

Abstract
G-protein receptor 84 (GPR84) is an orphan receptor that is induced markedly in monocytes/macrophages and microglia during inflammation, but its pathophysiological function is unknown. Here, we investigate the role of GPR84 in a murine model of traumatic nerve injury. Naive GPR84 knock-out (KO) mice exhibited normal behavioral responses to acute noxious stimuli, but subsequent to partial sciatic nerve ligation (PNL), KOs did not develop mechanical or thermal hypersensitivity, in contrast to wild-type (WT) littermates. Nerve injury increased ionized calcium binding adapter molecule 1 (Iba1) and phosphorylated p38 MAPK immunoreactivity in the dorsal horn and Iba1 and cluster of differentiation 45 expression in the sciatic nerve, with no difference between genotypes. PCR array analysis revealed that Gpr84 expression was upregulated in the spinal cord and sciatic nerve of WT mice. In addition, the expression of arginase-1, a marker for anti-inflammatory macrophages, was upregulated in KO sciatic nerve. Based on this evidence, we investigated whether peripheral macrophages behave differently in the absence of GPR84. We found that lipopolysaccharide-stimulated KO macrophages exhibited attenuated expression of several proinflammatory mediators, including IL-1β, IL-6, and TNF-α. Forskolin-stimulated KO macrophages also showed greater cAMP induction, a second messenger associated with immunosuppression. In summary, our results demonstrate that GPR84 is a proinflammatory receptor that contributes to nociceptive signaling via the modulation of macrophages, whereas in its absence the response of these cells to an inflammatory insult is impaired.
Keywords: cytokine, G-protein linked receptors, inflammation, macrophage, microglia, neuropathic pain
Introduction
Although there has been significant progress in the understanding of pain processes, chronic pain remains an area of considerable unmet medical need because of limited number and efficacy of treatments. Therefore, the search for new and more effective treatment continues. It has emerged that the immune system and its related molecules are important players in persistent pain and thus possess immense therapeutic potential. Unsurprisingly, the identification of novel immune-related targets has attracted the interest of many. Here we have explored the role of an orphan receptor, G-protein receptor 84 (GPR84), that was identified originally using an expressed sequence tag data mining strategy (Wittenberger et al., 2001). GPR84 consists of a generic heptahelical structure and is thought to belong to the rhodopsin superfamily, with little similarity to other known receptors. Expression analysis revealed that human and mouse GPR84 mRNA is found predominantly in hematopoietic tissues, including bone marrow, lung, spleen, and lymph node (Wittenberger et al., 2001; Wang et al., 2006). Its expression is restricted to peripheral immune cells and microglia in the CNS, and it is upregulated via appropriate immunostimulation [such as lipopolysaccharide (LPS) or TNF exposure; Wang et al., 2006; Bouchard et al., 2007; Lattin et al., 2008].
Although GPR84 was discovered well over a decade ago, very little is known about its functional role. So far, functional analyses have suggested that it is a free fatty acid (FFA) sensing GPCR that couples primarily to the Bordetella pertussis toxin-sensitive Gαi/o pathway (Wang et al., 2006; Suzuki et al., 2013). In particular, FFAs with medium carbon chain lengths of C9–C14 have shown efficacy in heterologous expression systems, but the endogenous ligand has yet to be identified. Several groups have postulated an immunomodulatory role that coincides with its restricted immune cell expression. It has been reported that GPR84 mediates chemotaxis and the release of TNF and IL-12 from macrophages and IL-8/chemokine ligand 8 from granulocytes (Wang et al., 2006; Suzuki et al., 2013). IL-12 plays a key role in promoting T-helper 1 (Th1)-driven immunity while inhibiting Th2 anti-inflammatory responses. Therefore, it is plausible that GPR84 is involved in regulating Th1 differentiation to promote an inflammatory response. Interestingly, these findings were consistent with augmented Th2 cytokine production in GPR84 null T cells (Venkataraman and Kuo, 2005). Furthermore, Bouchard et al. (2007) demonstrated that GPR84 mRNA is strikingly upregulated in cortical and spinal microglia in a model of endotoxemia and experimental autoimmune encephalomyelitis (EAE). Together, these findings are suggestive of an immunoregulatory and neuroinflammatory function.
Despite a lack of evidence of a role in nociceptive transmission, the upregulation of GPR84 mRNA has been observed in clinically relevant animal models of diabesity and EAE, which feature pain-associated pathologies (Bouchard et al., 2007; Tomlinson and Gardiner, 2008; Nagasaki et al., 2012; Khan et al., 2014). Moreover, its role in promoting a proinflammatory immune response and the release of cytokines, many of which can directly or indirectly modulate pain pathways (Kiguchi et al., 2012), supports the notion that GPR84 is a promising new target in the field.
Here we used GPR84 null mice to examine pain behaviors in a model of peripheral nerve injury. We focused on changes in the response of spinal microglia and peripheral macrophages with the aim of elucidating the contribution of GPR84 to neuropathic pain states.
Materials and Methods
Experimental animals.
GPR84 (GenBank accession number NM_030720, Ensembl identification number ENSMUSG00000063234) KO mice were provided by Deltagen under a GlaxoSmithKline license agreement. Targeting replaced 257 bp of coding sequence with an IRES–LacZ–poly(A) expression cassette and a positive selection cassette that contains the neomycin phosphotransferase gene driven by the PGK promoter (Neo). The insertion of the LacZ IRESLacZ introduces a premature translational stop signal that deletes the first three predicted transmembrane domain sequences. Disruption of the GPR84 gene was confirmed using PCR and standard agarose gel electrophoresis using the following primers: gene specific (endogenous), 5′-ACAGCTCAGATGCCAACTTCTCCTG-3′; gene specific (targeted, endogenous), 5′-TCCTAGAGCAATGAGACAGAGGGTG-3′; and Neo (targeted), 5′- GACGAGTTCTTCTGAGGGGATCGATC-3′.
Mice forming the initial breeding pairs were supplied by GlaxoSmithKline, which consisted of heterozygous (HET) F1 offspring from WT and KO breeding. HET pairs were bred in-house from 8 weeks old to produce litters of mixed genotypes according to Mendelian genetics. Mice were housed individually or in groups in standard environmental conditions (12 h light/dark cycle) with ad libitum access to food and water. Experiments were conducted in a blocked design on randomly selected, mixed sex- and age-matched WT and KO mice weighing 20–30 g (7–14 weeks old). HETs were only used for breeding. Animal husbandry and experiments were performed in a nonsterile housing environment in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986. For all studies, the experimenter was blinded to genotype and treatment.
Mechanical withdrawal threshold.
Static mechanical thresholds of alert and unrestrained mice were examined via von Frey hair application (0.008–1 g; Touch Test; Stoelting) to the plantar surface of the hindpaw using the “up–down” method (Chaplan et al., 1994). Before testing, mice were acclimatized individually for 1 h in acrylic testing cubicles (8 × 5 × 10 cm) on an elevated wire mesh floor to enable access to the lateral paw surface. Placement in testing cubicles was selected at random for each testing day. Briefly, calibrated von Frey hairs were applied in an alternate manner to the left or right hindpaw, starting with the 0.6 g filament. The flexible nylon hair was applied so that the fiber was bent for 3 s or until a paw-withdrawal reflex occurred. A positive withdrawal response is followed by a lower force hair and vice versa for a negative response until a change in behavior occurs. Using this up–down sequence, four subsequent hairs were assessed and the 50% paw-withdrawal threshold (PWT) was calculated according to the method described by Dixon (1980).
