

Abstract
Background
With the use of cultured human retinal pigment epithelial cells, we have previously described a number of cellular responses to oxidative stress caused by H2O2. We also demonstrated that the cytotoxicity caused by H2O2 could be prevented by the prostaglandin derivative, 15-deoxy-delta 12, 14-Prostaglandin J2 (15d-PGJ2).
Results
Further characterization of the experimental system indicated that the half-life of H2O2 in cultures was ~1 hour. At a fixed H2O2 concentration, the cytotoxicity was dependent on the volume of H2O2 solution used in the culture, such that higher volume caused more cytotoxicity. Most cells were committed to die if the culture was treated for 2 hours with a cytotoxic concentration of H2O2. The prostaglandin derivative, 15d-PGJ2, could prevent oxidative damage caused by t-butyl hydroperoxide, in addition to H2O2. Further studies indicated that both H2O2 and tBH caused an increase in reactive oxygen species and depolarization of mitochondrial membrane potential. Pretreatment of cells with 1 μM 15d-PGJ2 led to a modest decrease in reactive oxygen species generation, and a significant restoration of mitochondrial membrane potential.
Conclusion
This agent may be used in the future as a pharmacological tool for preventing cellular damage caused by oxidative stress.
Keywords: 15d-PGJ2; oxidative stress; ROS (reactive oxygen species); MMP (mitochondrial membrane potential), RPE (retinal pigment epithelium).
Background
Oxidative stress on retinal pigment epithelial cells (RPE) has been suggested as a cause of age-related macular degeneration [1,2]. With the use of primary cultures of human RPE and the ARPE-19 cell line, we reported earlier that oxidative stress caused a number of structural and biochemical changes [3]. For example, cytoskeletal changes were observed shortly after RPE cells were treated with H2O2. This was accompanied by activation of the ERK (Extracellular signal-regulated Kinase) MAP kinase, and formation of lipid peroxidation products (4-hydroxynonanel protein adducts). Nuclear condensation was observed 12 hours after H2O2 treatment, and cells died in a manner consistent with apoptosis.
In an effort to identify pharmacological agents capable of preventing oxidative stress-induced cytotoxicity in RPE, we found the prostaglandin derivative, 15-deoxy-delta 12, 14-Prostaglandin J2 (15d-PGJ2), could prevent H2O2-induced cytotoxicity both in primary human RPE cultures and in ARPE-19 cells [3]. Even though 15d-PGJ2 is a ligand for peroxisome proliferator-activated receptors (PPARs) [4,5], further studies indicated that the saving effect of 15d-PGJ2 was not shared by other agonists of PPARs. It was thus concluded that this agent saved these cells in a way not involving PPAR activation. In other experimental systems, 15d-PGJ2 has been shown to have a potent inhibitory effect on immune cell functions [6,7] and is effective in preventing disease progression of the Experimental Autoimmune Encephalomyelitis, an animal model of the autoimmune disease multiple sclerosis [8].
A series of intracellular events occur as a result of oxidative stress, which may eventually lead to apoptosis. For example, an intracellular increase in reactive oxygen species (ROS) is accompanied by depolarization of the mitochondrial membrane potential (MMP, ΔΨm) [9]. This is followed by the release of cytochrome c from mitochondria, caspase activation and other events that lead to apoptosis [10]. A study of mitochondrial alterations is an important aspect of apoptosis, and is a subject of recent reviews [11,12]. Oxidative stress-induced mitochondrial DNA damage in RPE has been suggested to be a cause of age-related macular degeneration [13,14]. The current study was designed to test the hypothesis that 15d-PGJ2 prevented oxidative stress-induced RPE cell death by preventing early events caused by oxidative stress, such as the generation of intracellular ROS and the depolarization of mitochondrial membrane potential. While we used H2O2 as the primary source of oxidative stress in most experiments, we also used t-butyl hydroperoxide (tBH) [15] in this study to test the validity of results obtained with H2O2.
Results
H2O2 degradation and its effect on cell viability
We have previously shown that H2O2 caused a dose- and time-dependent decrease in cellular viability in ARPE-19 cells. Furthermore, the cytotoxicity of H2O2 was dependent on the cell density in each well [3]. If ARPE-19 cells were plated at 20,000 cells/well in a 96-well plate for two days, 2 mM H2O2 (100 μl/well) decreased cell viability to ~15% of controls after a 1-day treatment while 1 mM H2O2 had little effect on cell viability under identical experimental conditions.
As a further analysis of the experimental system, experiments were performed to determine the degradation of H2O2 in the cultures, and its effect on cell viability. Cells were treated with 2 mM H2O2 for various periods of time to assess the H2O2 concentration that remained in each well. Results (Fig. 1A, open squares) indicated that the readings were so close to background that H2O2 degradation could not be adequately assessed. Subsequent experiments using 5 mM or 10 mM H2O2 indicated that H2O2 degraded in a time-dependent manner (Fig. 1A), with a half-life of ~1 hour (Fig. 1B).
Figure 1.
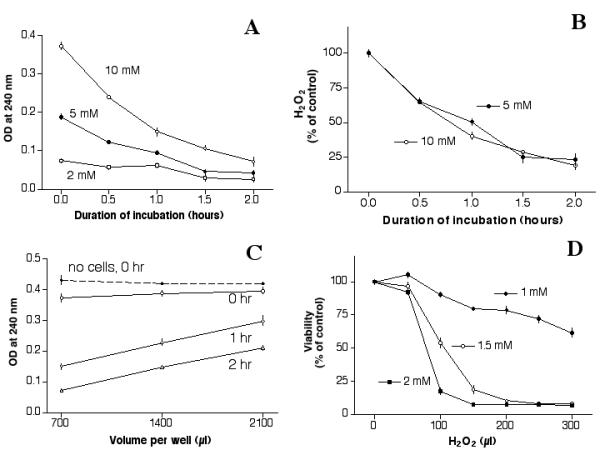
Degradation of H2O2 and its effect on cellular viability. (A): Degradation of H2O2 in cell cultures. ARPE-19 cells were plated in a 24-well plate (140,000 cells/well) for a day, fed with serum-free medium for a day, then treated with 700 μl of H2O2 (2 mM, 5 mM or 10 mM) for various periods of time, then the concentration of H2O2 remaining in each culture was determined. The OD at 240 nm is a measure of H2O2 concentration (See Methods). Results indicated that H2O2 decreased in each culture in a time-dependent manner, which could be observed clearly in cultures treated with 5 mM or 10 mM H2O2. Readings from cells treated with 2 mM H2O2 were very close to background levels. (B): Time-dependent degradation of H2O2. Results of cultures treated with 5 mM or 10 mM H2O2 were re-plotted and expressed as a percentage of its original concentration. The half-life for H2O2 degradation was ~1 hour under these experimental conditions. (C): Degradation of H2O2 as a function of volume. Cells grown in 24-well plates were treated with 10 mM H2O2 (700 μl/well, 1,400 μl/well or 2,100 μl/well), and then the H2O2 concentration in each well was determined. Results indicated that H2O2 degradation rate was affected by the volume used in each well, such that the concentration remained higher in cultures with higher volume of H2O2 solution. The H2O2 concentration remained constant in culture wells without cells as indicated by the upper-most line (solid diamonds), which was recorded at time zero. Almost identical readings were recorded 1 hour or 2 hours later (not shown). Note: Treatment of cells with H2O2 for 0 hour was determined by applying H2O2 solution to the cultures briefly (~2 min). The solution was then removed, and the OD measured. This short contact with cultures caused a ~10% loss of H2O2 from its original concentration. This can be seen from the difference between "no cells, 0 hr (solid diamonds, the upper-most line) and "0 hr" (open circles, the second line from the top). (D): Cytotoxicity of H2O2 on ARPE-19: Effects of concentration and volume. Cells plated in a 96-well plate for two days were treated with 1 mM, 1.5 mM or 2 mM H2O2 in various volume for 1 day, then the viability of each treatment was determined by the MTT assay. Results indicate that at a fixed concentration, the volume used in each well affected the cytotoxicity of H2O2.
