP self-renewal (Covey et al., 2011). To assess the expression of interleukin-6, LIF, and CNTF after neonatal mouse H-I, we microdissected the SVZs from controls and the injured IL and CL hemispheres at intervals of recovery spanning from 8 to 168 h. Quantitative PCR analyses revealed that interleukin-6 mRNA was elevated 32-fold at 8 h after injury within the IL SVZ and returned to baseline within 24–48 h (Fig. 4), while there was no change within the CL SVZ (Buono, 2011). LIF mRNA was elevated 31-fold within the IL SVZ at 8 h, peaked at 24 h, with a 69-fold increase, and then returned to baseline at 48 h after injury (Fig. 4). Within the CL SVZ, LIF mRNA increased 29-fold at 24 h and returned to baseline at 48 h of recovery (Buono, 2011). CNTF mRNA was unchanged at early time points after injury but increased 30-fold at 168 h post injury (Fig. 4). Within the CL SVZ, there was a fourfold increase in CNTF mRNA at 24 h, which returned to baseline by 48 h (data not shown). The CL SVZ showed a similar, but lower-magnitude response to the IL SVZ displaying a ninefold increase in CNTF mRNA at 168 h after injury (data not shown).
Neonatal H-I expands a multipotential precursor and glial-restricted progenitors
Our published and current data support the hypothesis that LIF plays a critical role in NP expansion after neonatal H-I. A previous study (Bauer and Patterson, 2006) showed that LIF, both in vitro and in vivo, will expand NPs. Using gain- and loss-of-LIF function studies on SVZ NPs, we have confirmed the results of the study by Bauer and Patterson (2006) and have extended them to demonstrate that LIF is needed to maintain the normal balance of NPs (Buono et al., 2012). However, before evaluating the specific role of LIF within the SVZ after brain injury, we first needed to establish which NPs were affected by neonatal H-I and used our recently developed multimarker flow cytometry method to do so. Since LIF mRNA peaked at 24 h after H-I, we microdissected SVZs from CL and IL hemispheres and from controls at 24 h postinjury. IL SVZs were injury matched, and two SVZs were combined before tissue dissociation. SVZs were dissociated with Liberase-DH and incubated with fluorescent probes for CD133-efluor 710, LeX-FITC, CD140a-PE, NG2-Alexa Fluor 700, and EGF-Alexa Fluor 647. Debris, dead cells, and doublets were excluded from analysis, using LIVE/DEAD Violet and forward scatter gates. Gates were set using isotype controls (Fig. 5). Flow cytometric analyses revealed that control and sham-operated animals displayed no difference; thus, these datasets were combined. Using this multicolor flow analysis, seven different types of NPs that are either MPs or glial restricted progenitors (GRPs) could be identified as resident with the neonatal SVZ (Buono et al., 2012). The nomenclature that we have used to describe these precursors defines the precursors based on their differentiation potential in vitro; for example, with a numerical designation (e.g., MP1, MP2). The table in Figure 6 provides the antigenic features of each precursor together with the acronym ascribed to it. We previously reported that there is an increase of NG2+EGFR+ cells after H-I in the neonatal rat (Alagappan et al., 2009). Our FACS studies have established that there are two sets of precursors that are NG2+ within the SVZ—one set is multipotential, whereas the other is glial restricted. As we have yet to establish a fifth surface marker to distinguish between these two precursors, we will refer to the population of cells that are CD133-LeX-NG2+CD140a− as MP3/GRP2 cells. A significant proportion of cells within the SVZ remained unstained for any of the markers used. It is likely that these are precursors committed to the astrocyte lineage, committed neuroblasts and late oligodendrocyte progenitors. Studies are underway to incorporate additional surface markers for these progenitors so that they may be studied (Buono et al., 2015).