Paw pressure.
Noxious mechanical thresholds were examined in the hindpaws of lightly restrained alert mice via an Analgesy-Meter (7200; Ugo Basile; Randall and Selitto, 1957). The plantar surface of the hindpaw was placed on a pedestal with a probe resting on the dorsal surface. Increasing pressure was applied via the probe up to a maximum of 120 g to prevent tissue damage. The nociceptive threshold was taken as the force at which the mouse responded.
Thermal withdrawal threshold.
Thermal thresholds in unrestrained and alert mice were determined with the Hargreaves method using the plantar test (7370; Ugo Basile; Hargreaves et al., 1988). Before testing, mice were acclimatized for 1 h in individual acrylic testing cubicles (8 × 5 × 10 cm) on a glass plate. Placement in testing cubicles was selected at random for each testing day. An infrared light source of an arbitrary intensity of 30 [calibrated to elicit a paw-withdrawal latency of 10–15 s in naive mice] was directed onto the plantar surface of the hindpaw through the glass plate. The left and right paws were tested alternately, and withdrawal reflex responses were recorded for each paw in seconds on three separate occasions with at least 2 min between stimuli. Each test had a maximum latency of 23 s to prevent tissue damage.
Tail immersion withdrawal.
Thermal thresholds of the tails of lightly restrained mice were examined via the tail immersion-withdrawal test (Mogil et al., 1999). The distal third of the tail was immersed directly in water at a set temperature of either 49°C or 52 ± 0.2°C (Grant SUB14; Grant Instruments). The thermal withdrawal latency was recorded to the nearest 0.01 s as a characteristic vigorous tail reflex response. A maximum latency of 20 and 10 s was permitted at temperatures of 49°C and 52°C, respectively, to prevent tissue damage.
Hot plate.
Noxious thermal thresholds of the hindpaws were examined via the hot plate test (Eddy and Leimbach, 1953) using an incremental hot/cold plate (IITC Life Sciences) set at a temperature of 49 ± 0.2°C. Mice were placed on the hot plate in a 10-cm-diameter acrylic testing box; jumping, licking, or stamping reflex responses were recorded to the nearest 0.01 s. A maximum latency of 20 s was permitted to prevent tissue damage.
Cold plate.
Noxious cold thresholds of the hindpaws of lightly restrained mice were examined using an incremental hot/cold plate (IITC Life Sciences) set at a temperature of 10 ± 0.2°C. Each paw was tested separately by placing the plantar surface on the plate. The latency to withdraw was recorded to the nearest 0.01 s. A 20 s cutoff was implemented to prevent tissue damage.
Locomotor function.
Balance and coordination were examined via the locomotor test. Mice were placed on a RotaRod accelerating from 2 to 40 rpm over a period of 300 s (7650; Ugo Basile). Mice were initially trained before testing, and unsuccessful test runs were performed again. None remained on the apparatus after 100 s.
Neuropathic pain model.
Animals were anesthetized with 2–3% isoflurane (Abbott Animal Health), and the left hindpaw was secured, shaved, and sterilized. The sciatic nerve was carefully exposed and isolated from neighboring connective tissue via a small incision midway of the left thigh. At a site within close proximity to the trochanter, distal to the posterior biceps semitendinosus nerve branch, a 5-0 Vicryl suture (Ethicon) was inserted into the nerve and ligated so that one-third to one-half of the nerve was held tightly within the ligature, as described previously (Seltzer et al., 1990). In sham operated mice, the same procedure was performed except that the nerve was not ligated. Mechanical (von Frey) and thermal (Hargreaves) withdrawal thresholds were examined on 3–5 separate days before surgery and on days 4, 7, 11, 14, 18, and 21 after surgery.
Tissue preparation and immunohistochemistry.
On days 7 or 21 after surgery, mice were terminally anesthetized with sodium pentobarbital (0.2 g/ml, i.p.; Euthatal; Merial Animal Health) and perfused transcardially with a 0.9% saline and 0.1% heparin solution (Leo Laboratories), followed by fixation with 4% paraformaldehyde (PFA; VWR) in 0.1 m phosphate buffer. Lumbar spinal cord and sciatic nerve were dissected and postfixed for 2 h in PFA and cryoprotected in a 20% sucrose/0.1 m PBS (VWR) for a minimum of 3 d at 4°C. Subsequently, tissue was embedded in Optimum Cutting Temperature medium (VWR), snap frozen with liquid nitrogen, and stored at −80°C. Before embedding, sutures were removed from injured sciatic nerves. Transverse spinal cord sections of the L4 and L5 lumbar region and longitudinal nerve sections were cut on the cryostat at 20 μm thickness and subsequently thaw mounted onto Superfrost Plus microscope slides (VWR). After drying, 7 d post-PNL or sham surgery spinal cord sections were incubated overnight with primary antibody solution for phosphorylated p38 MAPK (p-p38; rabbit anti-p-p38; 1:100; Sigma) and visualized with extrAvidin–FITC after two stages of signal amplification with avidin–biotin complex (Vector Laboratories) and biotinyl tyramide (PerkinElmer Life and Analytical Sciences) as shown previously (Clark et al., 2006). The sections were then incubated overnight with the second primary antibody, raised against rabbit anti-Iba1 (1:1000; Wako Chemicals). Spinal cord (21 d) and sciatic nerve sections were incubated with Iba1 and rat anti-cluster of differentiation 45 (CD45; 1:500; R&D Systems). Subsequently, sections were incubated with the appropriate secondary antibody solution for 2 h (1:1000; IgG-conjugated Alexa Fluor 488 or 546; Invitrogen). All antibodies were prepared in PBS supplemented with 0.1% Triton X-100 (VWR) and 0.2% sodium azide (Sigma). Slides were coverslipped with Vectashield Mounting Medium containing DAPI (Vector Laboratories), and images were captured using a Axioplan microscope (Zeiss).
Quantification of immunoreactivity.
In spinal cord sections, analysis of p-p38 and Iba1 immunoreactivity was performed by counting the number of positive profiles within three fixed 4 × 104 μm2 boxes in the lateral, central, and medial areas of the dorsal horn, using the nuclear marker DAPI to assist in determining positive cells, as described previously (Clark et al., 2007). A mean value was obtained for both the ipsilateral and contralateral dorsal horns. In sciatic nerve sections, two boxes of 4 × 104 μm2 area were placed proximal to the nerve lesion and 1 mm from the epicenter. For analysis of contralateral nerve sections, two boxes were placed randomly along the nerve. The number of Iba1- and CD45-positive cells was counted within the boxes, and a mean value was obtained. Again DAPI was used to assist in the identification of positive cell profiles. Throughout the quantification process, a minimum of three sections per animal was assessed, and scoring was performed blind to genotype and treatment.
Bio-Gel elicited peritoneal macrophage cell culture and stimulation.