The decrease in H2O2 concentration was inversely related to the volume of H2O2 solution in each well, such that cultures with more H2O2 solution maintained H2O2 concentration better (Fig. 1C). For example, the OD readings in cells treated with 2,100 μl, 1,400 μl or 700 μl (10 mM H2O2) per well were 0.30, 0.23 or 0.15, respectively after 1 hour (open diamonds), and 0.21, 0.15 or 0.07, respectively, after 2 hours (open triangles). Without cells, the H2O2 concentrations remained almost constant regardless of the volume added to each well (Fig. 1C, solid diamonds).
Based on these results, it was hypothesized that at a fixed H2O2 concentration and a fixed cell density, the volume of H2O2 solution added to each well should affect the cytotoxicity of H2O2. To test this, a set of cells grown in 96-well plates were treated with H2O2 of various volumes for one day, then the viability of each well was determined by the MTT assay. Results indicated that while 100 μl of 2 mM H2O2 decreased the viability to ~20% of controls, 50 μl of 2 mM H2O2 had little effect on the viability (Fig. 1D, solid squares). Similar volume-dependent cytotoxicity was observed with 1.5 mM and 1 mM H2O2. At 1.5 mM H2O2, it took 100 μl to decrease the viability to ~50% of controls (Fig. 1D, open circles).
In view of these results, in addition to maintaining a constant cell density, the volume of H2O2 solution used in this study was adjusted proportionally throughout according to the surface area of the culture wells. For example, while 100 μl/well was used in a 96-well plate, 700 μl/well was used in a 24-well plate.
Temporal commitment point of oxidative stress induced cytotoxicity
Results from Fig. 1D indicated that if cells were treated with 100 μl of 2 mM H2O2, the majority of cells would die by 24 hours after treatment. The length of time required for cells to commit to death under these conditions was determined. Cultures were treated with 2 mM H2O2 at time zero, and then H2O2 was replaced with fresh medium from a set of cells at 30-minute intervals. Viability was then determined 24 hours later. Results (Fig. 2, solid circles) indicated that if cells were treated with 2 mM H2O2 for 1 hour followed by fresh medium (without H2O2) for 23 hours, the viability decreased to ~75% of control. If the cells were treated with H2O2 for 2 hours, then with fresh medium for 22 hours, the viability decreased to ~10% of control. It was thus concluded that with 2 mM H2O2 treatment, most cells were committed to die 2 hours after treatment.
Figure 2.
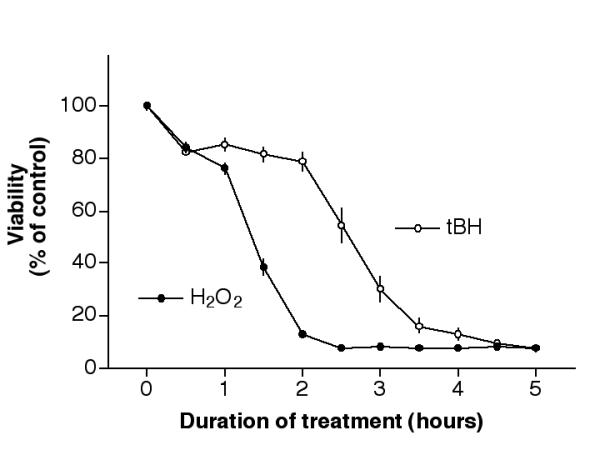
Temporal commitment point of H2O2 and tBH. Cells plated in 96-well plates for 2 days were treated with 2 mM H2O2 (solid circles) or 600 μM tBH (open circles) for various duration as indicated, then with fresh medium (without the testing agent) until 24 hours after initial treatment. Cell viability was determined by the MTT assay. Note: 2 mM H2O2 and 600 μM tBH were used to produce effective cell death after a 24-hour treatment. (See Fig. 1, 100 μl/well and Fig. 6A, solid circles)
In a set of analogous experiments, the commitment point of cells after tBH treatment was determined. Approximately 80% of cells survived 600 μM tBH treatment if this agent was removed within 2 hours of treatment (Fig. 2, open circles). On the other hand, most cells were committed to die if the treatment continued for 4 hours (see below for more experiments involving tBH).
Effect of 15d-PGJ2 on H2O2-induced ROS generation
We have previously shown that pretreatment of ARPE-19 cells with 15d-PGJ2 greatly reduced the cytotoxicity caused by H2O2 [3]. For example, while H2O2 at 1.3 mM, 1.4 mM or 1.5 mM reduced the viability to 64%, 46% or 28% of controls, pretreatment of these cells with 1 μM 15d-PGJ2 raised the viability to 95%, 84% or 68% of control, respectively. Similar saving effects were also observed in primary cultures of human RPE. Since H2O2 could induce intracellular generation of ROS, experiments were performed to determine whether 15d-PGJ2 could reduce intracellular ROS levels after H2O2 treatment, thereby reducing the H2O2-induced cytotoxicity.
Initial experiments indicated that H2O2 treatment of cells caused a dose-dependent increase in ROS levels in ARPE-19 cultures, as indicated by the conversion of H2DCF-DA to its oxidized form, DCF-DA. The maximal ROS generation was caused by 1 mM H2O2, which reached ~3× of basal levels (Fig. 3A). It is of interest to note that total DCF-DA generated in each well was ~2× as much as that associated with cells. For example, while H2O2 at 250, 500 or 1000 μM generated ~55 FU, 67 FU or 70 FU of DCF-DA in each well (Fig. 3A, solid circles), the "cell associated" DCF-DA was ~28 FU, 33 FU or 35 FU, respectively (Fig. 3A, open circles). DCF-DA formation appears to occur intracellularly, because without cells, H2O2 did not cause conversion of H2DCF-DA to DCF-DA (data not shown). Together, these results indicate that approximately half of the DCF-DA generated inside the cells diffused out of the cells.