Within the ipsilateral SVZ, there was a fivefold decrease in cells with the phenotypic characteristics of NSCs (CD133+LeX+NG2−CD140a−) and a sixfold decrease in one set of multipotential precursors (MP1s; CD133−LeX+NG2−CD140a−; Fig. 6A,B). There was also a threefold increase in MP2s (CD133+LeX+NG2+CD140a−); a threefold increase in MP3/GRP2s, which became the largest population (CD133−LeX−NG2+CD140a−EGFR−); and a threefold increase in GRP3s (CD133−LeX-NG2+CD140a+; Fig. 6C,F,G). There was a subpopulation within the MP3/GRP2s that expressed high levels of EGFR and that increased twofold by 48 h of recovery (referred to as MP3/GRP2E; Fig. 6H). The CL SVZ data were similar to those of the control SVZ, but displayed slight changes compared with the IL SVZ. Also within this uninjured SVZ, there was a threefold increase in a mixed bipotential progenitor population (referred to as BNAP/GRP1) that was not seen in the control or IL SVZs (Fig. 6E). These data suggest that secreted molecules from the injured hemisphere exert a more global effect.
To assess whether the changes in the frequencies of the different NPs of the SVZ after H-I could be attributed to changes in their proliferation, we modified the flow cytometry protocol to accommodate the incorporation of EdU, which is a nucleoside analog of thymidine and is incorporated into DNA during active DNA synthesis (Buck et al., 2008). Mice were injected intraperitoneally with EdU at 20 and 22 h after injury, and SVZs were dissociated and labeled for flow cytometry 24 h postinjury, and the proportions of EdU+ NPs were quantified (Table 1). A depiction of the gating strategy and examples of the flow data are provided in Figure 5. Within the ipsilateral SVZ, there was a threefold increase in EdU+ MP2s (CD133+LeX+NG2+CD140a−), a threefold decrease in EdU+ platelet-derived growth factor (PDGF)-fibroblast-derived growth factor (FGF)-responsive progenitors (CD133−LeX+NG2+CD140a+), a fivefold increase in EdU+ GRP3s (CD133−LeX−NG2+CD140a+), and a twofold increase in EdU+ MP3/GRP2s (CD133−LeX−NG2+CD140a−). The changes in proliferation of these populations 24 h after injury directly correlated with the changes in relative frequency at 48 h after injury (Fig. 6). EdU+ NSCs (CD133+LeX+NG2−CD140a−) were decreased threefold in the ipsilateral SVZs compared with controls, but these data did not reach significance due to the infrequent numbers of NSCs. This decreased trend in NSC proliferation correlated with the decrease in relative NSC numbers 48 h after H-I (Fig. 6). EdU+ MP1s (CD133−LeX+NG2−CD140a−) were not altered 24 h following H-I despite the overall numbers of MP1s being decreased fivefold 48 h after injury.
Table 1.
Neonatal H-I increases proliferation of multipotential and glial-restricted progenitors
| WT | CD133+LeX+NG2−/CD140a−EdU+ (NSC) | CD133−LeX+NG2−/CD140a−EdU+ (MP1) | CD133+LeX+NG2+/CD140a−EdU+ (MP2) | CD133−LeX+NG2+/CD140a+EdU+ (PFMP) | CD133−LeX+NG2+/CD140a−EdU+ (BNAP/GRP1) | CD133−LeX−NG2+/CD140a+EdU+ (GRP) | CD133−LeX−NG2+/CD140a−EdU+ (MP3/GRP2) |
|---|---|---|---|---|---|---|---|
| Naive | 0.60 ± 0.28 | 1.49 ± 0.29 | 4.72 ± 0.34 | 3.99 ± 0.81 | 28.63 ± 10.34 | 0.98 ± 0.14 | 19.87 ± 4.67 |
| Sham | 0.70 ± 0.39 | 1.17 ± 0.31 | 4.97 ± 0.65 | 3.10 ± 0.45 | 35.70 ± 12.14 | 0.49 ± 0.21 | 22.57 ± 2.81 |
| IL | 0.19 ± 0.35 | 1.54 ± 0.57 | 13.44 ± 2.15* | 0.00 ± 0.00* | 50.33 ± 10.92 | 5.16 ± 0.99* | 42.83 ± 5.80* |
Data are presented as the mean ± SEM. SVZs from WT C57BL/6 mice were analyzed by flow cytometry at 24 h of recovery using the Click-iT EdU system for labeling proliferating cells and using fluorescently labeled probes to the following cell surface antigens: CD133/LeX/NG2/CD140a. A total of 100,000 cell events were analyzed per group after excluding debris and dead cell by DAPI. Two injury-matched SVZs were pooled per sample, and three samples were analyzed per condition.