Adult GPR84 WT and KO mice were given an intraperitoneal injection (1 ml) of Bio-Gel P-100 2% polyacrylamide beads (Bio-Rad). Four days later, mice were culled via cervical dislocation, and the layer of skin covering the peritoneum was wiped with 70% ethanol. A total volume of 20 ml of sterile cold PBS (Invitrogen) containing EDTA (3 mm; Invitrogen) was injected into the peritoneal cavity using a 25 gauge needle. After gentle massaging, the buffer was retrieved in 14 ml Falcon tubes, filtered to remove the polyacrylamide beads, and spun to obtain a pellet. Cells were then resuspended and plated in cAMP-Screen Direct precoated 96-well assay plates (Invitrogen) at a density of 1.5 × 105 cells per well for the assessment of cAMP production. For mRNA analysis, peritoneal macrophages were collected in the same way as described above without previous administration of Bio-Gel beads and were plated in six-well plates at a density of 2 × 106 cells per well for RNA extraction.
Cells were maintained in DMEM with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Invitrogen, Sigma) and incubated at 37°C (5% CO2/95% O2). Two hours later, cells were washed, and the medium was replaced. Twenty four hours later, the medium was replaced with FBS-free medium for 2 h, followed by 3 h of LPS stimulation (1 μg/ml; Sigma). The culture medium of control wells was replaced with fresh FBS-free medium without subsequent stimulation.
Bone marrow-derived macrophage cell culture and stimulation.
Adult GPR84 WT and KO mice were culled via cervical dislocation. The lower body was sterilized with 70% ethanol, and the skin of both hindlimbs was removed. Muscle and fat tissue surrounding the femur, tibia, and fibula was removed carefully, and the bones were collected in ice-cold DMEM. Subsequently, the bones were flushed with 5–10 ml of cold PBS and the cells were collected, resuspended and plated in DMEM containing 10% FBS, 1% penicillin–streptomycin (Sigma) and macrophage-colony stimulating factor (Peprotech). Cultures were maintained for 1 week at 37°C (5% CO2/95% O2). After 1 week, cells were incubated with non-enzymatic cell dissociation buffer (Millipore) at 37°C for 10 min and were scraped carefully and replated in cAMP-Screen Direct precoated 96-well assay plates (Invitrogen) at a density of 1.0 × 105 cells per well to assess cAMP production. Twenty-four hours later, the medium was replaced with FBS-free medium for 2 h, followed by 3 h of LPS stimulation (1 μg/ml; Sigma). The culture medium of control wells was replaced with fresh FBS-free medium without subsequent stimulation.
RNA extraction and cDNA synthesis.
On days 7 or 21 after PNL or sham surgery, mice were terminally anesthetized with sodium pentobarbital (0.2 g/ml, i.p.; Euthatal; Merial Animal Health). The lumbar region of the spinal cord was dissected, separated into the ipsilateral and contralateral sides, and placed into separate RNase free 1.5 ml Eppendorf tubes. The ipsilateral and contralateral sciatic nerves of ∼1 cm in length were also collected and stored. The collected tissue was immediately snap frozen in liquid nitrogen and stored at −80°C for additional processing. RNA was extracted via homogenizing the tissue samples using a hybrid method of phenol extraction (TRIzol; Invitrogen) and column purification (RNeasy; Qiagen). After purification, the RNA was eluted using RNase-free water, and its concentration and purity were estimated using a NanoDrop ND-100 Spectrophotometer (Thermo Fisher Scientific). All 260:280 absorbance ratios were in the range of 1.96–2.15. During RNA isolation, samples were also treated with DNase (Qiagen) to prevent genomic contamination. RNA integrity was confirmed with a RNA 6000 Nano Chip Bioanalyzer (Agilent Technologies). cDNA was subsequently synthesized from RNA using the SuperScript II reverse transcriptase kit (Invitrogen) according to the guidelines of the manufacturer.
After LPS stimulation of cultured peritoneal macrophages, cells were homogenized by removing the media and directly adding TRIzol to the well. RNA was extracted, and cDNA was synthesized as described above.
Taqman array setup and quantitative real-time PCR.
Taqman PCR array cards were custom designed using the Applied Biosystems website (http://www.appliedbiosystems.com). The cards use micro-fluidic technology, comprising 384 wells and four sets of 96 different primer/probe pairs against specific genes within the mouse genome, including four reference genes: glyceraldehyde-3-phosphate dehydrogenase, 18S ribosomal RNA, β-actin, and hypoxanthine phosphoribosyltransferase. Spinal cord and sciatic nerve cDNA samples from 7 and 21 d post-PNL or sham surgery and control or LPS-stimulated macrophages were diluted and added in a 1:1 ratio to Taqman universal PCR master mix (Applied Biosystems; contains DNA Taq polymerase and dNTPs). DNase-free water was added to the sample mix, producing a final [cDNA] of 2 ng/μl in a total volume of 100 μl. The sample mix was loaded into the appropriate port, and the cards were centrifuged so that 1 μl was channeled into each well. Cards were then sealed and placed into a 7900 HT Fast Real-Time PCR system (Applied Biosystems), and cDNA samples underwent 40 amplification cycles. For each sample, changes in gene expression was calculated using the Delta Delta cycling time (ΔΔCT) method as described previously (Schmittgen and Livak, 2008) and normalized against four reference genes with the R package ReadPCR and NormqPCR (Perkins et al., 2012). Transcripts with undetermined values in >50% of the samples were assigned an average default CT value of 38. When this occurred in both PNL and sham or control and LPS-stimulated sample groups, no fold change (FC) value was calculated (FC = treatment/control). Transcripts that were undetermined in <50% of samples obtained an average CT value based on the remaining data values.
cAMP-screen direct chemiluminescent ELISA.
GPR84 WT and KO Bio-Gel elicited peritoneal macrophages (B-GEPMs) or bone marrow-derived macrophages (BMDMs) were harvested as described above and plated in cAMP-Screen Direct precoated 96-well assay plates (Invitrogen) at a density of 1.0–1.5 × 105 cells per well. Two hours later, the cells were washed, replenished with fresh DMEM (Invitrogen), and left overnight to settle. All cell stimulation protocols were performed 24 h later and done in FBS-free DMEM (Sigma) at 37°C. WT and KO cells were incubated for 20 min with 50 μm forskolin (FSK; R&D Systems), and control wells were incubated in fresh media containing 0.002% ethanol. Cells that underwent LPS stimulation were incubated with 1 μg/ml LPS (Sigma) for 3 h before FSK stimulation. After the stimulation procedure, the assay was terminated, and samples were processed according to the guide of the manufacturer (Invitrogen). Subsequently, the luminescence signal for each well was measured using a standard luminometer (1 s/well), and the cAMP concentration was calculated via extrapolation from a standard curve.
Data and statistical analysis.
All data were analyzed using SigmaPlot 12.3 and SigmaStat software. For single comparisons between two groups, a paired Student's t test was applied (behavioral data). For multiple comparisons, one-way (immunohistochemical data) or two-way (behavioral data) ANOVA was used, followed by Student–Newman–Keuls (SNK) post hoc test to determine individual group differences. For the Taqman mouse PCR array card data, two-sided Welch's t tests were run in the R program on the ΔΔCT values. The p values were adjusted using the false discovery rate (FDR) to correct for multiple hypothesis testing, as described previously (Benjamini et al., 2001). Nonparametric tests were applied when the data were not normally distributed, and a Kruskal–Wallis one-way ANOVA on ranks was used, followed by Tukey's method. In all cases, the data are presented as the mean ± SEM, and p < 0.05 was set as the statistical significance level.
