

Abstract
The p38 mitogen-activated protein kinase (MAPK) signaling pathway can be activated in response to a wide range of extracellular signals. As a consequence, it can generate many different biological effects that depend on the stimulus and on the activated cell type. Therefore, this pathway has been found to regulate many aspects of tissue development and homeostasis. Recent work with the aid of genetically modified mice has highlighted the physiological functions of this pathway in skeletogenesis and postnatal bone maintenance. In this review, emphasis is given to the roles of the p38 MAPK pathway in chondrocyte, osteoblast and osteoclast biology. In particular, we describe the molecular mechanisms of p38 MAPK activation and downstream targets. The requirement of this pathway in physiological bone development and homeostasis is demonstrated by the ability of p38 MAPK to regulate master transcription factors controlling geneses and functions of chondrocytes, osteoblasts and osteoclasts.
Introduction
Skeletal development starts from condensation of mesenchymal progenitor cells at the sites of future skeletal elements, and early patterning events determine where and when mesenchymal cells condense and differentiate.1,2 A number of craniofacial bones and the lateral part of clavicles are formed through intramembranous ossification during which mesenchymal cells directly differentiate into bone-forming osteoblasts. In contrast, the axial and appendicular skeletons develop by endochondral ossification during which mesenchymal progenitors differentiate into chondrocytes to produce a cartilaginous template.1,2 In this so-called anlagen, chondrocytes undergo a program of proliferation and progressive maturation, terminating with hypertrophic differentiation. Hypertrophic chondrocytes produce a calcified matrix and then undergo apoptosis, attracting blood vessels, osteoclasts and osteoblasts from the perichondrium to remodel the cartilaginous template into bone.1,2 After completing its formation, bone is maintained throughout life by continuous remodeling, which is critical for its structural, mechanical and metabolic functions.3 This active process is controlled by osteoclasts that are derived from hematopoietic progenitors and resorb bone, by osteoblasts that replace resorbed structures by an equivalent amount of new bone, and by osteocytes that are matrix-embedded terminally differentiated osteoblasts functioning as mechanical and metabolic sensors.3 A growing body of evidence shows that the p38 mitogen-activated protein kinase (MAPK) signaling pathway regulates many aspects of bone development and maintenance. Here, we review the functions of this signaling pathway in chondrocytes, osteoblasts and osteoclasts.
Overview of the p38 MAPK Signaling Pathway
MAPKs are evolutionarily conserved enzymes that convert extracellular stimuli into a wide range of cellular responses. Extracellular signal-regulated kinases (ERK), p38 MAPK, Jun N-terminal kinases (JNK) and ERK5 represent the four groups of the MAPK subfamily.4,5 Among them, the p38 MAPK group is composed of four members encoded by their respective gene: p38α (encoded by MAPK14), p38β (encoded by MAPK11), p38γ (encoded by MAPK12) and p38δ (encoded by MAPK13).4,5 p38α and p38β are closely related proteins that are expressed in most cell types and have distinct and overlapping functions. In contrast, p38γ and p38δ show very selective tissue distribution and are likely to have specialized functions.4,5 The p38 MAPK have key roles in tissue development and homeostasis, as well as in neoplastic transformation and inflammation, as they control cell proliferation, differentiation, apoptosis, senescence and cytokine production.4,5
All p38 members share a similar activation loop sequence, i.e., threonine–glycine–tyrosine, which is targeted for dual phosphorylation by upstream MAPK kinases (MKK). MKK3 only activates p38α, p38γ and p38δ, whereas MKK6 activates all p38 members.4,5 In addition, MKK4, an upstream activator of JNK, can also activate p38α or p38δ in specific cell types. Finally, p38 MAPK can also be activated by an MKK-independent mechanism achieved by autophosphorylation of p38 after interaction with transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1)-binding protein 1.4,5 Several MAPK kinase kinases (MAP3K) can activate MKK3 and MKK6, including apoptosis signal-regulating kinase 1, mixed-lineage kinase 3 (MLK3), transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1), dual-leucine-zipper-bearing kinase 1 and MAP3K4.4,5 This diversity of MAP3K that can activate MKK3 and MKK6 provides the ability to p38 MAPK to transduce signals from a large variety of extracellular stimuli. p38 MAPK signaling can be inactivated by both generic phosphatases and dual-specificity phosphatases.4,5 Although p38 MAPK activity is mainly controlled by phosphorylation–dephosphorylation, it can also be regulated by changes in expression levels of p38 pathway components, scaffolding proteins and autophagy.5 The substrates of p38 MAPK are either transcription factors such as activating transcription factor 2 (ATF2), ELK1, myocyte-specific enhancer 2 (MEF2), CCAAT/enhancer-binding protein β (C/EBPβ) and C/EBP homologous protein, or other protein kinases including MAPK-activated protein kinases 2 and 3 (MAPKAPK2/3), mitogen- and stress-activated protein kinase 1 and MAPK-interacting kinases 1 and 2.4,5
The p38 MAPK Signaling Pathway in Chondrocytes
Role of p38 MAPK in chondrocyte differentiation
A role of p38 MAPK in chondrogenesis has been first documented with the use of different cell culture systems and selective p38 MAPK inhibitors. The p38 MAPK phosphorylation and activity are increased during the course of the chondrogenic program, and inhibition of p38 blocks chondrocyte differentiation and cartilage nodule formation,6 suggesting that this signaling pathway has an important role in chondrogenesis. Moreover, p38 MAPK has been demonstrated to be activated in response to and to mediate the effects of several growth factors essential for chondrocyte differentiation and function, including TGF-β, growth/differentiation factor 5 (GDF5), bone morphogenetic proteins (BMP) and connective tissue growth factor7,8,9,10 (Figure 1). Indeed, the p38 MAPK pathway is involved in the regulation of collagen 2 and aggrecan expressions, proteoglycan synthesis and cartilage nodule formation in response to those growth factors in vitro.7,8,9 Notably, p38 signaling has been shown to positively regulate hypertrophic chondrocyte differentiation in a micromass system of mesenchymal cells recapitulating the entire chondrogenic program.11 In this context, treatment of micromass cultures with p38 inhibitors results in a delay of hypertrophic differentiation as evidenced by reduced expressions of hypertrophic marker genes (collagen 10, tissue nonspecific alkaline phosphatase, bone sialoprotein and matrix metalloproteinase 13) and impaired mineralization.11 Consistent with this finding, parathyroid hormone (PTH) and its related peptide, known to inhibit chondrocyte differentiation toward hypertrophy, have been shown to exert this negative effect by decreasing p38 MAPK activity through protein kinase C (PKC).12 In addition, chondrocytes undergoing terminal differentiation inexorably progress toward apoptosis, and elevation of reactive oxygen species (ROS) level is probably a major mechanism governing this process.13 It is now well recognized that those by-products of cellular oxidative metabolism can activate certain signaling pathways. Indeed, increased ROS level during chondrocyte differentiation has been found to activate ERK and p38 MAPK, resulting in chondrocyte hypertrophy13 (Figure 1).