*p < 0.05 versus naive and sham by one-way ANOVA and Tukey's post hoc test.
Having established which NPs increase or decrease in frequency due to neonatal H-I, we next assessed the role of LIF within the SVZ after injury using the same flow cytometric panel. Unfortunately, the LIF-null neonates sustained either extremely severe brain damage or did not survive H-I (often dying within 15 min of exposure to hypoxia). As the neonatal LIF Hets displayed a similar cellular phenotype (Buono et al., 2012), we compared the effects of neonatal H-I on LIF Hets versus wild-type controls. Unlike LIF nulls, LIF Hets survived H-I. Flow cytometric analyses revealed that untouched control and sham-operated neonates again were not different; thus, these data were combined. The SVZs from the IL hemisphere revealed no change in NSCs, but a decrease in all multipotential precursors, particularly MP2s (which showed a threefold increase within wild-type injured SVZs; Figs. 6C, 7C). The BNAP/GRP1 population in IL SVZ also decreased slightly compared with the threefold increase seen in the wild-type injured SVZ. It is important to note that BNAPs/GRP1s are LIF responsive (Buono et al., 2012). As seen previously in uninjured LIF Hets, MP3/GRP2s were more abundant in the wild-type SVZ and, interestingly, were less prevalent after H-I (Fig. 7G). However, the subpopulation of EGFR+ cells found within the MP3/GRP2s increased by 32%, which is similar to the wild-type response (Figs. 6H, 7H). However, unlike the wild-type neonatal H-I response, the CL SVZ was almost identical to control neonates and did not show a significant increase in BNAPs/GRP1s (Fig. 6E, 7E). These data support the conclusion that LIF or a molecule that is controlled by LIF was absent; and thus, the injury response was compromised versus wild-type neonates.
Discussion
The rat SVZ is known to expand in size during acute recovery from neonatal H-I with an increase in tripotential NPs (Felling et al., 2006; Yang and Levison, 2006; Alagappan et al., 2009) that is followed at later time points with an increase in the production of new neuroblasts, neurons, astrocytes, oligodendrocyte precursors, and oligodendrocytes (although the latter are rarer; Back et al., 2002; Plane et al., 2004; Zaidi et al., 2004; Ong et al., 2005; Yang and Levison, 2007; Yang et al., 2008; Bain et al., 2010; Dizon et al., 2010). However, it has not been clear with any level of precision which NPs are expanding after H-I and which injury signals are necessary for their expansion. To better understand which NPs expand after neonatal H-I, we directly assessed the heterogeneous population of precursors within the SVZ after acute insult using a novel multimarker flow cytometry panel. This powerful tool enabled us to quantify the relative proportions of eight unique NPs after injury. Surprisingly, we discovered that NSCs (CD133+LeX+NG2−CD140a−) are significantly diminished as a consequence of H-I, whereas there is an expansion of intermediate progenitors. The multipotential progenitors designated MP2s and three types of GRPs increased significantly by 48 h after injury, which can be attributed to a greater proportion of these cells entering the cell cycle. In other studies, we have established that LIF increases commensurate with the increase in multipotential NPs and that LIF is one of the major regulators of NP stemness (Buono et al., 2012). Thus, we tested the hypothesis that LIF is essential for the expansion of NPs after neonatal H-I by assessing how LIF haplodeficiency affects the SVZ NP response after injury. Our studies demonstrate that LIF is a key regulator of NP expansion within the SVZ during acute recovery from neonatal H-I.