Figure 3.
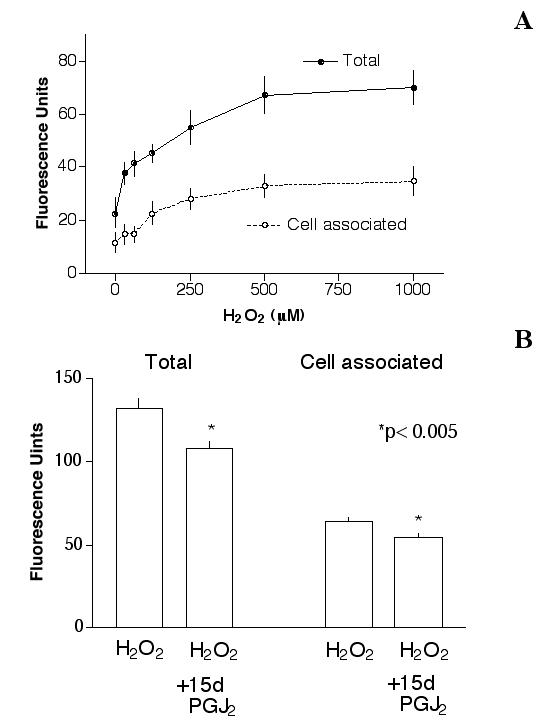
Effect of 15d-PGJ2 on H2O2-induced ROS generation. (A): ROS generation by H2O2. Cells were treated with 10 μM H2DCF-DA for 20 min, followed by various concentrations of H2O2 for 20 min, then the "total" and "cell-associated" ROS generated was determined as described in Materials and Methods. Results indicated that H2O2 caused a dose-dependent ROS generation. At 1 mM H2O2, the "total" ROS production was ~70 FU, which was ~3× of basal levels (~23 FU). Similarly, this concentration of H2O2 caused an increase of "cell-associated" ROS to ~3× of basal level (35 FU vs. 12 FU, respectively). The "cell-associated" ROS was ~50% of the "total" ROS generated in each well. (B): 15d-PGJ2 reduced H2O2-induced ROS generation. Cells were pretreated with 1 μM 15d-PGJ2 overnight, then with 10 μM H2DCF-DA for 20 min followed by 1 mM H2O2 for 20 min (15d-PGJ2 was not present in cultures after the overnight incubation). Results indicated that 15d-PGJ2 caused a statistically significant decrease of "total" and "cell-associated" ROS generation. Without H2O2, short-term (20 min) or overnight treatment of cells with 1 μM 15d-PGJ2 did not alter cellular ROS levels (not shown).
15d-PGJ2 was tested to see if it could prevent H2O2 induced ROS elevation. Cells were pretreated with 1 μM 15d-PGJ2 overnight, challenged with 1 mM H2O2 the next day for 20 minutes, then ROS in each well was determined. Results (Fig. 3B) indicated that while 1 mM H2O2 generated ~132 FU "total" ROS in this set of experiments, 15d-PGJ2 pretreatment reduced ROS to ~108 FU (p < 0.005). Similarly, the "cell associated" ROS was reduced by 15d-PGJ2 from ~64 to ~54 FU (p < 0.005). The decrease in ROS levels caused by 15d-PGJ2 may be partially responsible for its ability to prevent H2O2-induced cytotoxicity.
H2O2-induced mitochondrial membrane depolarization in ARPE-19 cells
Other than ROS generation, H2O2 treatment of cells leads to depolarization of mitochondrial membrane potential, which can be measured by the JC-1 dye [12]. Depolarization of mitochondrial membrane causes a shift in the emission spectrum from red to green color, which can be quantified by a fluorescence plate reader. Initial experiments indicated that the emission intensity of the green peak and the red peak could be determined optimally at ~545 nm and ~595 nm, respectively (see Fig. 5A). Treatment of cells with H2O2 led to an alteration of the relative intensity of these two peaks (See Fig. 5C). The ratio of readings at 545 nm and 595 nm was thus used to assess mitochondrial membrane potential. A higher 545/595 emission intensity ratio suggests more mitochondrial depolarization.
Figure 5.

Prevention of H2O2-induced mitochondrial membrane depolarization by 15d-PGJ2. A-D: The JC-1 emission spectra between 520 nm to 620 nm were determined for cells under various conditions. (A): Untreated cells; (B): Cells treated with 1 μM 15d-PGJ2 overnight; (C): Cells treated with 1.5 mM H2O2 for 2 hours; (D): Cells treated with 1 μM 15d-PGJ2 overnight, then with 1.5 mM H2O2 (without 15d-PGJ2) for 2 hours. Note H2O2 caused a shift of the relative intensity of the peaks, and 15d-PGJ2 pre-treatment restored membrane potential to a condition closer to untreated cells. E-F: Cells were pretreated with 1 μM 15d-PGJ2 overnight, then with 1.5 mM H2O2 (without 15d-PGJ2) for 2 hours (E) or 4 hours (F), then the 545/595 emission intensity ratios were determined. Note in either 2-hour or 4-hour treatment, H2O2 caused an increase of the 545/595 emission intensity ratio, 15d-PGJ2 pre-treatment restored the ratio to that similar to control value (p < 0.001 between H2O2-treated and 15d-PGJ2+H2O2-treated cells in E and F).
The relative intensity of the 545/595 peaks as a function of H2O2 concentration and duration of exposure was determined. Results indicated that H2O2 treatment for 30 min caused a slight dose-dependent increase of both peaks (Fig. 4A). Similar observations were made in cells treated with various concentrations of H2O2 for 1 hour (Fig. 4B). There was little change in 545/595 emission intensity ratio (not shown).
Figure 4.

H2O2 caused depolarization of mitochondrial membrane potential. Cells were treated with various concentrations of H2O2 for 0.5 hour (A), 1 hour (B), 2 hours (C) or 4 hours (D), then processed for the JC-1 assay as described in the Materials and Methods. Emission readings at 545 nm and 595 nm were plotted as a function of H2O2 concentration. Results from (A-D) were re-plotted and presented as (E) to illustrate a dose- and time-dependent increase of 545/595 emission intensity ratio. (F): Cells were treated with various concentrations of FCCP for 4 hours, then the 545/595 emission intensity ratio of each treatment was determined. Results indicated that FCCP caused a dose-dependent increase of the ratio, which reached a maximum at ~20 μM. Note the degree of mitochondrial depolarization caused by 2 mM H2O2 was similar to that caused by 10 μM FCCP. Results of H2O2 treatment were derived from 4 experiments (total 12 replicates), and those of FCCP treatment were from 3 experiments (total 9 replicates).