Figure 1.
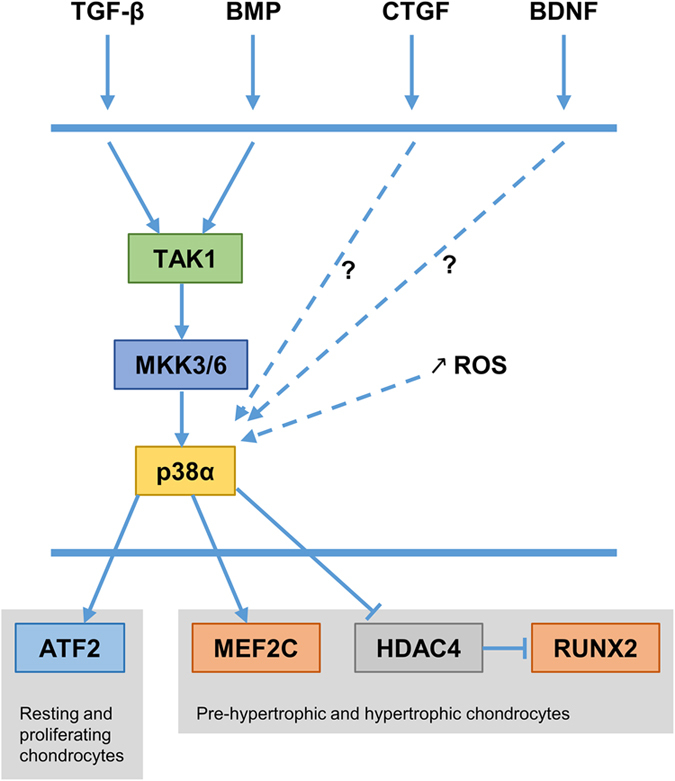
p38 MAPK signaling in chondrocytes. p38α is the predominant p38 member in chondrocytes. TGF-β and BMP activate p38α signaling through TAK1. CTGF and BNDF activate p38α signaling through unknown mechanisms. During terminal differentiation of chondrocytes, the level of reactive oxygen species (ROS) increases and induces p38α activation. p38α regulates ATF2 activity in resting/proliferating chondrocytes. Moreover, it regulates MEF2C, HDAC4 and RUNX2 in pre-hypertrophic and hypertrophic chondrocytes.
Furthermore, the brain-derived neurotrophic factor and its receptor TRKB, which regulate neuronal differentiation and survival and maintain synapse plasticity in the hippocampus and hypothalamus, have been shown to be expressed in non-neuronal cells, including growth plate chondrocytes.14 Here, their interaction promotes hypertrophic differentiation through increased p38 MAPK activity14 (Figure 1). To confirm the in vivo significance of these observations, the author has generated mice with targeted disruptions of TrkB or p38alpha in chondrocytes by using the col2-cre mouse strain.15 Both mutant mice exhibit a similar degree of dwarfism with reduced growth-plate width and delayed hypertrophic differentiation as evidenced by altered expressions of Sox9 and Runx2, two crucial transcription factors required for skeletogenesis.15 p38α is the predominant p38 member in chondrocytes. It is localized throughout the growth-plate cartilage, but its expression is the greatest in the pre-hypertrophic zone, which is coherent with its proposed role in hypertrophic chondrocyte differentiation.15 Interestingly, p38α phosphorylation has been found to be decreased in mice lacking TrkB in chondrocytes, further indicating that this pathway acts downstream of this receptor.15 Although both mutant mice show diminished hypertrophic zone width, p38alpha mutant mice also exhibit narrowing and disorganization of the proliferating chondrocyte columns in growth plates, suggesting that this pathway acts downstream of other ligand/receptor interactions and also regulates other aspects of chondrocyte biology than the transition toward hypertrophy.15
Upstream Activators of p38 MAPK in Chondrocytes
TGF-β superfamily members such as TGF-β, BMP and GDF5 are critical regulators of chondrogenesis and exert their actions through canonical SMAD signaling and non-canonical MAPK signaling. TAK1 is a MAP3K that directs TGF-β- and BMP-induced signaling to MAPK activation16,17 (Figure 1). Its in vivo function in cartilage development has been recently discovered by two independent groups with the use of chondrocyte-specific Tak1 knockout mice.16,17 TAK1 expression mainly occurs in pre-hypertrophic and hypertrophic chondrocytes during endochondral bone development.16,17 Tak1 mutant mice display severe chondrodysplasia with dwarfism (shorter axial and appendicular skeleton), impaired formation of secondary ossification centers and joint abnormalities. Deletion of Tak1 in chondrocytes results in decreased chondrocyte proliferation, disorganized proliferating zone in growth plates and increased hypertrophic chondrocyte apoptosis.16,17 Alteration of chondrocyte proliferation results from reduced expression of Ihh in pre-hypertrophic chondrocytes.16,17 Only one group has observed a delayed chondrocyte hypertrophic differentiation in the absence of TAK1.17 BMP signaling has been found to be impaired in Tak1-deficient chondrocytes, as shown by reduced phosphorylation of SMAD1/5/8 and p38 MAPK.16 Therefore, it is likely that decreased p38 activity accounts for some of the skeletal abnormalities caused by Tak1 ablation in chondrocytes.