NSCs are diminished after H-I to generate an increase in MP2s and GRPs
Neonatal H-I occurs coincident with the second wave of gliogenesis, and particularly with oligodendrogenesis. NPs show differential vulnerability to neonatal H-I with the rapidly dividing oligodendrocyte precursors that are especially vulnerable to H-I (Back et al., 2001; Levison et al., 2001; Ness et al., 2001; Back et al., 2002). Three weeks after H-I, Levison et al. (2001) demonstrated that there is extensive dysmyelination and subsequent astrogliosis in the periventricular white matter along with depletion of NPs within the SVZdl. Interestingly, however, studies in both rat and mouse have shown that, although the ependymal cells and progenitors in the medial SVZdl cells are under considerable stress, this region, and in particular the cells with the features of NSCs, are resistant to damage (Rothstein and Levison, 2002; Romanko et al., 2004; Brazel et al., 2014; Chen et al., 2015).
Using multimarker flow cytometry to evaluate the composition of the mouse SVZ during the acute recovery period from H-I, we report here that the NSCs (CD133+LeX+Lin−) are significantly reduced by 48 h of recovery with a concomitant increase in multipotential progenitors (MP2s) and glial-restricted progenitor (GRP3, GRP2 and GRP2E) frequency. Previous studies have demonstrated that NSCs are resilient to cytotoxic stress, and, therefore, it is unlikely that they are dying. Since there was a strong trend toward decreased EdU incorporation in the NSCs, whereas there was a significant increase in EdU incorporation in the MPs and GRPs, the most parsimonious explanation for the decreased frequency of the NSCs is that they are proliferating more slowly after H-I. Alternatively, there could be a change in the mode of division after injury as suggested by Zhang et al., (2004), who showed that, after ischemia, precursors within the SVZ display an increase in symmetric divisions (Zhang et al., 2004). However, our data would suggest that these symmetrical divisions result in the production of two MPs rather than two NSCs, with a depletion of the NSC pool and an expansion of multipotential progenitors. That our flow cytometry data are not in accord with the neurosphere assay data is not surprising given that the NSA cannot distinguish between sphere-forming NSCs and progenitors, let alone multipotential or bipotential progenitors, whereas the flow cytometric assay method that we have developed has significantly greater sensitivity and robustness.
Our studies on LIF-null mice reveal essential functions of LIF after neonatal brain injury. Unfortunately, the majority of the LIF-null mice could not withstand the H-I paradigm; therefore, they could not be analyzed. This outcome was not totally surprising as LIF has been reported to have a protective role in endotoxic shock and host defense (Weber et al., 2005), and LIF has been shown to reduce cutaneous inflammation (Zhu et al., 2001). By contrast, the LIF Hets had a similar SVZ cellular phenotype to the LIF nulls (Buono et al., 2012), and they survived the injury paradigm. In LIF Hets, the expansion of MP2s, MP3/GRP2s, and GRP3s after H-I was inhibited, indicating that these cells require LIF signaling to expand after injury (Table 2).
Table 2.
In vivo effects of neonatal H-I on neural precursor population dynamics within the SVZ at 48 h of recovery
| Cell | H-I |
|
|---|---|---|
| WT | LIF Hets | |
| NSC | Down | UC |
| MP1 | Down | Down |
| MP2 | Up | Down |
| PFMP | Down | Down |
| BNAP/GRP1 | UC | Down |
| GRP3 | Up | UC |
| GRP2 | Up | UC |
| GRP2E | Up | Up |
Results summarize changes in SVZ NPs in WT and LIF Hets. UC, No change between respective control and IL hemispheres.
