

Abstract
Background
Microarray-based comparative genomic hybridisation (array CGH) is a technique by which variation in relative copy numbers between two genomes can be analysed by competitive hybridisation to DNA microarrays. This technology has most commonly been used to detect chromosomal amplifications and deletions in cancer. Dedicated tools are needed to analyse the results of such experiments, which include appropriate visualisation, and to take into consideration the physical relation in the genome between the probes on the array.
Results
M-CGH is a MATLAB toolbox with a graphical user interface designed specifically for the analysis of array CGH experiments, with multiple approaches to ratio normalization. Specifically, the distributions of three classes of DNA copy numbers (gains, normal and losses) can be estimated using a maximum likelihood method. Amplicon boundaries are computed by either the fuzzy K-nearest neighbour method or a wavelet approach. The program also allows linking each genomic clone with the corresponding genomic information in the Ensembl database http://www.ensembl.org.
Conclusions
M-CGH, which encompasses the basic tools needed for analysing array CGH experiments, is freely available for academics http://www.uio.no/~junbaiw/mcgh, and does not require any other MATLAB toolbox.
Background
In cancer, gene amplification and deletion frequently contribute to alterations in the expression of oncogenes and tumour-suppressor genes, respectively. Thus, detection and mapping of these DNA copy number changes are important for both the basic understanding of cancer and its diagnosis [1]. Comparative genomic hybridisation to DNA microarrays (array CGH) allows efficient, genome-wide analyses of relative genome copy number in a single experiment. In array CGH [1,2], copy numbers would be related to the Cy3:Cy5 fluorescence ratios (hereafter called CGH ratios) of the microarray targets bound to each probe spot. There are some public available tools for array CGH analysis, but they either only run in Excel [3] or the software does not support the pre-processing (filtering or normalization) of array CGH data [4]. Therefore, there is a need for tools, preferably platform independent, which are capable of assessing the quality of CGH arrays as well as identifying the DNA copy number changes and link these with relevant genome information. We describe here the development of M-CGH, a MATLAB toolbox for analysing CGH ratios, which has the ability to reliably locate the copy number changes.
Implementation
M-CGH can directly load clone chromosomal location files and data export files from microarray image analysis packages, such as QuantArray or GenePix, in tab-delimited text format. Microarray experiments contain many sources of errors, i.e. human operations, array printer performance, labelling and hybridisation efficiency [5]. Therefore, reliable pre-processing of CGH ratios is essential for the later detection of copy number changes. In M-CGH, we first exclude all empty and manually flagged spots, and then filter out spots with intensities lower than background in both channels and net signal intensities below two times standard deviations of the local background intensities (figure 1a). Multiple ratio normalization functions (simple normalization, intensity dependent normalization and sub-array position normalization) are available in M-CGH, which help to correct various errors that affect CGH ratios [5]. We assess the quality of spot reproducibility by calculating the mean and standard deviation for the CGH ratios of repeated spots, and eliminate probes for which standard deviation exceed 0.2, or if the ratio measurement is based on a single spot.
Figure 1.
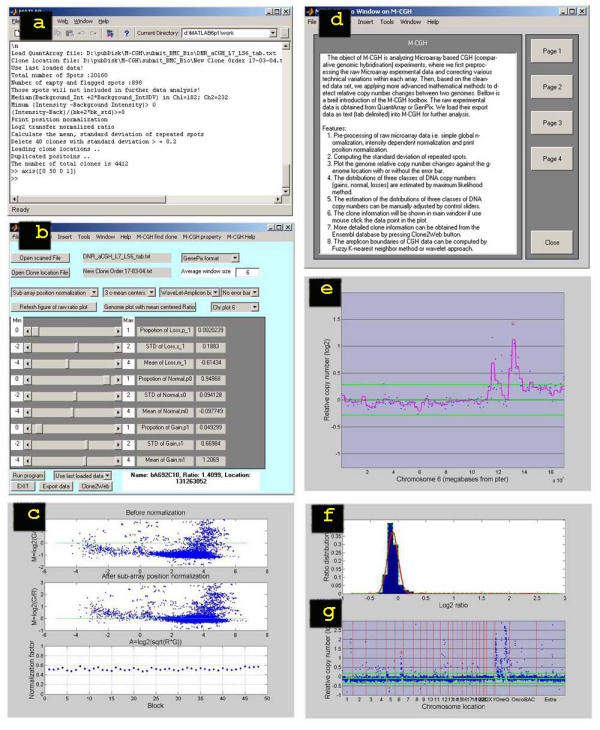
Illustrations of the M-CGH user interface, 1a) Summary information of each array CGH experiment is listed in the MATLAB command window; 1b) M-CGH main window, showing the parameters used, allowing manual adjustments, and spot information with a link to the Ensembl database (Clone2Web); 1c) Sub-array position normalization of CGH ratios, where the lower panel show the normalization factor used in each sub-array, indicating lack of discrepancies in this case; 1d) M-CGH help documentation; 1e) An interactive plot of log2 CGH ratios as a function of their relative chromosomal locations, with estimated amplicon boundaries (pink smooth line); 1f) An interactive histogram plot of CGH ratios, with fitted Gaussian distributions (red smooth line); 1g) An interactive plot of log2 CGH ratios as a function of their genome location, showing all chromosomes in numerical sequence, delimited by red lines.