Targets of p38 MAPK in chondrocytes
Several transcription factors that are known to be regulated by p38 MAPK are expressed in chondrocytes. For instance, p38 has been found to regulate the transcriptional activity of SOX9, the master transcription factor controlling chondrocyte differentiation.18 Moreover, ATF2 is expressed in resting and proliferating chondrocytes during endochondral bone development. Atf2-deficient mice display a uniform dwarfism due to reduced chondrocyte proliferation, disorganized chondrocyte columns and impaired endochondral ossification.19 Consistent with these observations, ATF2 regulates chondrocyte proliferation by modulating cyclin D1 levels and by inhibiting hypertrophic differentiation.20 As ATF2 is a direct target of p38 in response to TGF-β in chondrocytes,21 it is conceivable that p38 regulates proliferating chondrocytes through this transcription factor (Figure 1). On the other hand, p38 MAPK has also been found to favor hypertrophic chondrocyte differentiation both in vitro and in vivo,11,12,15 and this, in part, by stimulating the transcriptional activity of MEF2C11,22 (Figure 1). Further, it has been shown that p38 MAPK promotes histone deacetylase 4 (HDAC4) degradation and subsequent release of RUNX2 from the repressive influence of HDAC4 resulting in chondrocyte hypertrophy23 (Figure 1). Therefore, it is possible that p38 signaling can exert distinct functions, depending on the developmental stage of chondrocytes and particularly on targeted transcription factors. This notion is further supported with the patterns of expression of ATF2, MEF2C and RUNX2, with ATF2 restricted to resting/proliferating chondrocytes19 and MEF2C and RUNX2 to pre-hypertrophic/hypertrophic chondrocytes.22,23
The p38 MAPK Signaling Pathway in Osteoblasts
Role of p38 MAPK in osteoblast differentiation and function
Previous in vitro investigations using cell lines or primary osteoblasts with selective p38 inhibitors have shown that p38 MAPK regulates osteoblast differentiation, extracellular matrix deposition and mineralization in response to different osteogenic ligands such as TGF-β,24 BMP2,24,25,26 Wnt proteins,27,28 PTH29,30 and epinephrine31 (Figure 2). p38 signaling regulates the commitment of mesenchymal cell lines to the osteoblast lineage in response to the canonical Wnt3a27 or the non-canonical Wnt4,28 indicating that this pathway is integrated in Wnt signaling in osteoblasts. In addition, p38 MAPK has been found to cooperate with the canonical Wnt/β–catenin pathway in response to Wnt3a, by enhancing β-catenin transcriptional activity.27 p38 is activated by BMP in osteoblasts, and, in this situation, cooperates with the canonical SMAD signaling by favoring SMAD1 phosphorylation and its translocation to the nucleus.26 Moreover, PTH can activate p38 MAPK through a protein kinase A (PKA)-dependent mechanism in osteoblasts.29 In this context, p38 has been shown to be required for PTH-induced osteoblast differentiation. β-arrestin signaling, another component of PTH signaling with PKA and PKC, has been found to modulate PTH-induced p38 target gene expression.30
Figure 2.
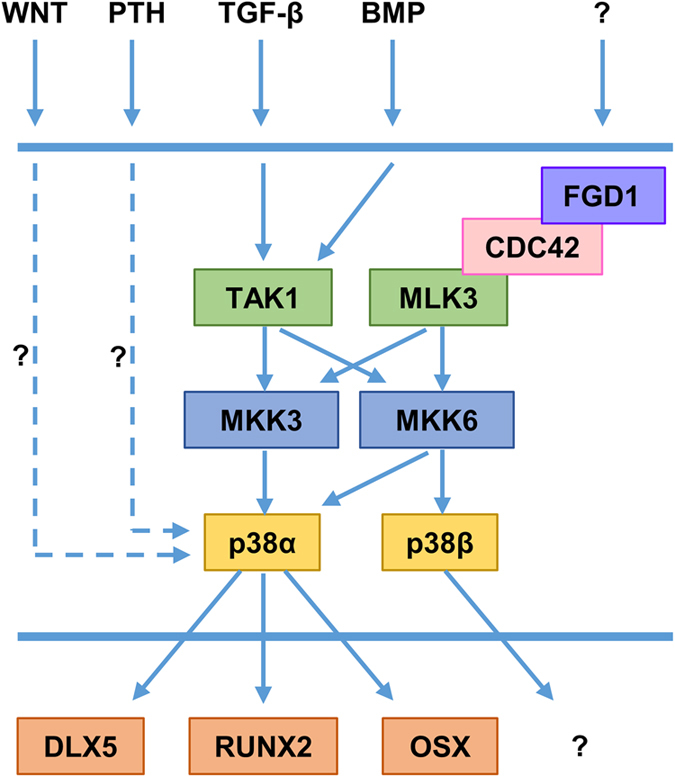
p38 MAPK signaling in osteoblasts. p38α regulates osteoblast differentiation and functions during bone development and adult bone homeostasis, whereas p38β regulates late osteoblast differentiation during bone development. TGF-β and BMP activate p38 signaling through TAK1-MKK3/6. PTH and WNT proteins activate p38 signaling through unknown mechanisms. MLK3 acting downstream of FGD1/CDC42 activates p38 signaling, but signals activating FGD1/CDC42 are currently unknown. MKK6 can activate both p38α and p38β, whereas MKK3 can activate only p38α. p38α positively regulates the transcriptional activity of DLX5, RUNX2 and OSX. Transcription factors regulated by p38β remain to be identified.