Results
GPR84 KO mice have normal acute pain thresholds, but neuropathic pain hypersensitivity is absent
We first investigated whether GPR84 function affects acute pain, and WT and KO mice were assessed behaviorally after acute peripheral application of a range of mechanical and thermal stimuli (Fig. 1). KO mice exhibited normal thermal pain thresholds to varying levels of intensity applied to the hindpaw or tail (Fig. 1A–E) compared with WT littermates. KO mice also exhibited normal mechanical thresholds of low and high intensity (Fig. 1F,G) and showed no deficits in locomotor function (Fig. 1H).
Figure 1.

GPR84 KO mice display normal acute pain responses and locomotor ability. To assess acute nociception in naive GPR84 WT and KO mice, responses to a range of thermal and mechanical acute pain tests were compared. There were no significant differences between genotypes in the temperature threshold tests [Hargreaves (A); tail immersion withdrawal at 49°C (B) or 52°C (C); hotplate at 49°C (D); cold plate at 10°C (E)] or mechanical threshold tests [von Frey (F); paw pressure (G)]. GPR84 KO mice also displayed normal locomotor function using the RotaRod apparatus (H). Data are presented as mean ± SEM. p > 0.05, independent Student's t test, n = 8–14.
To determine whether GPR84 plays a role in chronic pain, we tested mechanical and thermal thresholds of WT and KO mice after PNL or sham surgery. It is well established that peripheral nerve injury results in the development of mechanical and thermal hypersensitivity in the ipsilateral hindpaw. Accordingly, nerve injured WT mice showed an average reduction of 68.8% in mechanical thresholds from baseline that was present on day 4 and persisted until day 21 (Fig. 2A). WT mice also exhibited an increase in thermal sensitivity, with an average reduction of 25.4% in thresholds from baseline over the 21 testing days (Fig. 2B). In contrast, nerve injured KO mice did not develop mechanical (Fig. 2C) or thermal (Fig. 2D) hypersensitivity over the 21 testing days, and thresholds did not drop from baseline or differ from KO sham controls at any point. For clarity, genotype data are presented separately, but statistical analysis was conducted on all four experimental groups. Together, these data indicate that GPR84 deletion impairs the development and persistence of neuropathic pain hypersensitivity, but under normal physiological conditions, GPR84 is not involved in acute nociception.
Figure 2.

Reduced neuropathic pain in GPR84 KO mice. Mechanical (A, C) and thermal (B, D) withdrawal responses of GPR84 WT (A, B) and KO (C, D) mice were measured before (day 0) and up to 21 d after PNL or sham surgery. WT mice developed significant mechanical hypersensitivity and showed reduced thermal thresholds compared with sham controls. Mechanical and thermal hypersensitivity were absent in KO mice and did not differ from sham values at any time. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus baseline (day 0); #p < 0.05, ##p < 0.01, ###p < 0.001 versus sham control, two-way repeated measure ANOVA with SNK post hoc test, n = 9–12.
GPR84 does not modulate microglial responses under neuropathic conditions
Since microglia play a key role in the initiation of pain-associated behaviors and GPR84 is expressed exclusively on these cells in the CNS (Bouchard et al., 2007), we examined the microglial phenotype to elucidate whether deletion of GPR84 alters nerve injury-induced microgliosis. Quantification of Iba1 immunoreactivity at 7 d after PNL revealed that both genotypes showed a 3.2-fold increase in Iba1-positive cells in the ipsilateral dorsal horn (WT PNL, 21.1 ± 1.7; KO PNL, 21.6 ± 2.4) compared with sham controls (WT, 6.6 ± 2.0; KO, 6.7 ± 0.7; Fig. 3A,B). This change persisted at 21 d after PNL for both genotypes, with a 1.8-fold increase compared with sham (WT PNL, 9.3 ± 0.7; KO PNL, 11.1 ± 0.8 vs WT, 5.3 ± 0.6; KO, 6.1 ± 0.9; Fig. 3D,E). Notably, at both time points, KO microglia exhibited typical de-ramified and amoeboid morphology in response to injury to that seen in the WT.
Figure 3.
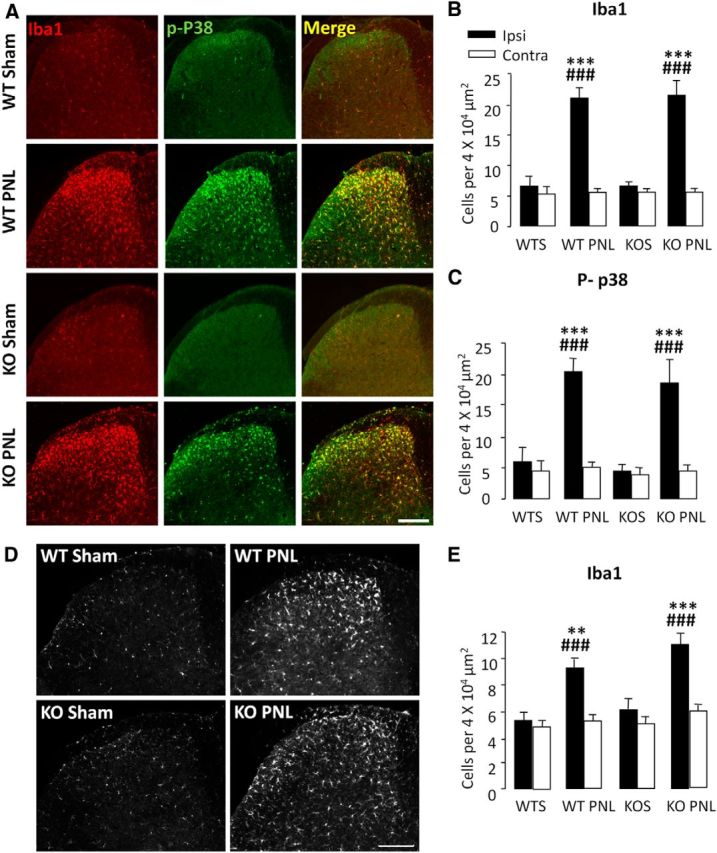
Nerve injury-induced microgliosis is normal in GPR84 KO mice. At 7 d (A–C) and 21 d (D, E) after PNL, there was a significant increase in Iba1- and p-p38-positive cells in the ipsilateral dorsal horn of GPR84 WT (WT PNL) and KO (KO PNL) mice compared with control sham groups (WTS and KOS) (A, D; quantified in B, C, E). There were no significant differences between genotypes. Data are presented as mean ± SEM. **p < 0.01, ***p < 0.001 versus contralateral; ###p < 0.001 versus sham, one-way ANOVA with SNK post hoc test, n = 4–6. Scale bar, 200 μm.
We also assessed whether GPR84 deletion affects the activation of microglial cells as revealed by p-p38 MAPK, a key kinase in nociceptive pathways (Ji and Suter, 2007; Ji et al., 2009). At day 7, WT and KO mice showed a 3.4- and 4.1-fold increase in p-p38 positive cells, respectively, in the ipsilateral dorsal horn (WT PNL, 20.4 ± 2.1; KO PNL, 18.6 ± 3.7) compared with sham controls (WT, 6.0 ± 2.3; KO, 4.5 ± 1.3; Fig. 3A,C), and there were no significant differences between genotypes. In summary, GPR84 does not play a role in modulating microglial numbers or activation via Iba1 and p-p38 markers, respectively, in response to peripheral neuropathy.