A change of the relative intensities of the 545 nm peak and 595 nm peak occurred when cells were treated with higher concentrations of H2O2 for 2 hours. Untreated cells at this time had an emission reading of 173 FU at 545 nm (Fig. 4C, open square at 0 mM H2O2), and 377 FU at 595 nm (Fig. 4C, solid circle at 0 mM H2O2). The 545/595 ratio was ~0.46 (i.e., 173/377). Cells treated with 0.5 mM or 1 mM H2O2 had emission readings at 545 nm or 595 nm slightly higher than those of controls. The 545/595 emission intensity ratio was similar to that in untreated cells (Fig. 4C). However, cells treated with 1.5 mM H2O2 had an increased emission reading at 545 nm and a decreased reading at 595 nm, such that these two readings were almost identical (Fig. 4C at 1.5 mM). With 2 mM H2O2 treatment, the reading at 545 nm was higher than that of 595 nm (Fig. 4C).
At 4 hours after treatment, there was a further decrease in the emission readings at 595 nm in cells treated with 1.5 mM or 2 mM H2O2 (as compared to untreated cells, see Fig. 4D, open circles), and a corresponding increase in the emission readings at 545 nm (Fig. 4D, solid circles).
Results from Fig. 4A,4B,4C,4D were re-plotted to show the change of 545/595 emission intensity ratio as a function of H2O2 concentrations and duration of treatment. There was little change of the ratio when cells were treated with 1 mM H2O2 up to 4 hours (Fig. 4E, open circles). Most cells survived this concentration of H2O2 after 1-day treatment (Fig. 1D, solid circle at 100 μl). In contrast, 2 mM H2O2 treatment led to a time-dependent increase in the 545/595 emission intensity ratio with a significant increase (over untreated cells) occurring at 2 hours after treatment (Fig. 4E, solid circle at 2-hour treatment point), and a further increase at 4 hours after treatment. Most of the cells were committed to die at this time (see Fig. 2, solid circle at 2-hour time point).
The mitochondrial uncoupler, FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) is commonly used as a positive control that causes mitochondrial depolarization [9]. This agent caused a dose-dependent increase of the 545/595 emission intensity ratio under the same experimental conditions. Cells treated with 0, 5, 10, 20 or 40 μM FCCP for 4 hours had a 545/595 emission intensity ratio of 0.8, 1.3, 2.1, 2.9 or 3.1, respectively (Fig. 4F).
Effect of 15d-PGJ2 on H2O2-induced mitochondrial membrane depolarization
We determined whether 15d-PGJ2 pretreatment could prevent H2O2-induced mitochondrial membrane potential depolarization. Cells were pretreated with 1 μM 15d-PGJ2 overnight, challenged with 1.5 mM H2O2 for 2 hours, then the emission spectrum of each treatment was determined. Results indicated that untreated cells showed two peaks at 545 nm and 595 nm (Fig. 5A). Similar peaks were observed in cells treated with 15d-PGJ2 overnight (Fig. 5B). Treatment of cells with H2O2 caused a shift in the relative intensity of these two peaks (Fig. 5C). However, overnight pretreatment of cells with 15d-PGJ2 restored the relative intensity to that close to the untreated cells (Fig. 5D).
This set of experiments was performed 4 times and the results (Fig. 5E) indicated that H2O2 led to an increase of 545/595 emission intensity ratio from 0.62 ± 0.06 (control) to 1.77 ± 0.11. Pretreatment of cells with 15d-PGJ2 had little effect on the 545/595 emission intensity ratio (0.65 ± 0.05, p > 0.05 between control and 15d-PGJ2), but the pretreatment greatly reduced the 545/595 emission ratio of H2O2-treated cells to 0.85 ± 0.06. Similar results were obtained when H2O2 challenge was extended to 4 hours (Fig. 5F). The 545/595 emission intensity ratios for control, H2O2-treated, 15d-PGJ2-treated or 15d-PGJ2/H2O2-treated cells were 0.82 ± 0.07, 2.74 ± 0.22, 0.85 ± 0.05 or 1.46 ± 0.09, respectively. The difference between H2O2-treated cells and 15d-PGJ2+H2O2-treated cells was statistically significant (p < 0.001). These results suggested that the inhibition of mitochondrial membrane depolarization by 15d-PGJ2 might be partially responsible for its ability to prevent H2O2-induced cytotoxicity.
We reported earlier that ciglitazone (a PPARγ agonist, tested between 1–10 μM) and WY14643 (a PPARα agonist, tested between 10–40 μM) had no saving effect in this experimental setting [3]. Subsequent experiments indicated that these two agents could not restore H2O2-induced mitochondrial membrane depolarization (see Additional file: 1).
Prevention of tBH-induced cell death by 15d-PGJ2
Given the saving effect of 15d-PGJ2 against H2O2-induced oxidative stress, we determined whether 15d-PGJ2 could prevent the oxidative stress caused by tBH. Results indicated that tBH at 400 μM or less had little cytotoxicity for ARPE-19 cells; however, 600 μM tBH killed most of the cells (Fig. 6, solid circles). Overnight pre-treatment of cells with 1 μM 15d-PGJ2 greatly prevented the cytotoxicity caused by tBH (Fig. 6A, open circles). For example, while only ~21% of the cells remained viable after 600 μM tBH treatment, 15d-PGJ2 pre-treatment raised the viability to ~78%.
Figure 6.