The in vivo function of p38 MAPK kinase signaling has been recently highlighted with different genetically modified murine models (also reviewed in Caverzasio and Thouverey32; Greenblatt et al.33). Three-week-old Mkk3−/− and Mkk3−/−;Mkk6+/− mice display decreased mineralization of parietal and frontal bones, reduced trabecular bone volume and cortical thickness in long bones and impaired mineralization of secondary centers of ossification.34 In comparison, Mkk6−/− mice show a low bone mass in limbs but no alteration of calvarial mineralization, indicating that those two MKKs exert anatomically selective roles.34 Consistent with these findings and the selectivity of MKK3 and MKK6 for the four p38 members (with MKK6 activating all four members and MKK3 activating only p38α, γ and δ; Figure 2), 3-week-old p38beta-deficient mice only exhibit a low bone mass phenotype in long bones, whereas injections of cre-expressing lentivirus over the calvarium of p38alpha-floxed mice retard calvarial mineralization.34 Those data further support the notion that p38α and p38β functions depend on the anatomical location and the ossification mechanism (endochondral or intramembranous) during bone development. In addition, these results also reveal that loss of p38β is not compensated by p38α and thus that p38α and p38β exert non-redundant functions. This has been confirmed by in vitro analyses demonstrating that p38α regulates early osteoblast differentiation, whereas p38β controls late osteoblast maturation.34
The p38α MAPK represents the most abundant p38 member in bone, and its specific individual contribution in bone development and postnatal maintenance has been elucidated by using the Cre-LoxP technology.35,36 Mice in which p38alpha is deleted under the control of the osterix (Osx) promoter exhibit impaired mineralization of frontal and parietal bones of the calvaria, delayed posterior fontanel ossification, and abnormal development of maxilla and mandible at birth, and decreased calvarial thickness, reduced trabecular bone volume and cortical thickness in long bones at 12 weeks of age,35 which is in accordance with abnormal skeletal development of Mkk3−/− and Mkk3−/−;Mkk6+/− mice.34 Lack of p38α in pre-osteoblasts results in defective osteoblast differentiation, as evidenced by reduced expressions of Osx (encoding another master transcription factor regulating osteoblast differentiation), collagen 1, alkaline phosphatase, bone sialoprotein and osteocalcin.35 Mice lacking p38α in mature osteoblasts (disruption under the control of the osteocalcin promoter) do not display bone alterations until 5 weeks of age, but then demonstrate a progressive decrease in bone mineral density and trabecular and cortical bone mass in both axial and appendicular skeletons.36 Those abnormalities correlate with defective osteoblast function and impaired endosteal and trabecular bone formation.36 Those data demonstrate the critical role of p38 MAPK in postnatal bone acquisition and remodeling, which has been also confirmed by the inducible ablation of p38alpha in pre-osteoblasts of adult mice.35 In contrast, 4-month-old Mkk6−/− mice, in which p38β cannot be activated, have normal osteoblast number and bone formation,37 indicating that p38β does not have a significant role in adult bone homeostasis. Interestingly, absence of p38α in mature osteoblasts completely inhibits the bone anabolic effect of intermittent PTH treatment in mice, further supporting that p38α is an important component of PTH signaling in osteoblasts.38 Disruption of either p38α or p38β MAPK signaling does not affect expressions of receptor activator of nuclear factor kappa-B ligand (Rankl) and osteoprotegerin, or bone resorption in physiological conditions.34,35,36 Moreover, p38γ expression is very low in osteoblasts, whereas p38δ is expressed at the same level as p38α;35,36 however, its role in osteoblast biology remains to be elucidated.
Upstream activators of p38 MAPK in osteoblasts
Tak1 is expressed in bone-lining osteoblasts and in matrix-embedded osteocytes.34 In osteoblasts, TAK1 has been shown to trigger p38 activation and only modest effects on ERK and JNK downstream of either BMP or TGF-β stimulation34 (Figure 2). Mice with an Osx-cre-mediated excision of Tak1 in pre-osteoblasts display clavicular hypoplasia, hypomineralization of the calvarium and delayed fontanel fusion, a phenotype resembling that of Runx2+/− mice and humans with cleidocranial dysplasia.34 Those observations clearly indicate that TAK1 signaling is required for adequate intramembranous ossification. In addition to the clavicular and calvarial phenotypes, Tak1 mutant mice also exhibit reduced cortical and trabecular bone mass in limbs, and decreased mineralization of secondary ossification centers.34 This global phenotype resembles that of Mkk3−/−;Mkk6+/−34 and Osx-cre;p38alphaf/f mutant mice,35 except that the clavicular hypoplasia is not seen in those mice, suggesting that another signaling pathway contributes to clavicle development. Pre-osteoblasts lacking TAK1 fail to differentiate into mature osteoblasts and have defective function.34
Another MAP3K, MLK3, has been found to act downstream of faciogenital dysplasia protein 1 (FGD1) and has been identified as a physiologic regulator of osteoblastogenesis and bone development by modulating ERK and p38 MAPK activity39 (Figure 2). Mlk3−/− mutant mice show dental abnormalities, defective mineralization of the calvarium and osteopenia, an equivalent skeletal phenotype in comparison with that of patients with mutations in FGD1. This skeletal phenotype is associated with a significant decrease in bone formation and reduced expressions of osteoblast maker genes such as collagen 1 and osteocalcin.39 In the absence of MLK3, the defective osteoblast differentiation is associated with impaired activation of ERK and p38 MAPK, and reduced RUNX2 phosphorylation.39
Targets of p38 MAPK in osteoblasts
The similar cleidocranial dysplasia and osteopenia observed in mice lacking TAK1 in pre-osteoblasts and Runx2+/− mice have suggested that signaling pathways downstream of TAK1 may regulate RUNX2 activity.34 Then, p38 MAPK has been identified as a positive regulator of osteoblast differentiation and function, in part by regulating the transcriptional activity of RUNX234 (Figure 2). In fact, p38 can phosphorylate RUNX2 on Ser28, Ser31, Ser244, Ser301, Ser319 and Ser472, and thus increases its transcriptional activity by promoting its association with the transcriptional co-activator CREB-binding protein.34 DLX5 is a member of the distal-less homeobox transcription factor family. Its expression is induced by canonical SMAD signaling downstream of BMP stimulation in osteoblasts.40 In turn, p38 MAPK phosphorylates DLX5 and therefore increases its transcriptional activity by promoting its association with the transcriptional co-activator p30040 (Figure 2). DLX5 directly stimulates osteoblast differentiation by inducing Osx expression. Dlx5 knockout mice display craniofacial malformations and impaired ossification of the calvarium, but normal development of limbs and axial skeleton.41 Thus, it is likely that p38 MAPK regulates craniofacial bone development in part through DLX5. Furthermore, p38 MAPK has also been reported to phosphorylate OSX on Ser73 and Ser77 and to stimulate its transcriptional activity by promoting its association with SWI/SNF and p30042 (Figure 2). OSX and RUNX2 have also been found to interact and exert a transcriptional cooperation that is regulated by p38- and ERK-mediated phosphorylations.43
Modulator of p38 MAPK in osteoblasts
Finally, only one molecular mechanism involved in the regulation of p38 MAPK activity in osteoblasts has been described so far.44 A neighbor of Brca1 gene protein 1 (NBR1), a receptor for selective autophagosomal degradation of ubiquitinated target proteins, has been described as a negative regulator of p38 MAPK in osteoblasts.44 Indeed, global genetic truncation of Nbr1 (trNbr1) results in progressive increase in bone mass with age due to enhanced bone formation, osteoblast differentiation and p38 activity.44 Unlike the full-length protein, trNBR1 could not bind activated p38 MAPK and target it for degradation.44 Interestingly, NBR1 truncation leads to negligible effect on osteoclastogenesis and bone resorption, indicating a specific regulation of p38 activity in osteoblasts.44 Therefore, pharmacological manipulation of NBR1 may represent a conceivable option to increase bone mass in osteoporotic patients.