GPR84 activation promotes a proinflammatory macrophage phenotype
As we found no evidence of an altered microglia response in association with the KO behavioral phenotype, we sought to examine the possible contribution of peripheral macrophages. Macrophages are the key phagocytic immune cell of the PNS and are also reported to play a role in nerve injury-induced behavioral hypersensitivity (Myers et al., 1996; Liu et al., 2000; Barclay et al., 2007). To investigate whether deletion of GPR84 may alter the peripheral inflammatory response to nerve damage mediated by infiltrating macrophage cells, longitudinal sciatic nerve sections containing the injury site were immunostained for Iba1 and CD45 at 7 d after PNL. Positive cell profiles costained with DAPI were counted proximal to the lesion site and 1 mm from the epicenter as illustrated in Figure 4I. Quantification of immunoreactivity revealed that, in WT and KO mice, there was a 93.4 and 92.8% increase in Iba1-positive cells, respectively, compared with contralateral controls (WT ipsilateral, 43.0 ± 6.6; KO ipsilateral, 42.8 ± 1.7 vs WT contralateral, 3.1 ± 0.6; KO contralateral, 3.3 ± 0.2; Fig. 4A,B,E,F,J). Similarly, there was an equivalent increase in CD45 expression in WT (88.5%) and KO (87.0%) mice compared with contralateral controls (WT ipsilateral, 11.1 ± 3.5; KO ipsilateral, 11.0 ± 1.9 vs WT contralateral, 1.4 ± 0.4; KO contralateral 1.6 ± 0.2; Fig. 4C,D,G,H,J). This confirms that GPR84 does not play a role in regulating the infiltration of monocytes or the local proliferation and migration of resident macrophages to the injury site.
Figure 4.
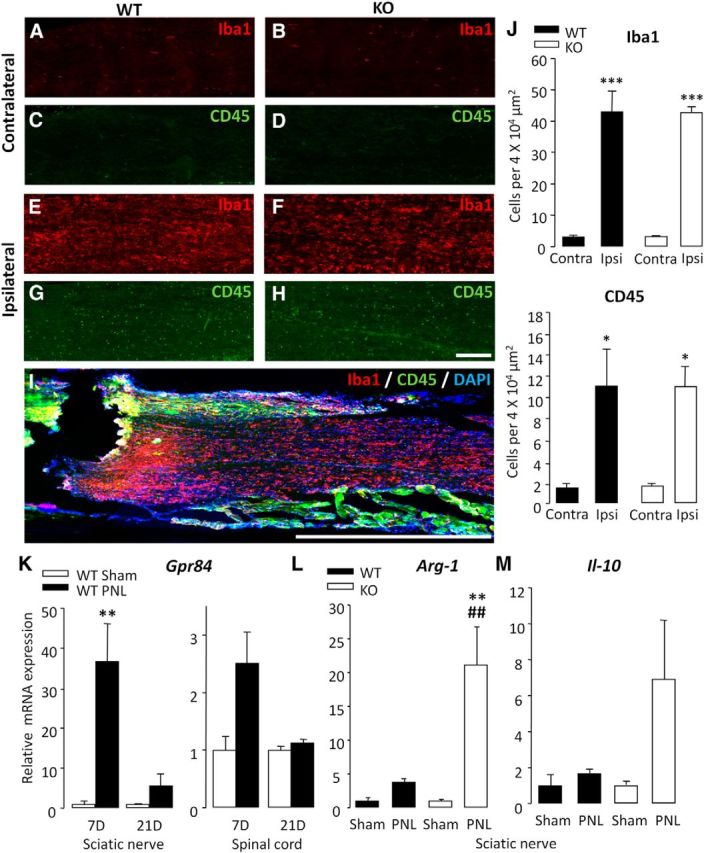
After injury, GPR84 KO mice exhibit normal peripheral macrophage recruitment but elevated anti-inflammatory marker expression in the sciatic nerve. There was a significant increase in Iba1- and CD45-positive cells in the ipsilateral (ipsi) sciatic nerve 7 d after PNL in GPR84 WT (E, G) and KO (F, H) mice compared with contralateral (contra) controls (A, C and B, D, respectively; quantified in J). There were no significant differences between genotypes. Longitudinal sections of nerve were counted, proximal to the nerve injury and 1 mm from the epicenter as illustrated in I. Scale bar, 1000 μm. Data are presented as mean ± SEM. *p < 0.05, ***p < 0.001 versus contralateral, one-way ANOVA with SNK post hoc test, n = 3. Scale bar, 200 μm. Transcriptional analysis revealed an induction of GPR84 expression in WT sciatic nerve and spinal cord at 7 and 21 d after PNL compared with sham (K). In the sciatic nerve of KO mice 7 d after PNL, there was a greater induction of Arg-1 (L) and Il-10 (M) compared with WT littermates. Changes in mRNA expression are normalized to reference genes. Data are presented as mean ± SEM. **p < 0.01 versus sham; ##p < 0.01 versus WT PNL, t test with FDR for multiple testing correction, n = 4.
We next wanted to determine whether deletion of GPR84 correlates with an altered nerve injury-induced inflammatory mediator expression. Here we used high-throughput custom-made Taqman array cards to profile the transcriptional regulation of a range of immune-related genes (cytokines, chemokines, and growth factors) in the nerve and spinal cord of WT and KO mice. We found that a number of genes were differentially regulated between the genotypes (data not shown), several of which were of particular interest to us, including arginase-1 (Arg-1), Il-10, and Gpr84 itself. Genes of interest were identified via implementing strict threshold criteria. This included an FC threshold of ≥2 so that genes with marginal expression changes as a result of noise were excluded. An FC ratio (KO FC/WT FC) of ≥1.5 was also set to omit genes that were not transcriptionally regulated by GPR84. Together, these criteria enabled the identification of genes that were considerably differentially regulated between genotypes.
In agreement with our hypothesis that GPR84 is a valid target in chronic pain, its expression was upregulated in the sciatic nerve and spinal cord of neuropathic WT mice at days 7 and 21 compared with sham controls; however, this was only significant in the nerve at day 7 (36.7 ± 9.4; Fig. 4K). Interestingly, Arg-1, a marker associated specifically with alternatively activated macrophages, was among the top upregulated genes in the sciatic nerves of KO mice 7 d after PNL compared with WT controls (WT, 3.8 ± 0.4; KO, 21.2 ± 5.6; Fig. 4L). Furthermore, the expression of the anti-inflammatory cytokine Il-10 was more greatly induced in the injured nerves of KOs than WTs (WT, 1.7 ± 0.2; KO, 6.9 ± 3.2; Fig. 4M). These results indicate that, although the number of monocytes/macrophages in the injured sciatic nerve does not differ between genotypes, it is possible that GPR84 regulates the activation state of these cells instead. In addition, GPR84 mRNA upregulation was greater in the nerve than the spinal cord of WT mice, and there were no differences in microgliosis between genotypes, suggesting that GPR84-mediated signaling may be more important in the periphery. Based on this evidence and the documented role of macrophages in models of inflammation and traumatic nerve injury-induced pain, we then tested whether GPR84 deletion impairs the ability of macrophages to launch an inflammatory response.