Following this analysis, we use data from within each experiment to determine the upper and lower threshold for scoring amplified and deleted chromosomal segments. This is not trivial, since some aneuploid samples may have many copy number deviations, complicating the determination of the "normal" ratio. In particular, dedicated arrays may be enriched for genes that are targets of amplification or deletion, thus making normalization difficult. We accomplish this by using a maximum likelihood method to fit a mixture of three Gaussian distributions (representing amplifications, normal and deletion) to a histogram of normalized log2 CGH ratios from the array CGH analysis [3]. The fitted distribution is
![]()
where Pi, μi and σi are the relative proportion, mean and standard deviation of the ith distribution respectively (figure 1b) and 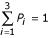 . CGH ratios representing normal DNA copy numbers are centered at a mean closest to zero, decreased copy numbers less than zero, and increased copy numbers greater than zero. We obtain the fit by first applying the Expectation Maximization (EM) algorithm [6] to estimate Pi, μi and σi for each Gaussian distribution, and then plotting an interactive histogram (figure 1f) of normalized log2 CGH ratios, with initial fitting of the three Gaussian distributions. The interactive histogram provides visual feedback with a smooth fit line superimposed on the observed data histogram. Users can assess the quality of the initial fitting, and manually adjust the control sliders of Pi, μi and σi in the M-CGH main window (figure 1b) until the sum of the three Gaussian distributions fits the histogram of CGH ratios (as in figure 1f, a solid red line shows the result of the fit of the sum of the three distributions). After a fit is obtained, we use the distribution fitted to the part of the histogram representing normal DNA copy number to determine the 3σ upper and lower thresholds for determining amplifications and deletions (as in figure 1g, 3σ upper and lower thresholds are represented by two solid green lines). Array CGH experiments in which the central distribution has a σ greater than 0.2 will be considered unreliable, and may be eliminated from further studies. M-CGH also provides an interactive plot of log2 CGH ratios as a function of its location in the genome (figure 1g) or chromosome (figure 1e). When a data point is clicked on, the CGH ratio, clone name and location will be displayed in the main window (figure 1b). Detailed clone information, such as gene content, can be obtained from the Ensembl database by pressing the Clone2Web button (figure 1b).
. CGH ratios representing normal DNA copy numbers are centered at a mean closest to zero, decreased copy numbers less than zero, and increased copy numbers greater than zero. We obtain the fit by first applying the Expectation Maximization (EM) algorithm [6] to estimate Pi, μi and σi for each Gaussian distribution, and then plotting an interactive histogram (figure 1f) of normalized log2 CGH ratios, with initial fitting of the three Gaussian distributions. The interactive histogram provides visual feedback with a smooth fit line superimposed on the observed data histogram. Users can assess the quality of the initial fitting, and manually adjust the control sliders of Pi, μi and σi in the M-CGH main window (figure 1b) until the sum of the three Gaussian distributions fits the histogram of CGH ratios (as in figure 1f, a solid red line shows the result of the fit of the sum of the three distributions). After a fit is obtained, we use the distribution fitted to the part of the histogram representing normal DNA copy number to determine the 3σ upper and lower thresholds for determining amplifications and deletions (as in figure 1g, 3σ upper and lower thresholds are represented by two solid green lines). Array CGH experiments in which the central distribution has a σ greater than 0.2 will be considered unreliable, and may be eliminated from further studies. M-CGH also provides an interactive plot of log2 CGH ratios as a function of its location in the genome (figure 1g) or chromosome (figure 1e). When a data point is clicked on, the CGH ratio, clone name and location will be displayed in the main window (figure 1b). Detailed clone information, such as gene content, can be obtained from the Ensembl database by pressing the Clone2Web button (figure 1b).
Fuzzy K-nearest neighbour method and wavelet approach
For computing the amplicon boundaries from the CGH array analysis, we first use the nearest neighbour interpolation to smooth observed CGH ratios (users may modify the window size for the interpolation, figure 1b). Then we apply the fuzzy K-nearest neighbour method [7] to classify the data points into three classes (gains, normal and losses) based on estimated means μi of the three Gaussian distributions, and the fuzzy membership values will represent the level of copy number changes. Alternatively, we may use the wavelet approach [8] to estimate the amplicon boundaries (as in figure 1e, the pink smooth line is the amplicon boundaries computed by wavelet approach).