Interestingly, the EGFR+ subpopulation within the MP3/GRP2s (MP3/GRP2Es) increased after H-I in the LIF Hets, which is similar to the wild-type H-I response, indicating that these cells respond to injury independent of LIF. We previously reported that NG2+EGFR+ cells increase in abundance after H-I in the neonatal rat (Alagappan et al., 2009). Studies have suggested that EGFR plays a critical role in the proliferation, migration, and differentiation of progenitors, especially during gliogenesis. Aguirre et al. (2004) FACS purified cells that were NG2+/EGFR+, and demonstrated that these cells were tripotential both in vitro and after transplantation in vivo. In 2005, this same group showed that the acquisition of EGFR by NG2+ neonatal cells correlated with their ability to proliferate and migrate out of the SVZ. They also showed that 4 weeks after transplantation, NG2+EGFR+ cells differentiated into astrocytes and oligodendrocytes (Aguirre et al., 2005, 2007). In mice with constitutive EGFR expression, the proliferation and migration of neonatal NG2+olig2+PDGFRα+nestin+GFAP−CC1− cells was enhanced and their differentiation inhibited (Ivkovic et al., 2008). Although these two groups reported apparently disparate results, our data suggest that they could have been studying two different NG2+ precursors. In fact, our flow cytometric data reveal seven phenotypically distinct NG2+ precursors, three of which are multipotential, 4 of which are glial restricted, and one of which is bipotential for neurons and astrocytes. Thus, our data along with the data from these studies suggest that there are different types of NG2+ precursors that may respond differentially to injury. Further experiments will be needed to test this hypothesis. Additional data would enable us to better understand why there is replacement and maturation of oligodendrocytes after some injuries, whereas in other injuries there is a lack of differentiation with an accumulation of NG2+ cells at the lesion (Levine, 1994; Chang et al., 2000; Watanabe et al., 2002).
Reactive astrocytes secrete LIF to regulate NP expansion within neonatal SVZ after H-I
In response to neonatal H-I, rats and mice show increased proliferation of NPs that occurs preferentially in the medial and lateral compartments of the SVZ (Felling et al., 2006; Yang and Levison, 2006; present study). We have previously shown that cell death is extensive in the adjacent striatum and within the lateral region of the SVZ, whereas there is minimal cell death in the most medial aspect of the SVZ (Brazel et al., 2004; Romanko et al., 2004). These deaths are a mixture of necrotic, excitotoxic, and apoptotic (Romanko et al., 2004). When cells die necrotic or excitotoxic deaths, they release the contents of their cytoplasm, and, notably, they release ATP. Interestingly, under normal conditions, LIF is released by astrocytes in response to ATP liberated from electrical impulse activity in axons, which in turn promotes myelination of oligodendrocytes (Ishibashi et al., 2006). LIF is likely secreted subsequent to astrocyte activation as a consequence of extracellular ATP release from dying cells. Within the IL SVZ, there is a robust increase in nestin and mushashi-1 expression, reflecting an increase in reactive astrocytes (data not shown). ATP also induces quiescent astrocytes to become reactive by increasing the expression of EGFR due to the intracellular increase of calcium (Liu et al., 2006; Xia and Zhu, 2011). In culture, astrocytes secrete twofold more LIF than untreated control upon stimulation with EGF (data not shown). Alagappan et al. (2009) showed that SVZ cells acquire higher levels of EGFR after H-I that requires the transcription factor egr-1 (Alagappan et al., 2013).
Whereas our studies demonstrate that LIF is necessary for the normal regenerative response to H-I, LIF is not generally regarded as a mitogen; therefore, other factors must be promoting proliferation within the medial and lateral regions of the SVZ. Sources of these factors include infiltrating microglia and reactive astrocytes. Reactive astrocytes are known to be a source of a plethora of growth factors, including FGF-2. Although it has not been directly shown, it is likely that LIF increases the production of FGF-2 in astrocytes, like CNTF (Albrecht et al., 2003), and that this production of FGF-2 enhances NP expansion after H-I. Ongoing studies will test this hypothesis.
Footnotes
This work was presented in part at the International Society for Cerebral Blood Flow and Metabolism meeting (Levison et al., 2009) and was supported by grants from the National Institutes of Health (NIH; MH-59950 and HD-052064) and the Leducq Foundation awarded to S.W.L., along with a training Fellowship awarded to K.D.B. on a NIH-National Institute of Neurological Disorders and Stroke Institutional Training Grant (NS-051157). We thank C. Stewart for permission to use his leukemia inhibitory factor (LIF)-null mice; R.D. Fields for shipping the LIF mice; S. Singh and D. Stein for flow cytometry assistance; T. Galenkamp for cell sorting; J. Goldman for manuscript suggestions; and A. White for editing this manuscript.