As before, we used custom-made Taqman array cards to analyze the relative expression of a range of mediators in peritoneal macrophages after a potent immune stimulus. LPS stimulation of cultured macrophages increases cytokine synthesis in a Toll-like receptor-4 (TLR4) dependent manner, which is a key molecule involved in generating responses to pathogen-associated molecular patterns (PAMPs). PAMPs, such as HSP60 and HSP70, necrotic cells, and components of the extracellular matrix (ECM) are abundantly found at sites of injury. These factors activate TLR signaling and nuclear factor-κB (NF-κB)-mediated transcription of many proinflammatory mediators (Nguyen et al., 2002; Willis et al., 2005). Accordingly, application of LPS to the injured sciatic nerve enhances the recruitment of macrophages and the process of Wallerian degeneration (Boivin et al., 2007), whereas deletion of TLR4 attenuates pain-associated behaviors (Tanga et al., 2005). Based on this evidence, we believe that our in vitro paradigm was a relevant representation of the in vivo situation and would enable us to examine genotypic-dependent differences in the macrophage response.
Among the 92 immune-related genes tested after LPS exposure, several proinflammatory mediators associated with pro-nociceptive signaling showed a greater upregulation in WT than KO macrophages. These were IL-1β (WT, 155.4 ± 51.3; KO, 123.8 ± 50), IL-6 (WT, 570.4 ± 67.2; KO, 198.6 ± 35.6), Tnf-α (WT, 74.5 ± 7.6; KO, 51.5 ± 7.5), prostaglandin E synthase 2 (Ptges2; WT, 408.2 ± 80.6; KO, 237.1 ± 99.4), nitric oxide synthase 2 (Nos2; WT, 85.8 ± 22.7; KO, 26.3 ± 7.5), Il-12b (WT, 81.3 ± 19.3; KO, 29.1 ± 7.9), chemokine (C-C motif) ligand 2 (Ccl2; WT, 37.9 ± 6.7; KO, 18.3 ± 3.1), and Ccl3 (WT, 101.4 ± 9.3; KO, 47.3 ± 5.7). Conversely, anti-inflammatory Il-10 was more greatly induced in the KO (WT, 27.2 ± 20.9; KO, 44.0 ± 37.1; Fig. 5A,B). LPS stimulation also increased Gpr84 expression in WT cells (3.3 ± 0.7) and was undetectable in the KO (Fig. 5A,B). These findings suggest that GPR84 regulates the release of a subset of proinflammatory mediators under neuropathic conditions. Therefore, it is plausible that the painless KO phenotype is attributable to a reduced capacity of peripheral macrophages to launch an inflammatory response.
Figure 5.

GPR84 KO macrophages exhibit an attenuated response to LPS stimulation and elevated levels of cAMP. The heat map (A) displays LPS-induced transcriptional changes of a subset of mediators in peritoneal macrophages. In response to 3 h LPS stimulation (1 μg/ml) there was a greater upregulation of a range of mediators in WT than KO cells. Transcripts of interest (Gpr84, Il-1β, Il-6, Tnf-α, Ptges2, Nos2, Il-12b, Ccl2, and Ccl3) are presented separately (B). Changes in mRNA expression are normalized to reference genes and are relative to control, which is presented as an FC (FC = LPS/control). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01 versus WT, t test, n = 4. GPR84 KO B-GEPMs and BMDMs exhibited greater FSK-induced cAMP levels than WT cells under unstimulated and LPS-stimulated (3 h; 1 μg/ml) conditions (C). Data are presented as mean ± SEM. **p < 0.01, ***p < 0.001 versus appropriate control, Kruskal-Wallis one-way ANOVA on ranks with Tukey's test, n = 4–6.
In light of our transcriptional data, we wanted to ascertain whether KO macrophages are immunosuppressed by examining the synthesis of cAMP under FSK- and LPS-stimulated conditions. Elevated intracellular cAMP has been shown to suppress innate immune functions, such as phagocytosis, microbe killing, and production of proinflammatory mediators, but promote the release of anti-inflammatory mediators (Bourne et al., 1974; Serezani et al., 2008; Peters-Golden, 2009). We demonstrated that, in two separate populations of macrophages (B-GEPMs and BMDMs), KO cells generated greater levels of intracellular cAMP than WT cells compared with control (B-GEPMs WT, 121.2 ± 6.4 fmol/ml vs KO, 342.5 ± 37.9 fmol/ml; BMDMs WT, 1960.3 ± 559.8 fmol/ml vs KO, 6995.1 ± 5225.3 fmol/ml; Fig. 5C). This intriguing greater capacity of KO macrophages to synthesize cAMP was also maintained under LPS-stimulated conditions, which is known to reduce cAMP production (BMDMs: WT, 2965.9 ± 1425.3 fmol/ml; KO, 6791.5 ± 3615.1 fmol/ml; Fig. 5C). In accordance with the literature, these findings support that GPR84 is coupled to the inhibitory Gαi/o family of G-proteins (Wang et al., 2006) and is essential for downregulating cAMP in peripheral macrophages to promote a proinflammatory response.
Discussion
To the best of our knowledge, this is the first study to demonstrate a critical role of GPR84 in experimental neuropathic pain. Consistent with this observation, GPR84 mRNA expression was increased in the sciatic nerve and spinal cord after PNL, and its deletion abolished the associated mechanical and thermal behavioral hypersensitivity in mice. Given that GPR84 is expressed selectively in immune cells (Wang et al., 2006; Bouchard et al., 2007), we studied microglia and macrophage responses to neuropathy in the presence and absence of this receptor. Interestingly, we did not detect any differences in spinal microglial responses or macrophage numbers in the sciatic nerve between genotypes. However, despite no deficit in macrophage recruitment, we observed a striking transcriptional upregulation of the anti-inflammatory macrophage marker Arg-1 and cytokine Il-10 in injured KO nerves compared with WT controls.
Arg-1 is upregulated in alternatively activated macrophages and metabolizes l-arginine into l-ornithine and urea. Due to its higher affinity for arginine, it outcompetes inducible NOS (iNOS), which metabolizes l-arginine in classically activated cells (Kreider et al., 2007). Subsequently, ornithine amino-transferase synthesizes proline from ornithine, a substrate crucial in collagen production, whilst ornithine decarboxylase generates polyamines that are involved in cellular proliferation. Together, these pathways contribute to growth, differentiation, and ECM formation. Thus, alternatively activated macrophages are involved in functions related to host defense, repair/wound healing and the dampening down of harmful inflammation. In contrast, classically activated macrophages are associated with an upregulation of iNOS and the production of NO and l-citrulline. Successive downstream pathways result in the production of reactive oxygen species, which provide host protection against bacteria, protozoa, and viruses (Mosser and Edwards, 2008). As ARG-1 and iNOS compete for the same substrate (l-arginine), the macrophage response is tightly regulated and highly dependent on external cues, such as the stimulus encountered and the microenvironment. In terms of both phenotype and function, macrophages possess remarkable heterogeneity. Thus, it is generally believed that macrophage activation encompasses a spectrum of phenotypes that are not limited to discrete subpopulations (Mosser and Edwards, 2008; Murray and Wynn, 2011).