The p38 MAPK Signaling Pathway in Osteoclasts
Role of p38 MAPK in osteoclastogenesis
Osteoclast precursors and mature osteoclasts highly express p38α but no other p38 members.45 Once again, first evidence highlighting the role of p38α in osteoclastogenesis has come from in vitro experiments employing the RAW264 cell line and primary bone marrow cells with selective p38 inhibitors and dominant negative forms of p38 signaling components.46,47,48 In those cells, p38α functions downstream of RANK after stimulation by RANKL (the pro-resorptive cytokine that is necessary and sufficient for osteoclastogenesis) to stimulate osteoclast formation, maturation and bone resorption46,47 (Figure 3). Inhibition of p38 MAPK completely blocks osteoclastogenesis induced by RANKL.46,47 In osteoclast precursors, p38α functions downstream of tumor necrosis factor (TNF) receptor after stimulation by TNF-α to stimulate early osteoclast differentiation48 (Figure 3). Furthermore, p38α has been shown to be a major regulator of nuclear factor for activated T cells 1 expression (Nfatc1, encoding a master transcription factor regulating osteoclastogenesis).47 Consistent with those data, mice with post-developmental deletion of the p38α-encoding gene under the control of the inducible Mx1 promoter display reduced osteoclast number, low bone resorption and increased bone mass under physiological conditions, and are protected against TNF-α-induced arthritis and systemic bone loss.45 Although the Mx1-cre deleter strain is known to induce complete gene excisions in hepatocytes and hematopoietic cells, and variable deletions in other cell types,49 this decreased osteoclastogenesis has been shown to be cell autonomous, as bone marrow hematopoietic cells lacking p38α cannot differentiate into mature osteoclasts in response to macrophage colony-stimulating factor (M-CSF) and RANKL treatment in vitro.45
Figure 3.
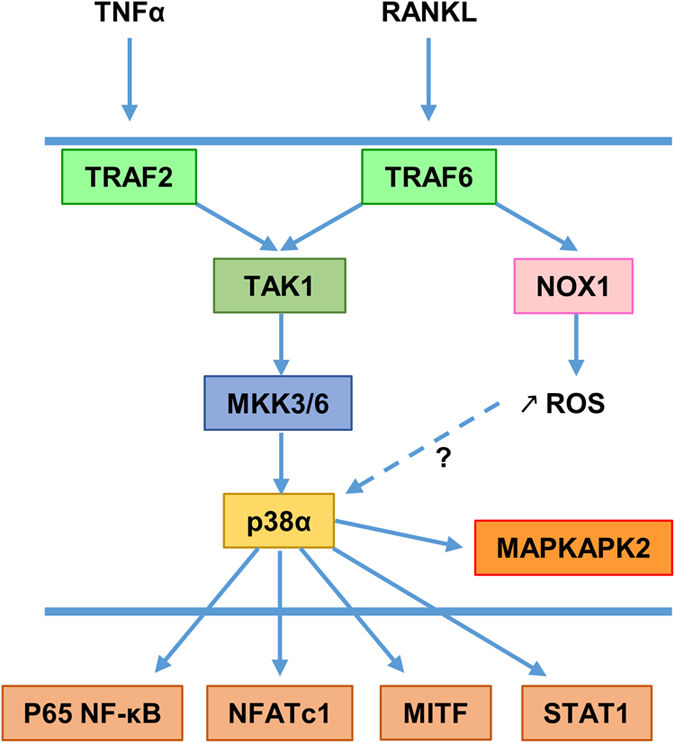
p38 MAPK signaling in osteoclasts. Osteoclasts and their precursors express only the p38α member. TNF-α and RANKL activate p38α MAPK signaling through TRAF2 and TRAF6, respectively. Both TRAF2 and TRAF6 regulate p38α signaling through TAK1. In response to RANKL, TRAF6 also triggers production of ROS through NAPDH oxidase 1 (NOX1), resulting in enhanced p38α activation. Finally, p38α phosphorylates the p65 NF-κB subunit, NFATc1, MITF and STAT1 to stimulate their transcriptional activity.
MKK3 and MKK6 have been found to be required for osteoclastogenesis in vitro.37,46,50 In accordance with this, both Mkk3−/− and Mkk6−/− mice exhibit a higher trabecular bone mass with increased trabecular number and decreased trabecular spacing in comparison with control littermates at 4 months of age.37 In those mice, osteoclast number is reduced, whereas bone formation is normal.37 In addition, Mkk3−/− and Mkk6−/− mice are partially protected against bone loss induced by estrogen deficiency due to diminished bone resorption.37 Absence of MKK337 or MKK646,50 in primary bone marrow cell cultures stimulated by M-CSF and RANKL results in reduced p38 MAPK activation, decreased expressions of Nfatc1 and other osteoclast marker genes (cathepsin K (CtsK), Oscar and matrix metalloproteinase 9) and impaired bone resorption.
Upstream activators of p38 MAPK in osteoclasts
The intracellular mechanisms by which RANKL activates RANK signaling is complex (reviewed in Wada et al.51). RANK lacks intrinsic enzymatic activity in its intracellular domain and transduces signals by recruiting adapter molecules such as TNF receptor-associated factor (TRAF) family of proteins. TRAF6 has been identified as the main adapter molecule that links RANK to osteoclast differentiation and function.51 In accordance with this notion, mice lacking TRAF6 exhibit severe osteopetrosis due to impaired osteoclast function.52 RANKL binding to RANK induces trimerization of TRAF6, which leads to the activation of nuclear factor κB (NF-κB) and MAPK signaling.51 Functional domains of TRAF6 involved in MAPK activation have been characterized.53 Among the various domains investigated, the RING finger and the first zinc finger domains are required for activation of TAK1 and downstream p38 and JNK MAPK53 (Figure 3). Moreover, TRAF6 has also been demonstrated to activate p38 and JNK MAPK during osteoclast differentiation by favoring ROS production through increased NAPDH oxidase activity54 (Figure 3).