The authors declare no competing financial interests.
References
- Aguirre AA, Chittajallu R, Belachew S, Gallo V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol. 2004;165:575–589. doi: 10.1083/jcb.200311141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A, Rizvi TA, Ratner N, Gallo V. Overexpression of the epidermal growth factor receptor confers migratory properties to nonmigratory postnatal neural progenitors. J Neurosci. 2005;25:11092–11106. doi: 10.1523/JNEUROSCI.2981-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Alagappan D, Lazzarino DA, Felling RJ, Balan M, Kotenko SV, Levison SW. Brain injury expands the numbers of neural stem cells and progenitors in the SVZ by enhancing their responsiveness to EGF. ASN Neuro. 2009;1:e00009. doi: 10.1042/AN20090002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagappan D, Balan M, Jiang Y, Cohen RB, Kotenko SV, Levison SW. Egr-1 is a critical regulator of EGF-receptor-mediated expansion of subventricular zone neural stem cells and progenitors during recovery from hypoxia-hypoglycemia. ASN Neuro. 2013;5:183–193. doi: 10.1042/AN20120032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht PJ, Murtie JC, Ness JK, Redwine JM, Enterline JR, Armstrong RC, Levison SW. Astrocytes produce CNTF during the remyelination phase of viral-induced spinal cord demyelination to stimulate FGF-2 production. Neurobiol Dis. 2003;13:89–101. doi: 10.1016/S0969-9961(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain JM, Ziegler A, Yang Z, Levison SW, Sen E. TGFbeta1 stimulates the over-production of white matter astrocytes from precursors of the “brain marrow” in a rodent model of neonatal encephalopathy. PLoS One. 2010;5:e9567. doi: 10.1371/journal.pone.0009567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Patterson PH. Leukemia inhibitory factor promotes neural stem cell self-renewal in the adult brain. J Neurosci. 2006;26:12089–12099. doi: 10.1523/JNEUROSCI.3047-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, McGuire T, Hu M, Kan L, Samanta J, Kessler JA. LIF and BMP signaling generate separate and discrete types of GFAP-expressing cells. Development. 2005;132:5503–5514. doi: 10.1242/dev.02166. [DOI] [PubMed] [Google Scholar]
- Brazel CY, Rosti RT, 3rd, Boyce S, Rothstein RP, Levison SW. Perinatal hypoxia/ischemia damages and depletes progenitors from the mouse subventricular zone. Dev Neurosci. 2004;26:266–274. doi: 10.1159/000082143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazel CY, Alaythan AA, Felling RJ, Calderon F, Levison SW. Molecular features of neural stem cells enable their enrichment using pharmacological inhibitors of survival-promoting kinases. J Neurochem. 2014;128:376–390. doi: 10.1111/jnc.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST, Salic A. Detection of S-phase cell cycle progression using 5-ethynyl-2′-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2′-deoxyuridine antibodies. Biotechniques. 2008;44:927–929. doi: 10.2144/000112812. [DOI] [PubMed] [Google Scholar]
- Buono KD. University of Medicine and Dentistry of New Jersey; 2011. Analyses of mouse neural precursor responses to leukemia inhibitor factor and hypoxia/ischemia. PhD thesis. [Google Scholar]
- Buono KD, Vadlamuri D, Gan Q, Levison SW. Leukemia inhibitory factor is essential for subventricular zone neural stem cell and progenitor homeostasis as revealed by a novel flow cytometric analysis. Dev Neurosci. 2012;34:449–462. doi: 10.1159/000345155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buono KD, Goodus MT, Moore L, Ziegler AN, Levison S. Multimarker flow cytometric characterization, isolation and differentiation of neural stem cells and progenitors of the normal and injured mouse subventricular zone. In: Prusac J, editor. Neural surface antigens: from basic biology towards biomedical applications. New York: Elsevier; 2015. pp. 175–185. [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Goodus M, de Toledo S, Azzam E, Levison S, Souayah N. Ionizing radiation perturbs cell cycle progression of neural precursors in the subventricular zone without affecting their long-term self-renewal. ASN Neuro. 