The high expression of Gpr84 in the nerve along with the striking injury-induced upregulation of Arg-1 in the KO provided a strong rationale to examine peripheral macrophages. This was on the basis that Arg-1 is exclusively expressed by macrophages in the injured nerve (Ydens et al., 2012) and is a specific marker associated with alternative activation of these cells, as discussed above. In addition, we found no evidence of an altered microglia response in the KO and observed greater LPS-induced Gpr84 expression in WT cultured macrophages than microglia (data not shown). On closer examination, we found that KO macrophages exhibited attenuated LPS-induced expression of several well documented proinflammatory mediators, particularly Il-1β, Il-6, Tnf-α, Ptges2, Nos2, Il-12b, Ccl2, and Ccl3 (Austin and Moalem-Taylor, 2010; Kiguchi et al., 2012), while showing a greater induction of anti-inflammatory Il-10 expression. We also observed greater intracellular FSK-induced cAMP production in two populations of peripheral KO macrophages than WT. cAMP is upstream of ARG-1 activation, and elevated levels are associated with a broad range of immunosuppressive functions, including the downregulation of proinflammatory mediators and the upregulation of anti-inflammatory mediators, as well as a reduction in phagocytic activity (Bourne et al., 1974; Aronoff et al., 2005; Serezani et al., 2008; Peters-Golden, 2009; Wall et al., 2009). In light of this evidence, we postulate that, under pathological conditions, the ability of KO macrophages to release a subset of mediators in response to an inflammatory insult is compromised, resulting in diminished pain behaviors. We propose that GPR84 is a proinflammatory receptor that breaks intrinsic inhibitory mechanisms by suppressing intracellular cAMP through a Gαi/o-coupled signaling pathway. This results in the release of pro-nociceptive mediators that directly and/or indirectly contribute to the development of peripheral and central sensitization.
Considering that GPR84 mRNA in the nerve was significantly upregulated at day 7 and to a lesser extent at day 21 in correlation with IL-10 and Arg-1 expression, the persistent absence of pain behaviors suggests that it may be more important in the initiation phase of neuropathic pain. It is well documented that early transcriptional changes are critical in generating long-term changes in nervous system function. Furthermore, GPR84 is most certainly not the only molecular player in neuropathic pain, and there is substantial evidence supporting the notion of several mechanisms underlying the initiation and maintenance of central sensitization (Zimmermann, 2001; Latremoliere and Woolf, 2009).
In animal models of nerve injury, the infiltration of classically activated macrophages (iNOS+/ARG−) into the sciatic nerve has been reported as soon as day 1 (Komori et al., 2011). Treatment with anti-inflammatory cytokines, such as IL-4 or TGF-β, attenuates pain-associated behaviors via the modulation of macrophages. For example, in one study, perineural administration of TGF-β decreased macrophage infiltration, which correlated with reduced behavioral hypersensitivity (Echeverry et al., 2013). Likewise, the analgesic effects of IL-4 coincided with a shift in the macrophage phenotype as indicated by upregulated phosphorylated signal transducer and activator of transcription 6 and CD206 expression (Kiguchi et al., 2015). The beneficial effects of inhibiting classical activation while promoting alternative activation have also been demonstrated in models of post-incisional pain and experimental autoimmune neuritis (EAN). One group showed that local morphine administration into the incised sites alleviated pain attributable to increased infiltration of wound healing F4/80+ CD206+ macrophages (Godai et al., 2014). Similarly, genetic deletion of TNF-α or treatment with compound A, a plant-derived glucocorticoid receptor ligand, in EAN mice correlated with decreased severity scores and progression of mechanical allodynia. This was found to be a result of increased numbers of alternatively activated ED2+ (CD163) macrophages in the sciatic nerve (Zhang et al., 2009, 2012). In agreement with these studies, our results suggest that the absence of pain behaviors in the KO concurred with an alternatively activated macrophage phenotype. Unsurprisingly, this notion is not limited to chronic pain because classically activated macrophages have been implicated in numerous other diseases, such as obesity and diabetes (Bastard et al., 2006; Mosser and Edwards, 2008). Therefore, selective targeting of these cells poses to be a viable option for the therapeutic treatment of various human diseases.
An interesting observation from our research was the significant microgliosis in the spinal cord of injured KO mice, despite the absence of pain behaviors. GPR84 is expressed exclusively on spinal microglia, and there is an independent body of evidence supporting the contribution of these cells to chronic pain pathways (Xanthos and Sandkuhler, 2014). Nevertheless, the number of microglia in the dorsal horn did not differ in GPR84 null mice compared with WT littermates, nor could we see any difference in the degree of p-p38, known to be critical for microglial–neuron interactions under neuropathic conditions. Dissociation between microglia and their contribution to behavioral hypersensitivity in animal models of peripheral neuropathy have been documented previously (Colburn et al., 1997; Hashizume et al., 2000; Winkelstein and DeLeo, 2002; Shi et al., 2011). However, although we were unable to identify microglial involvement, our analysis was limited to gross immunophenotypic differences; thus, we cannot rule out a potential contribution of these cells. Furthermore, in contrast to our findings, a recent study demonstrated a beneficial role of GPR84 in a mouse model of Alzheimer's disease (AD). The authors showed that GPR84 KO mice exhibited accelerated cognitive decline that correlated with reduced microgliosis, hence indicating that microglial recruitment is protective in amyloid pathology (Audoy-Rémus et al., 2015). Evidence of microgliosis has been demonstrated in a number of different neurodegenerative diseases (Parkinson's disease, multiple sclerosis, and amyotrophic laterals sclerosis) including AD, as well as other types of neurological disorders (stroke, inflammation, and epilepsy). This response is generally believed to have detrimental effects, and it has not been until recently that more studies have begun to take into account both the harmful and protective effects of these multitalented cells. As a result, it remains unclear whether microglia activation is a causative (“bad for”) or reactive (“good for”) factor in disease pathology.
Currently, the signaling pathway of GPR84 is unknown. In any case, considering that its expression is highly regulated by LPS and other stimulators of the NF-κB pathway (IL-1β and TNF-α; Bouchard et al., 2007), it is plausible that it shares signaling pathways similar to that of chemokine receptors. The endogenous ligand also remains unknown, but of course, it too is likely to have an interesting biology and be important in pain processing. Some data suggest that short-chain fatty acids can activate the receptor, and these may turn out to be the endogenous ligand. Whatever the actual ligand is, we postulate that, under pathological conditions, GPR84 can be activated to evoke the release of proinflammatory mediators that contribute to pro-nociceptive signaling.
In conclusion, our findings point toward a peripherally driven role for GPR84 signaling in the context of neuropathic pain. The ability of GPR84 to induce pain hypersensitivity may provide scope for the modulation of peripheral macrophage activation and hence the alleviation of pain. We believe that the specific immune cell expression and proinflammatory profile warrants GPR84 as an exciting, new, non-neuronal target for the treatment of chronic pain.