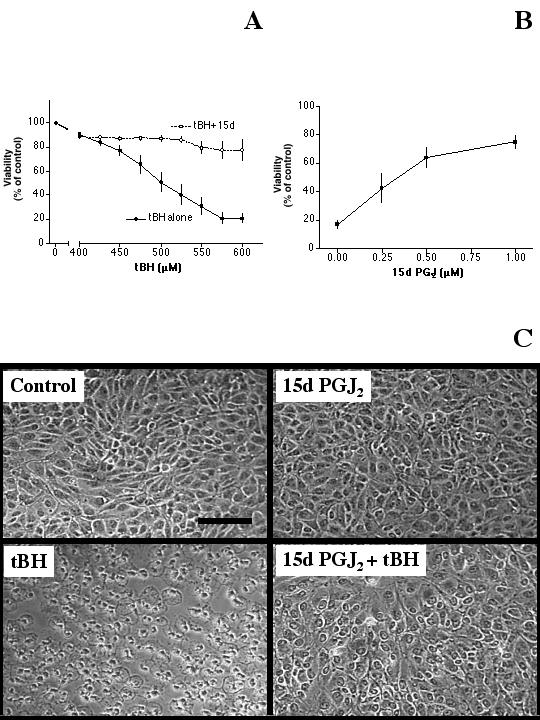
Prevention of tBH-induced cytotoxicity by 15d-PGJ2. (A): Cells plated in 96-well plates were pre-treated with serum-free medium (solid circles) or 1 μM 15d-PGJ2 (prepared in serum-free medium, open circles) overnight, then with various concentrations of tBH (without 15d-PGJ2) for one day. The viability of each treatment was determined by the MTT viability assay. Results indicated that 15d-PGJ2 pretreatment greatly reduced tBH cytotoxicity. (B): Cells were pre-treated with various concentrations of 15d-PGJ2 overnight followed by 450 μM tBH (without 15d-PGJ2) for one day, then the viability of each treatment was determined. Results indicated that 15d-PGJ2 prevented tBH induced cell death in a dose-dependent manner. (C): Cells were treated with serum-free medium (control), 1 μM 15d-PGJ2, 450 μM tBH or 1 μM 15d-PGJ2 overnight pre-treatment followed by 450 μM tBH for one day (15d-PGJ2 + tBH). Results indicated that 15d-PGJ2 greatly prevented tBH-induced cell death. Bar: 50 μm.
Experiments were performed to determine the dose-dependent saving effect of 15d-PGJ2. In this set of experiments, 1-day treatment of cells with 450 μM tBH decreased cell viability to ~17% of control. Pre-treatment of cells with 0.25, 0.5 or 1 μM 15d-PGJ2 raised the viability to ~43%, 64% or 75%, respectively (Fig. 6B). This saving effect was consistent with morphological observations with a microscope (Fig. 6C).
Effect of 15d-PGJ2 on tBH-induced ROS generation and mitochondrial membrane depolarization
Treatment of cells with tBH caused a dose-dependent increase of ROS generation. At 1000 μM tBH, the "total" ROS was ~3× control (Fig. 7A, solid squares). There was also a parallel increase of "cell-associated" ROS (Fig. 7A, open squares). To test whether 15d-PGJ2 pre-treatment could reduce tBH-induced ROS, cells were pre-treated with 1 μM 15d-PGJ2 overnight followed by 1000 μM tBH challenge, then the "total" and "cell-associated" ROS were determined. Results (Fig. 7B) indicated that tBH generated ~190 FU of "total" ROS, which was decreased to ~149 FU by 15d-PGJ2 pre-treatment (p < 0.001). Similarly, the "cell-associated" ROS was reduced from ~139 FU (tBU alone) to ~100 FU (with 15d-PGJ2 pre-treatment, p < 0.001).
Figure 7.
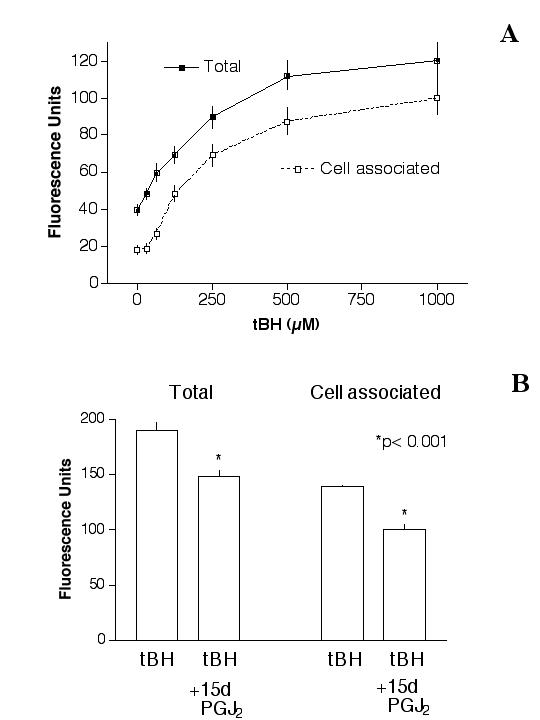
Effect of 15d-PGJ2 on tBH-induced ROS generation. (A): ROS generation by tBH. Cells were treated with H2DCF-DA for 20 min, followed by tBH for 20 min, and then the "total" (solid squares) and "cell-associated" (open squares) ROS generation was determined. Results indicated that tBH caused a dose-dependent ROS generation. At 1000 μM tBH, the "total" ROS production was ~121 RU, which was ~3× of basal levels (~40 FU). (B): 15d-PGJ2 reduced tBH-induced ROS generation. Cells were pretreated with 1 μM 15d-PGJ2 overnight, then challenged with 1000 μM tBH (without 15d-PGJ2) for 20 min. Results indicated that 15d-PGJ2 caused a statistically significant decrease of ROS generation. *p < 0.001.
We next determined whether tBH caused mitochondrial membrane depolarization and if 15d-PGJ2 could prevent the depolarization. Cells were treated with 400 μM, 500 μM or 600 μM tBH for 4 hours, and then processed for the JC-1 assay. Results indicated that tBH at these concentrations increased 545/595 emission intensity ratio from 0.76 ± 0.17 (control) to 1.03 ± 0.11, 1.23 ± 0.14 and 1.42 ± 0.19, respectively (Fig. 8, solid circles). Pretreatment of cells with 1 μM 15d-PGJ2 greatly decreased the 545/595 emission intensity ratio resulting from the tBH challenge (Fig. 8, open circles). The inhibition of mitochondrial membrane depolarization by 15d-PGJ2 may be partially responsible for its ability to prevent tBH-induced cytotoxicity.
Figure 8.
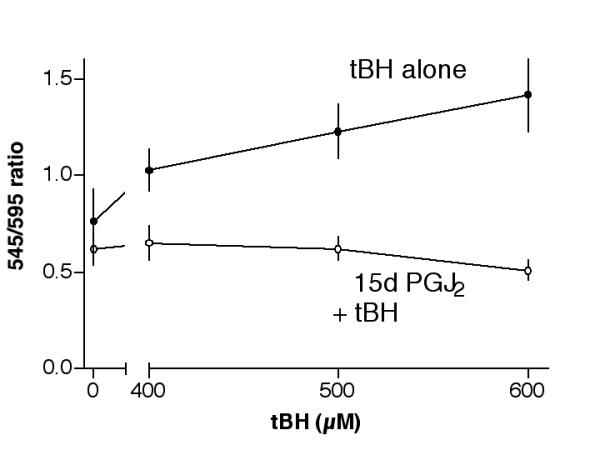
Prevention of tBH-induced mitochondrial membrane depolarization by 15d-PGJ2. Cells were pretreated with serum-free medium (control) or with 1 μM 15d-PGJ2 (prepared in serum-free medium) overnight, then with 400 μM, 500 μM or 600 μM tBH (without 15d-PGJ2) for 4 hours. The 545/595 emission intensity ratio of each treatment was determined. Results indicated that tBH caused an increase of the 545/595 emission intensity ratio, which was completely prevented by 15d-PGJ2 pre-treatment. Preliminary results indicated that tBH treatment for 4 hours was required to see a significant increase of 545/595 emission intensity ratio (data not shown).