2015;7:1759091415575845. doi: 10.1177/1759091415575845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey MV, Levison SW. Leukemia inhibitory factor participates in the expansion of neural stem/progenitors after perinatal hypoxia/ischemia. Neuroscience. 2007;148:501–509. doi: 10.1016/j.neuroscience.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covey MV, Loporchio D, Buono KD, Levison SW. Opposite effect of inflammation on subventricular zone versus hippocampal precursors in brain injury. Ann Neurol. 2011;70:616–626. doi: 10.1002/ana.22473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dizon M, Szele F, Kessler JA. Hypoxia-ischemia induces an endogenous reparative response by local neural progenitors in the postnatal mouse telencephalon. Dev Neurosci. 2010;32:173–183. doi: 10.1159/000313468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felling RJ, Snyder MJ, Romanko MJ, Rothstein RP, Ziegler AN, Yang Z, Givogri MI, Bongarzone ER, Levison SW. Neural stem/progenitor cells participate in the regenerative response to perinatal hypoxia/ischemia. J Neurosci. 2006;26:4359–4369. doi: 10.1523/JNEUROSCI.1898-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodus MT, Guzman AM, Calderon F, Jiang Y, Levison SW. Neural stem cells of the immature, but not the mature, subventricular zone mount a robust regenerative response to traumatic brain injury. Dev Neurosci. 2015;37:29–42. doi: 10.1159/000367784. [DOI] [PubMed] [Google Scholar]
- Gregg C, Weiss S. CNTF/LIF/gp130 receptor complex signaling maintains a VZ precursor differentiation gradient in the developing ventral forebrain. Development. 2005;132:565–578. doi: 10.1242/dev.01592. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, Fields RD. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivkovic S, Canoll P, Goldman JE. Constitutive EGFR signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J Neurosci. 2008;28:914–922. doi: 10.1523/JNEUROSCI.4327-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JM. Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci. 1994;14:4716–4730. doi: 10.1523/JNEUROSCI.14-08-04716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ. Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev Neurosci. 2001;23:234–247. doi: 10.1159/000046149. [DOI] [PubMed] [Google Scholar]
- Levison SW, Felling RJ, Yang Z, Ziegler A, Covey M, Alagappan D, Buono KD, Bain JM. Signals coordinating CNS regeneration from resident neural stem cells after neonatal hypoxia/ischemia. Paper presented at the 24th International Symposium on Cerebral Blood Flow, Metabolism, & Function, along with the 9th International Conference on Quantification of Brain Function with PET, sponsored by the International Society for Cerebral Blood Flow and Metabolism; June; Chicago, IL. 2009. [Google Scholar]
- Liu B, Chen H, Johns TG, Neufeld AH. Epidermal growth factor receptor activation: an upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J Neurosci. 2006;26:7532–7540. doi: 10.1523/JNEUROSCI.1004-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–158. doi: 10.1016/S1474-4422(04)00679-9. [DOI] [PubMed] [Google Scholar]
- Ness JK, Romanko MJ, Rothstein RP, Wood TL, Levison SW. Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev Neurosci. 2001;23:203–208. doi: 10.1159/000046144. [DOI] [PubMed] [Google Scholar]
- Ong J, Plane JM, Parent JM, Silverstein FS. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res. 2005;58:600–606. doi: 10.1203/01.PDR.0000179381.86809.02. [DOI] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Pitman M, Emery B, Binder M, Wang S, Butzkueven H, Kilpatrick TJ. LIF receptor signaling modulates neural stem cell renewal. Mol Cell Neurosci. 2004;27:255–266. doi: 10.1016/j.mcn.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16:585–595. doi: 10.1016/j.nbd.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Rice JE, 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Romanko MJ, Rothstein RP, Levison SW. Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J Cereb Blood Flow Metab. 2004;24:814–825. doi: 10.1097/01.WCB.0000123906.17746.00. [DOI] [PubMed] [Google Scholar]