Results
The performance of M-CGH was tested on a genomic microarray containing approximately 4000 unique elements (BACs and PACs). The array included a representation of the human genome at 1 Mb resolution, as well as the tiling path of a segment of chromosome 1 (1q12-q25) and 600 genomic clones containing known oncogenes and tumour-suppressor genes. A panel of human sarcomas, malignant tumours of mesenchymal origin, was used to assess the performance of the software (Meza-Zepeda, Kresse, Wang, Myklebost et al., unpublished). Total genomic DNA from approximately 20 tumours was labelled by random priming using Cy3-dCTP, in parallel normal reference DNA was labelled using Cy5-dCTP. Labelled tumour and reference DNA was competitively hybridised to the genome representation present on the array. Hybridisation was performed using an automated hybridisation station, GeneTAC (Genomic Solutions/Perkin Elmer), agitating the hybridisation solution for 48 hours at 37 °C. After hybridisation, slides were washed and scanned using an Agilent G2565BA scanner (Agilent Technologies). Images were analysed using GenePix Pro 4.1 (Axon Laboratories) or QuantArray 3.0 (Packard Biosciences). The spots were automatically segmented and manually adjusted where necessary. Export files were generated, transformed to tab delimited format and imported to M-CGH for further analysis. Figure 1 shows a sarcoma sample analysed using M-CGH.
Conclusions
Overall, we have presented a package for analyzing array CGH experiments, interactive data analysis with a user friendly graphical interface is available, and an on-line genomic information database (Ensembl) is linked. M-CGH is entirely platform independent and only requires MATLAB installed (the student version will suffice).
M-CGH states the number of spots that pass the quality control, allows all plots to be saved as image files, and further export of the filtered dataset, with normalized ratios and clone location, as a text file. Our future development of M-CGH will include applying visualization methods capable of assessing the DNA copy number changes of multiple CGH arrays simultaneously, filtering of misbehaving clones, implementing change-point analysis techniques such as those described by Lucito et al. [9], i.e. representational oligonucleotide microarray analysis, and developing a web tool so that it can be run without MATLAB software.
Availability and requirements
• Project name: M-CGH: Analysing microarray-based CGH experiments
• Project home page: http://www.mikromatrise.no/arraycgh/index.html
• Operating system(s): Platform independent
• Programming language: MATLAB
• Other requirements: MATLAB Version 6.1
• License: GNU
• Any restrictions to use by non-academics: Please inform the corresponding author if you are a non-academic user
Authors' contributions
JBW designed and developed software and drafted manuscript. LAMZ attended part of design and tested program, and drafted part of manuscript. SHK provided experiment dataset and tested software. OM conceived of the study, participated in its design and coordination.
Acknowledgments
Acknowledgements
We thank one anonymous reviewer for constructive comments on the manuscript. This work was supported by the Norwegian Cancer Society http://www.kreft.no, the FUGE (functional genomics) program http://www.fuge.no, and the University of Oslo (EMBIO). The arrays were produced by the Norwegian Microarray Consortium.
Contributor Information
Junbai Wang, Email: junbaiw@radium.uio.no.
Leonardo A Meza-Zepeda, Email: leonardm@radium.uio.no.
Stine H Kresse, Email: stinek@radium.uio.no.
Ola Myklebost, Email: olam@radium.uio.no.
References
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional programs of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson G, Hager JH, Volik S, Hariono S, Wernick M, Moore D, Albertson DG, Pinkel D, Collins C, Hanahan D, Gray JW. Genome scanning with array CGH delineates regional alterations in mouse islet carcinomas. Nat Genet. 2001;29:459–464. doi: 10.1038/ng771. [DOI] [PubMed] [Google Scholar]
- Autio R, Hautaniemi S, Kauraniemi P, Yli-Harja O, Astola J, Wolf M, Kallioniemi A. CGH-Plotter: MATLAB toolbox for CGH-data analysis. Bioinformatics. 2003;19:1714–1715. doi: 10.1093/bioinformatics/btg230. [DOI] [PubMed] [Google Scholar]
- Churchill GA. Fundamentals of experimental design for cDNA microarray. Nat Genet Sup. 2002;32:490–495. doi: 10.1038/ng1031. [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J Roy Stat Soc. 1977;39:1–38. [Google Scholar]
- Keller JM, Gray MR, Givens JA., JR A fuzzy k-nearest neighbour algorithm. IEEE SMC. 1985;15:580–585. [Google Scholar]
- Mallat S. A wavelet tour of signal processing. 2. San Diego: Academic Press; 1999. [Google Scholar]
- Lucito R, Healy J, Alexander J, Reiner A, Esposito D, Chi M, Rodgers L, Brady A, Sebat J, Troge J, West AJ, Rostan S, Nguyen KCQ, Powers S, Ye QK, Olshen A, Venkatraman E, Norton L, Wigler M. Representational Oligonucleotide Microarray Analysis: A High-Resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–2305. doi: 10.1101/gr.1349003. [DOI] [PMC free article] [PubMed] [Google Scholar]