To date, TAK1 has been identified as the MAP3K transmitting signals between RANK/TRAF6 and NF-κB and MAPK50,55,56 (Figure 3). TAK1 is activated in osteoclasts in response to RANKL and TNF-α.50 Absence of TAK1 in primary bone marrow osteoclast precursors results in reduced activation of inhibitor of κB kinase, p38 and JNK, defective NOTCH signaling and impaired osteoclast differentiation in response to M-CSF and RANKL treatment in vitro.55,56 To investigate TAK1 function in osteoclastogenesis in vivo, different murine models have been generated with Tak1 inactivation in monocytes/osteoclast progenitors (under the control of the Lysozyme M or the CD11b promoter) or in differentiating osteoclasts (under the control of the CtsK promoter).55,56 All those mice exhibit growth retardation, severe osteopetrosis characterized by dense and brittle bone mass, and reduced osteoclast number and bone resorption,55,56 clearly demonstrating that TAK1 is critical for physiological osteoclastogenesis and bone homeostasis. Interestingly, the osteopetrotic phenotype of those mice can be fully rescued by restoring NF-κB activation, indicating that signaling pathways generated from TAK1 (including p38) converge to enhance NF-κB activity.56
Targets of p38 MAPK in osteoclasts
Several downstream p38 targets involved in osteoclast differentiation and function have been characterized. RANKL induces osteoclastogenesis in great part by increasing NF-κB transcriptional activity and subsequent Nfatc1 expression.50,51 The p38 MAPK pathway has been found to contribute to RANKL-induced osteoclast formation by phosphorylating the p65 NF-κB subunit on Ser536 and thus increasing NF-κB transcriptional activity50 (Figure 3). NFATc1, PU.1 and microphthalmia transcription factor (MITF) are important transcription factors implicated in osteoclast differentiation and can interact with each other to synergistically enhance osteoclast marker gene expressions.47,57 Activated p38 MAPK in response to RANKL has been shown to phosphorylate NFATc1 (Figure 3), which leads to translocation of NFATc1 in osteoclast nuclei, its association with PU.1 and increased CtsK and tartrate-resistant acid phosphatase type 5 (Acp5) expressions.47,57 In addition, p38 MAPK can directly phosphorylate MITF on Ser307 following RANKL stimulation (Figure 3), thus leading to increased MITF transactivation and enhanced CtsK and Acp5 expressions.58 Monokine induced by interferon γ (MIG) is secreted by osteoclast precursors upon RANKL stimulation and stimulates osteoclast adhesion and migration.59 It has been demonstrated that MIG induction by RANKL is dependent on signal transducer and activator of transcription 1 (STAT1) activity and that STAT1 phosphorylation on Ser727 by p38 is critical in this context59 (Figure 3). Finally, p38 MAPK is known to activate downstream kinases such as MAPKAPK24,5 (Figure 3). Interestingly, bone marrow osteoclast precursors lacking MAPKAPK2 fail to differentiate into mature osteoclasts in response to M-CSF and RANKL treatment in vitro, as evidenced by reduced expression of Acp5, CtsK, Oscar and Mmp9, and impaired DNA-binding capacity of c-FOS and NFATc1 in promoters of osteoclast-specific genes.60 Coherent with that, mice lacking MAPKAPK2 show increased trabecular bone mass and cortical thickness, fewer osteoclasts and lower bone resorption compared with control littermates.60 Altogether, those data highlight the critical role of p38α in osteoclastogenesis and bone remodeling.
Conclusion
A large body of evidence shows that the p38 MAPK signaling pathway is critical for skeleton development and bone homeostasis (Table 1). Mutations in genes affecting the p38 pathway can cause developmental bone disorders such as chondrodysplasia, cleidocranial dysplasia or faciogenital dysplasia. In addition, this signaling pathway is altered in the context of osteoporosis, inflammatory osteolysis and osteopetrosis. p38α is the most abundant p38 member in bone cells, but direct therapeutic intervention on this MAPK is unthinkable because of its ubiquitous nature and pleiotropic functions. Therefore, an accurate knowledge of p38 MAPK function and regulation is needed to potentially identify therapeutic targets with the goal of modulating this pathway in bone. In particular, its molecular regulatory mechanisms (inhibiting phosphatases, scaffold proteins, molecular targeting to autophaghy), which generally function in a cell type-specific manner, may represent better targets than p38α MAPK itself. For instance, NBR1, which negatively regulates p38 activity specifically in osteoblasts, may represent such existing therapeutic targets to increase bone mass in osteoporotic patients.
Table 1. Skeletal phenotypes of mice with genetic deletion of genes encoding components of the p38 MAPK signaling pathway.
| Affected cells | Deleted kinase | Type of deletion | Skeletal phenotype | Cell defects | Ref. |
|---|---|---|---|---|---|
| Chondrocytes | p38α | Col2-cre | Dwarfism, narrowing/disorganization of proliferative and hypertrophic zones in growth plates | Delayed hypertrophic differentiation | 15 |
| TAK1 | Col2-cre | Severe chondrodysplasia with dwarfism, impaired formation of secondary ossification centers, joint abnormalities | Decreased proliferation, increased apoptosis | 16,17 | |
| Delayed hypertrophic differentiation | 17 | ||||
| Osteoblasts | p38α | Osx-cre, inducible | Defective mineralization of craniofacial bones at birth, low bone mass in adulthood | Impaired early osteoblast differentiation | 35 |
| Decreased bone formation when deleted during adulthood | |||||
| Ocn-cre | Decreased bone formation, low bone mass getting worse with age | Defective osteoblast function | 36 | ||
| p38β | Global KO | Low bone mass only in limbs at 3 weeks of age | Impaired late osteoblast maturation | 34 | |
| MKK3 | Global KO | Decreased calvarial mineralization, low bone mass at 3 weeks of age | Impaired early osteoblast differentiation | 34 | |
| MKK6 | Global KO | Low bone mass only in limbs at 3 weeks of age | Impaired late osteoblast maturation | 34 | |
| TAK1 | Osx-cre, inducible | Cleidocranial dysplasia, decreased mineralization of secondary ossification centers, low bone mass at 3 weeks of age | Impaired early osteoblast differentiation | 34 | |
| MLK3 | Global KO | Dental abnormalities, defective calvarial mineralization, osteopenia | Impaired early osteoblast differentiation | 39 | |
| Osteoclasts | p38α | Mx-cre, inducible | Increased bone mass | Impaired osteoclastogenesis | 45 |
| MKK3 | Global KO | High trabecular bone mass at 16 weeks of age | Impaired osteoclastogenesis | 37 | |
| TAK1 | LysM-cre CtsK-cre | Growth retardation, severe osteopetrosis | Impaired osteoclastogenesis | 55,56 |
Abbreviations: CtsK, cathepsin K; KO, knockout; MKK, mitogen-activated protein kinase kinase; MLK, mixed-lineage kinase; Osx, osterix; TAK1, transforming growth factor-β-activated kinase 1.