Discussion
H2O2 cytotoxicity as a function of volume
There are several important factors that can determine the cytotoxicity of H2O2 in cultured cells. In our previous studies with 96-well plates, we noticed that the degree of H2O2 cytotoxicity at a fixed concentration was very much dependent on cell density [3]. If we performed experiments 2 days after plating 20,000 cells per well in 96-well plates, the cytotoxicity of H2O2 (100 μl/well) could be observed between 1–2 mM. H2O2 at concentrations lower than 1 mM had no apparent cytotoxicty. Cells were more sensitive to H2O2 if they were plated at a lower density. As a result, we paid much attention to cell density in subsequent experiments when we intended to use culture plates other than 96-well plates. While using a 6-well plate, we plated down 30× as many cells per well as that used in each well of a 96-well plate in order to maintain the appropriate density. With this density issue in mind, we were puzzled to see that results of a series of experiments in 6-well plates using 2 ml/well H2O2 solution yielded much less cytotoxicity than we expected. Comparable cytotoxicity could only be observed when we used 3 ml/well H2O2 solution regarding surface area versus volume ratio.
These observations prompted us to determine H2O2 degradation in the culture wells, and analyze its effect on cytotoxicity. Results (Fig. 1A and 1B) suggested that H2O2 (either 5 mM or 10 mM, fixed volume) degraded at a rate that reached ~50% of the original concentrations in one hour. Further experiments (Fig. 1C) indicated that the rate of degradation (at a fixed concentration) was dependent on the volume of H2O2 used in the culture well. It appeared that cells (at a fixed density) would consume a fixed mass amount of H2O2 molecules such that wells containing more volume (thus more H2O2 molecules) would have more remaining H2O2 molecules after a given time period. As a result, wells containing a higher volume maintained the concentration better than those with a lower volume. The resulting higher concentration led to a higher cytotoxicity, as illustrated by Fig. 1D.
These results indicated that it is very important to control the volume of H2O2 solution used in the experiments. The volume used in this study was adjusted proportionally according to the surface area in order to obtain a consistent H2O2 cytotoxicity. As a result, other than control for uniformed cell density, we used 100 μl/well in a 96-well plate, 700 μl/well in a 24-well plate and 3 ml/well in a 6-well plate for all of our experiments. Even though these results were obtained using H2O2 as the cytotoxic agent, there is a possibility that similar considerations should be made while testing other cytotoxic agents.
Temporal commitment point: H2O2 vs. tBH
H2O2 and tBH have been used in a number of studies testing the responses of RPE toward oxidative stress. For example, it was shown that these two agents could alter gene expression in a specific manner [15]. Results from the current study indicate that 2 mM of H2O2 killed most of the cells after a 1-day treatment (Fig. 1D, see the solid square at 100 μl/well) but it took only 600 μM tBH to achieve similar cytotoxicity (Fig. 6A, solid circles). Based on this comparison, it appeared that tBH was more potent than H2O2. It was reported that H2O2 could be metabolized ~3 times as fast as tBH [16], which may partially explain the observation that it took ~3× concentrations of H2O2 to achieve similar cytotoxicity as tBH.
It is generally believed that ROS are short-lived and damage to cellular structures occurs rapidly. With this in mind, experiments were performed to test how soon cells were committed to die following treatment with 2 mM H2O2 or 600 μM tBH, concentrations that killed most of the cells in 24 hours. Results indicated that tBH, even with its apparent higher potency, appeared to have a slower action. In fact, if it was removed within 2 hours, ~80% of the cells had survived the treatment one day later. It took 4 hours for most cells to commit to die following tBH treatment (Fig. 2, open circles). In contrast, H2O2 appeared to damage cells more rapidly (Fig. 2, solid circles). Some cell damage should have occurred during the first hour of treatment based on the viability curve. The damage appeared to increase in a time-dependent manner such that most of the cells committed to die if the cultures were treated with H2O2 for 2 hours.
DCF-DA generation caused by H2O2 vs. tBH: membrane leakiness
One possible explanation for the finding that cells treated with H2O2 had an earlier commitment point is that H2O2 treatment led to a faster or higher rise in intracellular ROS. Results from this study indicated that both H2O2 (Fig. 3A) and tBH (Fig. 7A) treatment led to an increase in cellular ROS within 20 min and the maximal "total" ROS levels were ~3× of control levels in both cases. It is of interest to note that the "cell-associated" ROS was ~50% of total ROS in H2O2-treated cells, while it was about ~80% in tBH-treated cells. These results suggest that H2O2 treatment made the cell membrane more leaky, and more of the intracellular DCF-DA generated by H2O2 treatment leaked out into the extracellular compartment. Significant plasma membrane injury caused by H2O2 has been reported earlier by others [17]. The membrane leakiness caused by H2O2 might be so severe that it partially contributed to the rapid commitment of cell death observed in Fig. 2 (solid circles).
These results also suggest that experiments designed to assess ROS generation should consider whether the agent of interest causes membrane leakiness. If it does, repeated washing of the cultures after treatment could remove much of the DCF-DA made. This would lead to an under-estimation of ROS generation in the experimental system.
Disparity between ROS curves and viability curves
It should be noted that the ROS generation caused by H2O2 did not fit its cytotoxicity profiles. ROS generation reached a plateau at 500–1000 μM (Fig. 3A), however, the cell death caused by H2O2 occurred at 1–2 mM (Fig. 1D, see 100 μl/well for each concentration). It is possible that the stability of H2O2 in the culture partially accounted for this observation. Even though H2O2 at 500 μM could generate large quantity of ROS, it might be short-lived and fell under the threshold of cytotoxicity as H2O2 degraded in the culture. In contrast, while H2O2 at 1 mM or higher made similar amount of ROS, a higher concentration of H2O2 in the culture could ensure that intracellular ROS remained elevated and continued to cause cellular damage. This idea is consistent with the results from Fig. 1, which suggested that more cytotoxicity was observed if H2O2 concentrations remained high for a longer period of time.
Similar observations regarding ROS generation and cytotoxicity occurred in cultures treated with tBH. This agent at 400 μM or less exhibited little cytotoxicity (Fig. 6, solid circles). However, short-term treatment of cells with 400 μM tBH generated large amount of ROS (Fig. 7A). Analogous to the discussion for H2O2 above, sustained intracellular ROS generation caused by high concentrations of tBH may partially account for these observations.