Footnotes
This work was funded by the Biotechnology and Biological Sciences Research Council and GlaxoSmithKline. GPR84−/− mice were generously provided by GlaxoSmithKline.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174:595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- Audoy-Rémus J, Bozoyan L, Dumas A, Filali M, Lecours C, Lacroix S, Rivest S, Tremblay ME, Vallières L. GPR84 deficiency reduces microgliosis, but accelerates dendritic degeneration and cognitive decline in a mouse model of Alzheimer's disease. Brain Behav Immun. 2015;46:112–120. doi: 10.1016/j.bbi.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Barclay J, Clark AK, Ganju P, Gentry C, Patel S, Wotherspoon G, Buxton F, Song C, Ullah J, Winter J, Fox A, Bevan S, Malcangio M. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain. 2007;130:225–234. doi: 10.1016/j.pain.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Bastard JP, Maachi M, Lagathu C, Kim MJ, Caron M, Vidal H, Capeau J, Feve B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur Cytokine Netw. 2006;17:4–12. [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/S0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Boivin A, Pineau I, Barrette B, Filali M, Vallières N, Rivest S, Lacroix S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27:12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Pagé J, Bédard A, Tremblay P, Vallières L. G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditions. Glia. 2007;55:790–800. doi: 10.1002/glia.20506. [DOI] [PubMed] [Google Scholar]
- Bourne HR, Lichtenstein LM, Melmon KL, Henney CS, Weinstein Y, Shearer GM. Modulation of inflammation and immunity by cyclic AMP. Science. 1974;184:19–28. doi: 10.1126/science.184.4132.19. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Clark AK, D'Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99:868–880. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- Clark AK, Gentry C, Bradbury EJ, McMahon SB, Malcangio M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur J Pain. 2007;11:223–230. doi: 10.1016/j.ejpain.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79:163–175. doi: 10.1016/S0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Wu Y, Zhang J. Selectively reducing cytokine/chemokine expressing macrophages in injured nerves impairs the development of neuropathic pain. Exp Neurol. 2013;240:205–218. doi: 10.1016/j.expneurol.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Eddy NB, Leimbach D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther. 1953;107:385–393. [PubMed] [Google Scholar]
- Godai K, Hasegawa-Moriyama M, Kurimoto T, Saito T, Yamada T, Sato T, Kojima M, Kanmura Y. Peripheral administration of morphine attenuates postincisional pain by regulating macrophage polarization through COX-2-dependent pathway. Mol Pain. 2014;10:36. doi: 10.1186/1744-8069-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hashizume H, DeLeo JA, Colburn RW, Weinstein JN. Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine. 2000;25:1206–1217. doi: 10.1097/00007632-200005150-00003. [DOI] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Woodruff TM, Smith MT. Establishment and characterization of an optimized mouse model of multiple sclerosis-induced neuropathic pain using behavioral, pharmacologic, histologic and immunohistochemical methods. Pharmacol Biochem Behav. 2014;126:13–27. doi: 10.1016/j.pbb.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Kobayashi Y, Kishioka S. Chemokines and cytokines in neuroinflammation leading to neuropathic pain. Curr Opin Pharmacol. 2012;12:55–61. doi: 10.1016/j.coph.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Kobayashi Y, Saika F, Sakaguchi H, Maeda T, Kishioka S. Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain. 2015;156:684–693. doi: 10.1097/j.pain.0000000000000097. [DOI] [PubMed] [Google Scholar]
- Komori T, Morikawa Y, Inada T, Hisaoka T, Senba E. Site-specific subtypes of macrophages recruited after peripheral nerve injury. Neuroreport. 2011;22:911–917. doi: 10.1097/WNR.0b013e32834cd76a. [DOI] [PubMed] [Google Scholar]
- Kreider T, Anthony RM, Urban JF, Jr, Gause WC. Alternatively activated macrophages in helminth infections. Curr Opin Immunol. 2007;19:448–453. doi: 10.1016/j.coi.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, Saijo K, Glass CK, Hume DA, Kellie S, Sweet MJ. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res. 2008;4:5. doi: 10.1186/1745-7580-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, van Rooijen N, Tracey DJ. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain. 2000;86:25–32. doi: 10.1016/S0304-3959(99)00306-1. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M. Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain. 1999;80:67–82. doi: 10.1016/S0304-3959(98)00197-3. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RR, Heckman HM, Rodriguez M. Reduced hyperalgesia in nerve-injured WLD mice: relationship to nerve fiber phagocytosis, axonal degeneration, and regeneration in normal mice. Exp Neurol. 1996;141:94–101. doi: 10.1006/exnr.1996.0142. [DOI] [PubMed] [Google Scholar]
- Nagasaki H, Kondo T, Fuchigami M, Hashimoto H, Sugimura Y, Ozaki N, Arima H, Ota A, Oiso Y, Hamada Y. Inflammatory changes in adipose tissue enhance expression of GPR84, a medium-chain fatty acid receptor: TNFalpha enhances GPR84 expression in adipocytes. FEBS Lett. 2012;586:368–372. doi: 10.1016/j.febslet.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- Perkins JR, Dawes JM, McMahon SB, Bennett DL, Orengo C, Kohl M. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT-qPCR quantification cycle (Cq) data. BMC Genomics. 2012;13:296. doi: 10.1186/1471-2164-13-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M. Putting on the brakes: cyclic AMP as a multipronged controller of macrophage function. Sci Signal. 2009;2:pe37. doi: 10.1126/scisignal.275pe37. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–419. [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39:127–132. doi: 10.1165/rcmb.2008-0091TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XQ, Zekki H, Zhang J. The role of TLR2 in nerve injury-induced neuropathic pain is essentially mediated through macrophages in peripheral inflammatory response. Glia. 2011;59:231–241. doi: 10.1002/glia.21093. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Takaishi S, Nagasaki M, Onozawa Y, Iino I, Maeda H, Komai T, Oda T. Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J Biol Chem. 2013;288:10684–10691. doi: 10.1074/jbc.M112.420042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson DR, Gardiner NJ. Diabetic neuropathies: components of etiology. J Peripher Nerv Syst. 2008;13:112–121. doi: 10.1111/j.1529-8027.2008.00167.x. [DOI] [PubMed] [Google Scholar]
- Venkataraman C, Kuo F. The G-protein coupled receptor, GPR84 regulates IL-4 production by T lymphocytes in response to CD3 crosslinking. Immunol Lett. 2005;101:144–153. doi: 10.1016/j.imlet.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, Liu J, Driver A, Bao XR, Sternweis PC, Simon MI, Fraser ID. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95–p105. Sci Signal. 2009;2:ra28. doi: 10.1126/scisignal.2000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu X, Simonavicius N, Tian H, Ling L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J Biol Chem. 2006;281:34457–34464. doi: 10.1074/jbc.M608019200. [DOI] [PubMed] [Google Scholar]
- Willis D, Li KW, Zheng JQ, Chang JH, Smit AB, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–791. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkelstein BA, DeLeo JA. Nerve root injury severity differentially modulates spinal glial activation in a rat lumbar radiculopathy model: considerations for persistent pain. Brain Res. 2002;956:294–301. doi: 10.1016/S0006-8993(02)03560-6. [DOI] [PubMed] [Google Scholar]
- Wittenberger T, Schaller HC, Hellebrand S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J Mol Biol. 2001;307:799–813. doi: 10.1006/jmbi.2001.4520. [DOI] [PubMed] [Google Scholar]
- Xanthos DN, Sandkühler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- Ydens E, Cauwels A, Asselbergh B, Goethals S, Peeraer L, Lornet G, Almeida-Souza L, Van Ginderachter JA, Timmerman V, Janssens S. Acute injury in the peripheral nervous system triggers an alternative macrophage response. J Neuroinflammation. 2012;9:176. doi: 10.1186/1742-2094-9-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Hassan MY, Zheng XY, Azimullah S, Quezada HC, Amir N, Elwasila M, Mix E, Adem A, Zhu J. Attenuated EAN in TNF-alpha deficient mice is associated with an altered balance of M1/M2 macrophages. PLoS One. 2012;7:e38157. doi: 10.1371/journal.pone.0038157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang ZY, Schluesener HJ. Compound A, a plant origin ligand of glucocorticoid receptors, increases regulatory T cells and M2 macrophages to attenuate experimental autoimmune neuritis with reduced side effects. J Immunol. 2009;183:3081–3091. doi: 10.4049/jimmunol.0901088. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/S0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]