- Rothstein RP, Levison SW. Damage to the choroid plexus, ependyma and subependyma as a consequence of perinatal hypoxia/ischemia. Dev Neurosci. 2002;24:426–436. doi: 10.1159/000069052. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Sedik C, Ferriero DM. Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res. 1998;810:114–122. doi: 10.1016/S0006-8993(98)00892-0. [DOI] [PubMed] [Google Scholar]
- Shimazaki T, Shingo T, Weiss S. The ciliary neurotrophic factor/leukemia inhibitory factor/gp130 receptor complex operates in the maintenance of mammalian forebrain neural stem cells. J Neurosci. 2001;21:7642–7653. doi: 10.1523/JNEUROSCI.21-19-07642.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CL, Kaspar P, Brunet LJ, Bhatt H, Gadi I, Köntgen F, Abbondanzo SJ. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 1992;359:76–79. doi: 10.1038/359076a0. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Toyama Y, Nishiyama A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J Neurosci Res. 2002;69:826–836. doi: 10.1002/jnr.10338. [DOI] [PubMed] [Google Scholar]
- Weber MA, Schnyder-Candrian S, Schnyder B, Quesniaux V, Poli V, Stewart CL, Ryffel B. Endogenous leukemia inhibitory factor attenuates endotoxin response. Lab Invest. 2005;85:276–284. doi: 10.1038/labinvest.3700216. [DOI] [PubMed] [Google Scholar]
- Wyatt JS, Gluckman PD, Liu PY, Azzopardi D, Ballard R, Edwards AD, Ferriero DM, Polin RA, Robertson CM, Thoresen M, Whitelaw A, Gunn AJ. Determinants of outcomes after head cooling for neonatal encephalopathy. Pediatrics. 2007;119:912–921. doi: 10.1542/peds.2006-2839. [DOI] [PubMed] [Google Scholar]
- Xia M, Zhu Y. Signaling pathways of ATP-induced PGE2 release in spinal cord astrocytes are EGFR transactivation-dependent. Glia. 2011;59:664–674. doi: 10.1002/glia.21138. [DOI] [PubMed] [Google Scholar]
- Yang Z, Levison SW. Hypoxia/ischemia expands the regenerative capacity of progenitors in the perinatal subventricular zone. Neuroscience. 2006;139:555–564. doi: 10.1016/j.neuroscience.2005.12.059. [DOI] [PubMed] [Google Scholar]
- Yang Z, Levison SW. Perinatal hypoxic/ischemic brain injury induces persistent production of striatal neurons from subventricular zone progenitors. Dev Neurosci. 2007;29:331–340. doi: 10.1159/000105474. [DOI] [PubMed] [Google Scholar]
- Yang Z, Covey MV, Bitel CL, Ni L, Jonakait GM, Levison SW. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann Neurol. 2007;61:199–208. doi: 10.1002/ana.21068. [DOI] [PubMed] [Google Scholar]
- Yang Z, You Y, Levison SW. Neonatal hypoxic/ischemic brain injury induces production of calretinin-expressing interneurons in the striatum. J Comp Neurol. 2008;511:19–33. doi: 10.1002/cne.21819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi AU, Bessert DA, Ong JE, Xu H, Barks JD, Silverstein FS, Skoff RP. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia. 2004;46:380–390. doi: 10.1002/glia.20013. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang Z, Zhang C, Zhang L, Robin A, Wang Y, Lu M, Chopp M. Stroke transiently increases subventricular zone cell division from asymmetric to symmetric and increases neuronal differentiation in the adult rat. J Neurosci. 2004;24:5810–5815. doi: 10.1523/JNEUROSCI.1109-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Oishi K, Lee SC, Patterson PH. Studies using leukemia inhibitory factor (LIF) knock-out mice and a LIF adenoviral vector demonstrate a key anti-inflammatory role for this cytokine in cutaneous inflammation. J Immunol. 2001;166:2049–2054. doi: 10.4049/jimmunol.166.3.2049. [DOI] [PubMed] [Google Scholar]