Temporal commitment point vs. 545/595 emission intensity ratio
The temporal commitment of cell death after H2O2 treatment correlated well with a change in mitochondrial membrane potential. Treatment of cells with 2 mM H2O2 for less than one hour had little effect on 545/595 emission intensity ratio. A significant increase in the ratio, signaling depolarization of mitochondrial membrane potential, occurred at 2 hours after treatment, and continued to increase at 4-hour after treatment (Fig. 4E, solid circles). This transition could be observed clearly in the 545 nm and 595 nm emission intensity plots in Fig. 4A,4B,4C,4D. A decrease of the 595 nm readings and an increase of the 545 nm readings began at 2 hours after 2 mM H2O2 treatment. Cells treated with 1 mM H2O2 maintained the mitochondrial membrane potential well up to 4 hours after treatment (Fig. 4E, open circles), and remained mostly viable one day later (Fig. 1, solid circle at 100 μl/well). Taken together, these results suggest that changes in mitochondrial membrane potential soon after H2O2treatment may be a predictor of cellular viability assessed 24 hours later.
Even though oxidative stress led to mitochondrial membrane depolarization in this study, it should be noted that RPE apoptosis is not always preceded by depolarization of mitochondria membrane potential. For example, Kim et al. reported that RPE apoptosis caused by proteosome inhibition was preceded by a sustained hyperpolarization of mitochondrial membrane potential [18].
15d-PGJ2 prevented oxidative-stress induced cytotoxicity, ROS generation and mitochondrial membrane depolarization
We previously reported that 15d-PGJ2 could prevent H2O2-induced cytotoxicity. Results from this study further indicated that 15d-PGJ2 blocked tBH-induced cytotoxicity (Fig. 6). Pre-treatment of cells with 15d-PGJ2 caused a modest decrease of ROS levels after either H2O2 (Fig, 3B) or tBH (Fig. 7B) treatment. This inhibition may be partially responsible for its saving effect.
In this respect, it is important to mention that 15d-PGJ2 at higher concentrations (5–10 μM) was shown to induce intracellular ROS formation and cell death in human hepatic myofibroblasts [19] and in human neuroblastoma SH-SY5Y cells [20]. In our experimental system, 10 μM 15d-PGJ2 treatment for several days could also lead to cell death. Therefore, the effect of this agent can differ according to the concentration used. Since in this study we focused on the protective effect of 15d-PGJ2, we used this agent at 1 μM or lower concentrations. In this concentration range 15d-PGJ2 did not increase intracellular ROS in ARPE-19 cells after either short-term (20 min) or long-term (overnight) treatment (results not shown).
In order to further determine the mechanism(s) responsible for the saving effect of 15d-PGJ2 against H2O2-and tBH-induced cytotoxicity, we tested the hypothesis that 15d-PGJ2 could block mitochondrial membrane depolarization caused by those agents. Results indicated that 15d-PGJ2 prevented mitochondrial membrane depolarization caused by either H2O2 (Fig. 5) or tBH (Fig. 8).
Given the observations that cells treated with H2O2 or tBH showed ROS generation within 20 min of treatment (Fig. 3 and Fig. 7) but detectable mitochondrial depolarization occurred at 2–4 hours after treatment (Fig. 4 and Fig. 8), there is a possibility that ROS generation could lead to mitochondrial damage and membrane depolarization. 15d-PGJ2 appeared to prevent both events, but with a more prominent inhibitory effect on mitochondrial membrane depolarization.
It was reported that thiazolidinedione activation of PPARγ could enhance mitochondrial membrane potential and promote cell survival [21]. Even though 15d-PGJ2 can activate PPARγ, we have previously reported that the saving effect of this agent in this experimental system does not appear to involve PPARγ activation [3]. For example, ciglitazone (a thiazolidinedione PPARγ agonist) did not prevent H2O2-induced cytotoxicity. Consistent with our previous findings, the results from this study indicated that ciglitazone did not prevent H2O2-induced depolarization of mitochondrial membrane potential. WY14643, a PPARα agonist that had no saving effect in this experimental system [3], also did not prevent mitochondrial membrane depolarization.
Conclusion
This study documented ROS generation and alteration of mitochondrial membrane potential resulting from retinal pigment epithelial cells exposed to H2O2 and tBH. The prostaglandin derivative, 15d-PGJ2, inhibited ROS generation and prevented mitochondrial membrane depolarization, which may contribute to the prevention of cell death caused by oxidative agents. In an earlier report we also demonstrated that 15d-PGJ2 prevented cell death caused by oxysterols, toxic oxidative products of cholesterol [22]. Thus, 15d-PGJ2 may be used as a pharmacological tool to protect cells against oxidative stress or toxic products of oxidative stress.
Methods
Materials
15d-PGJ2 was purchased from Cayman (Ann Arbor, MI). FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone), hydrogen peroxide, t-butyl hydroperoxide, Hank's Balanced Salt Solution (HBSS) and other general biochemical reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. H2DCF-DA (2', 7'-dichlorodihydrofluorescein diacetate) and JC-1 (5,5', 6,6'-tetrachloro-1, 1', 3,3'-tetraethylbenzimidazolocarbocyanine iodide) were from Molecular Probes (Eugene, OR).
Cell cultures
ARPE-19 cells [23] were obtained from the American Type Culture Collection and grown in DMEM/F12 containing 10% newborn calf serum and 2.5 mM glutamine. In order to maintain a comparable growth condition with regard to cell density and medium volume, cells were plated at 20,000 cells/well in 96-well plates (100 μl medium per well) or 140,000 cells/well in 24-well plates (700 μl medium per well).
H2O2 degradation
Cells were plated in 24-well plates for 2 days, rinsed with HBSS, then treated with 700 μl H2O2 solution (prepared in HBSS). Solutions from a set of 3 wells were removed at various periods of time after treatment, then 100 μl of solution were transferred to a quartz 96-well plate, and the concentration of H2O2 was estimated by measuring the optical density (OD) at 240 nm [24,25] using a plate reader (Spectra Max 190, Molecular Devices, Sunnyvale, CA). "Blank" readings, obtained from cultures treated with HBSS only, were subtracted before the final results were plotted. Experiments were performed 3 times with 3 replicates per treatment.
Cell viability
As a general protocol, cells were plated in 96-well plates (20,000 cells/well) overnight, then fed with serum-free medium or treated with a testing agent (prepared in serum-free medium) the next day. Cultures were subjected to oxidative stress (H2O2 or tBH) for a day, and then the viability from each treatment was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay [3,22]. The OD of each well was measured by a plate reader (Spectra Max 190, Molecular Devices, Sunnyvale, CA) with a filter setting at 570 nm (reference filter setting was 630 nm). See figure legends for specific descriptions regarding each experiment. Cells were plated in 24-well plates (140,000 cells/well) for microscopic observations. After treatment, cells were washed twice with phosphate-buffered saline (PBS), then fixed for 10 minutes with 3.7% buffered formaldehyde, examined under a phase contrast microscope Nikon Eclipse TE 200 (Nikon Instruments Inc., Melville, NY). Cells were photographed digitally at a fixed exposure time by using a Photometrics CoolSNAP fx camera (Roper Scientific, Inc., Tucson, AZ) and the software MetaVue, Meta Imaging Series version 4.6r9 (Universal Imaging Corporation, Downingtown, PA).