Footnotes
The authors declare no conflict of interest.
References
- Olsen BR, Reginato AM, Wang W. Bone development. Annu Rev Cell Dev Biol 2000; 16: 191–220. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell 2002; 2: 389–406. [DOI] [PubMed] [Google Scholar]
- Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem 2010; 285: 25103–25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005; 15: 11–18. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signaling. Biochem J 2010; 429: 403–417. [DOI] [PubMed] [Google Scholar]
- Oh CD, Chang SH, Yoon YM, Lee SJ, Lee YS, Kang SS et al. Opposing role of mitogen-activated protein kinase subtypes, erk-1/2 and p38, in the regulation of chondrogenesis of mesenchymes. J Biol Chem 2000; 275: 5613–5619. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Shirai T, Morishita S, Uchida S, Saeki-Miura K, Makishima F. p38 mitogen-activated protein kinase functionally contributes to chondrogenesis induced by growth/differentiation factor-5 in ATDC5 cells. Exp Cell Res 1999; 250: 351–363. [DOI] [PubMed] [Google Scholar]
- Yosimichi G, Nakanishi T, Nishida T, Hattori T, Takano-Yamamoto T, Takigawa M. CTGF/Hcs24 induces chondrocyte differentiation through a p38 mitogen-activated protein kinase (p38MAPK), and proliferation through a p44/42 MAPK/extracellular-signal regulated kinase (ERK). Eur J Biochem 2001; 268: 6058–6065. [DOI] [PubMed] [Google Scholar]
- Tuli R, Tuli S, Nandi S, Huang X, Manner PA, Hozack WJ et al. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J Biol Chem 2003; 278: 41227–41236. [DOI] [PubMed] [Google Scholar]
- Zuzarte-Luís V, Montero JA, Rodriguez-León J, Merino R, Rodríguez-Rey JC, Hurlé JM. A new role for BMP5 during limb development acting through the synergic activation of Smad and MAPK pathways. Dev Biol 2004; 272: 39–52. [DOI] [PubMed] [Google Scholar]
- Stanton LA, Sabari S, Sampaio AV, Underhill TM, Beier F. p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem J 2004; 378: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen X, Wei L, Wu Q, Zhang Y, Chen Q. Mitogen-activated protein kinase p38 mediates regulation of chondrocyte differentiation by parathyroid hormone. J Biol Chem 2001; 276: 4879–4885. [DOI] [PubMed] [Google Scholar]
- Morita K, Miyamoto T, Fujita N, Kubota Y, Ito K, Takubo K et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J Exp Med 2007; 204: 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison MR. BDNF alters ERK/p38 MAPK activity ratios to promote differentiation in growth plate chondrocytes. Mol Endocrinol 2012; 26: 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison MR. Mice with a conditional deletion of the neuro trophin receptor TrkB are dwarfed, and are similar to mice with a MAPK14 deletion. PLoS ONE 2013; 8: e66206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim JH, Greenblatt MB, Xie M, Schneider MD, Zou W, Zhai B et al. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J 2009; 28: 2028–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnell LM, Jonason JH, Loiselle AE, Kohn A, Schwarz EM, Hilton MJ et al. TAK1 regulates cartilage and joint development via the MAPK and BMP signaling pathways. J Bone Miner Res 2010; 25: 1784–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc Natl Acad Sci USA 2006; 103: 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold AM, Grusby MJ, Kosaras B, Fries JW, Mori R, Maniwa S et al. Chondrodysplasia and neurological abnormalities in ATF-2-deficient mice. Nature 1996; 379: 262–265. [DOI] [PubMed] [Google Scholar]
- Beier F, Lee RJ, Taylor AC, Pestell RG, LuValle P. Identification of the cyclin D1 gene as a target of activating transcription factor 2 in chondrocytes. Proc Natl Acad Sci USA 1999; 96: 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu AM, Schwarz EM, Zuscik MJ, Drissi H, Puzas JE, Rosier RN et al. ATF-2 cooperates with Smad3 to mediate TGF-beta effects on chondrocyte maturation. Exp Cell Res 2003; 288: 198–207. [DOI] [PubMed] [Google Scholar]
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X et al. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell 2007; 12: 377–389. [DOI] [PubMed] [Google Scholar]
- Zhou J, Li P, Chen Q, Wei X, Zhao T, Wang Z et al. Mitogen-activated protein kinase p38 induces HDAC4 degradation in hypertrophic chondrocytes. Biochim Biophys Acta 2015; 1853: 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CF, Cheng SL. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-beta in normal human osteoblastic cells. J Biol Chem 2002; 277: 15514–15522. [DOI] [PubMed] [Google Scholar]
- Guicheux J, Lemonnier J, Ghayor C, Suzuki A, Palmer G, Caverzasio J. Activation of p38 mitogen-activated protein kinase and c-Jun-NH2-terminal kinase by BMP-2 and their implication in the stimulation of osteoblastic cell differentiation. J Bone Miner Res 2003; 18: 2060–2068. [DOI] [PubMed] [Google Scholar]
- Nöth U, Tuli R, Seghatoleslami R, Howard M, Shah A, Hall DJ et al. Activation of p38 and Smads mediates BMP-2 effects on human trabecular bone-derived osteoblasts. Exp Cell Res 2003; 291: 201–211. [DOI] [PubMed] [Google Scholar]
- Caverzasio J, Manen D.. Essential role of Wnt3a-mediated activation of mitogen-activated protein kinase p38 for the stimulation of alkaline phosphatase activity and matrix mineralization in C3H10T1/2 mesenchymal cells. Endocrinology 2007; 148: 5323–5330. [DOI] [PubMed] [Google Scholar]
- Chang J, Sonoyama W, Wang Z, Jin Q, Zhang C, Krebsbach PH et al. Noncanonical Wnt-4 signaling enhances bone regeneration of mesenchymal stem cells in craniofacial defects through activation of p38 MAPK. J Biol Chem 2007; 282: 30938–30948. [DOI] [PubMed] [Google Scholar]
- Rey A, Manen D, Rizzoli R, Ferrari SL, Caverzasio J. Evidences for a role of p38 MAP kinase in the stimulation of alkaline phosphatase and matrix mineralization induced by parathyroid hormone in osteoblastic cells. Bone 2007; 41: 59–67. [DOI] [PubMed] [Google Scholar]
- Bianchi EN, Ferrari SL. Beta-arrestin2 regulates parathyroid hormone effects on a p38 MAPK and NFkappaB gene expression network in osteoblasts. Bone 2009; 45: 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Palmer G, Bonjour JP, Caverzasio J. Regulation of alkaline phosphatase activity by p38 MAP kinase in response to activation of Gi protein-coupled receptors by epinephrine in osteoblast-like cells. Endocrinology 1999; 140: 3177–3182. [DOI] [PubMed] [Google Scholar]
- Caverzasio J, Thouverey C. Discovering the multiple roles of the p38 pathway in bone biology. IBMS BoneKEy 2010; 7: 458–461. [Google Scholar]
- Greenblatt MB, Shim JH, Glimcher LH. Mitogen-activated protein kinase pathways in osteoblasts. Annu Rev Cell Dev Biol 2013; 29: 63–79. [DOI] [PubMed] [Google Scholar]
- Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest 2010; 120: 2457–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Carballo E, Gámez B, Sedó-Cabezón L, Sánchez-Feutrie M, Zorzano A, Manzanares-Céspedes C et al. The p38α MAPK function in osteoprecursors is required for bone formation and bone homeostasis in adult mice. PLoS ONE 2014; 9: e102032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thouverey C, Caverzasio J. The p38α MAPK positively regulates osteoblast function and postnatal bone acquisition. Cell Mol Life Sci 2012; 69: 3115–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle DL, Hammaker D, Edgar M, Zaiss MM, Teufel S, David JP et al. Differential roles of MAPK kinases MKK3 and MKK6 in osteoclastogenesis and bone loss. PLoS One 2014; 9: e84818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thouverey C, Caverzasio J. Osteoblast-specific ablation of p38a blunts the bone anabolic activity of parathyroid hormone. Bone Abstracts 2014; 3: HT2. [Google Scholar]
- Zou W, Greenblatt MB, Shim JH, Kant S, Zhai B, Lotinun S et al. MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J Clin Invest 2011; 121: 4383–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulsamer A, Ortuño MJ, Ruiz S, Susperregui AR, Osses N, Rosa JL et al. BMP-2 induces Osterix expression through up-regulation of Dlx5 and its phosphorylation by p38. J Biol Chem 2008; 283: 3816–3826. [DOI] [PubMed] [Google Scholar]
- Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S et al. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development 1999; 126: 3795–3809. [DOI] [PubMed] [Google Scholar]
- Ortuño MJ, Ruiz-Gaspà S, Rodríguez-Carballo E, Susperregui AR, Bartrons R, Rosa JL et al. p38 regulates expression of osteoblast-specific genes by phosphorylation of osterix. J Biol Chem 2010; 285: 31985–31994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artigas N, Ureña C, Rodríguez-Carballo E, Rosa JL, Ventura F. Mitogen-activated protein kinase (MAPK)-regulated interactions between Osterix and Runx2 are critical for the transcriptional osteogenic program. J Biol Chem 2014; 289: 27105–27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse CA, Waters S, Marchbank K, Horner A, McGowan NW, Jovanovic JV et al. Neighbor of Brca1 gene (Nbr1) functions as a negative regulator of postnatal osteoblastic bone formation and p38 MAPK activity. Proc Natl Acad Sci USA 2010; 107: 12913–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm C, Hayer S, Kilian A, Zaiss MM, Finger S, Hess A et al. The alpha-isoform of p38 MAPK specifically regulates arthritic bone loss. J Immunol 2009; 183: 5938–5947. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Sudo T, Saito T, Osada H, Tsujimoto M. Involvement of p38 mitogen-activated protein kinase signaling pathway in osteoclastogenesis mediated by receptor activator of NF-kappa B ligand (RANKL). J Biol Chem 2000; 275: 31155–31161. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S et al. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem 2004; 279: 45969–45979. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Sudo T, Maruyama M, Osada H, Tsujimoto M. Activation of p38 mitogen-activated protein kinase is crucial in osteoclastogenesis induced by tumor necrosis factor. FEBS Lett 2000; 486: 23–28. [DOI] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science 1995; 269: 1427–1429. [DOI] [PubMed] [Google Scholar]
- Huang H, Ryu J, Ha J, Chang EJ, Kim HJ, Kim HM et al. Osteoclast differentiation requires TAK1 and MKK6 for NFATc1 induction and NF-kappaB transactivation by RANKL. Cell Death Differ 2006; 13: 1879–1891. [DOI] [PubMed] [Google Scholar]
- Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 2006; 12: 17–25. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 1999; 13: 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S et al. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 2001; 20: 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005; 106: 852–859. [DOI] [PubMed] [Google Scholar]
- Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG. TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol 2013; 33: 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarnkar G, Karuppaiah K, Mbalaviele G, Chen TH, Abu-Amer Y. Osteopetrosis in TAK1-deficient mice owing to defective NF-κB and NOTCH signaling. Proc Natl Acad Sci USA 2015; 112: 154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SM, Bronisz A, Hu R, Patel K, Mansky KC, Sif S et al. MITF and PU.1 recruit p38 MAPK and NFATc1 to target genes during osteoclast differentiation. J Biol Chem 2007; 282: 15921–15929. [DOI] [PubMed] [Google Scholar]
- Mansky KC, Sankar U, Han J, Ostrowski MC. Microphthalmia transcription factor is a target of the p38 MAPK pathway in response to receptor activator of NF-kappa B ligand signaling. J Biol Chem 2002; 277: 11077–11083. [DOI] [PubMed] [Google Scholar]
- Kwak HB, Lee SW, Jin HM, Ha H, Lee SH, Takeshita S et al. Monokine induced by interferon-gamma is induced by receptor activator of nuclear factor kappa B ligand and is involved in osteoclast adhesion and migration. Blood 2005; 105: 2963–2969. [DOI] [PubMed] [Google Scholar]
- Braun T, Lepper J, Ruiz Heiland G, Hofstetter W, Siegrist M, Lezuo P et al. Mitogen-activated protein kinase 2 regulates physiological and pathological bone turnover. J Bone Miner Res 2013; 28: 936–947. [DOI] [PubMed] [Google Scholar]