Measurement of reactive oxygen species
Cells were plated in 96-well plates (20,000 cells/well) overnight, fed with serum-free medium or treated with a testing agent (prepared in serum-free medium) the next day then the assay was performed the following day. Medium from each well was removed and each culture was treated with 10 μM H2DCF-DA (100 μl/well prepared in HBSS) for 20 minutes at 37°C. After removal of H2DCF-DA, cells were treated with H2O2 (or tBH, 100 μl/well prepared in HBSS) for 20 minutes at 37°C. The fluorescence of each well was detected by a fluorescence plate reader (Spectra Max Gemini XS, Molecular Devices, Sunnyvale, CA) with the following settings: excitation 485 nm, emission 535 nm and cutoff 530 nm. This first reading was designated as "total" fluorescence. The H2O2 (or tBH) solution was then replaced with 100 μl HBSS, and the fluorescence from each well was determined again. This second reading was designated as "cell associated" fluorescence. A set of cells without H2DCF-DA treatment was used as "blank" in each experiment. The reading of blank was subtracted before the results were plotted, expressed as net fluorescence units (FU).
Measurement of mitochondrial membrane potential (MMP, ΔΨm)
Cells were plated in 24-well plates (140,000 cells/well) overnight, fed with serum-free medium or treated with a testing agent (prepared in serum-free medium) the next day. The assay was performed the following day. Each culture was treated with H2O2 or tBH (700 μl/well prepared in serum-free medium) for a period of time (indicated in each experiment) at 37°C. After H2O2 treatment, cells were washed once with 500 μl HBSS then incubated with 5 μM JC-1 (300 μl/well) for 15 minutes at 37°C. After this incubation period, cells from each well were rinsed with 500 μl HBSS, then scraped into 300 μl HBSS, transferred into a 96-well tissue culture plate. The fluorescence of each well was detected by a fluorescence plate reader with the following settings: excitation 485 nm, emission 545 nm and 595 nm, cutoff 530 nm. In some experiments, the emission spectra between 520–620 nm were obtained. To prepare JC-1, stock solution (5 mM prepared in DMSO) was diluted (1:4) into 5% BSA, then further diluted (1:199) into HBSS. After proper mixing, this solution was filtered through a 0.2 μm filter, shielded from light until use.
Statistical analysis
Unless otherwise stated, results were pooled from 12 replicate samples derived from 3 independent experiments (cell viability and ROS) or 4 independent experiments (mitochondrial membrane potential), and expressed as mean ± SEM. Statistical analyses were performed by unpaired, two-tailed t test or by analysis of variance (one-way ANOVA) followed by the Bonferroni test to determine the significance of difference among means.
Abbreviations
15d-PGJ2, 15-deoxy-delta 12, 14-Prostaglandin J2; FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone; FU, fluorescence units; H2DCF-DA, 2', 7'-dichlorodihydrofluorescein diacetate; HBSS, Hank's Balanced Salt Solution; JC-1, 5,5', 6,6'-tetrachloro-1, 1', 3,3'-tetraethylbenzimidazolocarbocyanine iodide; MMP, mitochondrial membrane potential (ΔΨm), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide); OD, optical density; PPARs, peroxisome proliferator-activated receptors; ROS, reactive oxygen species; RPE, retinal pigment epithelium; tBH, t-butyl hydroperoxide
Authors' contributions
TKG and JYC were both involved in the experimental design and data collection. Both authors read and approved the final manuscript.
Additional file 1
The PPARγ agonist Ciglitazone and the PPARα agonist WY14643 failed to prevent H2O2-induced mitochondrial membrane depolarization.
This set of experiments was performed under conditions as those described in Fig. 5. (A): Untreated cells; (B): Cells treated with 1.5 mM H2O2 for 2 hours; (C): Cells treated with 10 μM Ciglitazone overnight, then with 1.5 mM H2O2 (without 15d-PGJ2) for 2 hours. (D): Cells treated with 40 μM WY14643 overnight, then with 1.5 mM H2O2 (without 15d-PGJ2) for 2 hours. Note H2O2 caused a shift of the relative intensity of the peaks, indicating mitochondrial depolarization occurred. Pre-treatment of cells with either Ciglitazone or WY14643 failed to prevent H2O2-induced mitochondrial membrane depolarization.
Supplementary Material
{kind=link}
Acknowledgments
Acknowledgements
This work was supported by funds from Research to Prevent Blindness. Support by the Jones Eye Institute and the Pat & Willard Walker Eye Research Center is greatly appreciated. Suggestions provided by Dr. Joel Weinberg (University of Michigan) and Dr. Matt Whiteman (National University of Singapore) in design of the JC-1 assay, and Dr. John M. Ong (Cedars-Sinai Medical Center) in design of the ROS assay are highly appreciated. We also appreciate the helpful discussions provided by the research group led by Dr. Robert Reis (University of Arkansas for Medical Sciences).
Contributor Information
Tarun K Garg, Email: gargtarunk@uams.edu.
Jason Y Chang, Email: changjasony@uams.edu.
References
- Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis. 1999;5:32–42. [PMC free article] [PubMed] [Google Scholar]
- Cai J, Nelson KC, Wu M, Sternberg P., Jr., Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res. 2000;19:205–221. doi: 10.1016/S1350-9462(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Garg TK, Chang JY. Oxidative stress causes ERK phosphorylation and cell death in cultured retinal pigment epithelium: Prevention of cell death by AG126 and 15-deoxy-delta 12, 14-PGJ2. BMC Ophthalmol. 2003;3:5. doi: 10.1186/1471-2415-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- Koppal T, Petrova TV, Van Eldik LJ. Cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prostaglandin J(2) acts as a general inhibitor of inflammatory responses in activated BV-2 microglial cells. Brain Res. 2000;867:115–121. doi: 10.1016/S0006-8993(00)02270-8. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L. Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-deoxy-Delta(12,14)-prostaglandin J(2) in activated murine macrophages. Mol Cell Biol. 2000;20:1692–1698. doi: 10.1128/MCB.20.5.1692-1698.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–2515. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- Scanlon JM, Reynolds IJ. Effects of oxidants and glutamate receptor activation on mitochondrial membrane potential in rat forebrain neurons. J Neurochem. 1998;71:2392–2400. doi: 10.1046/j.1471-4159.1998.71062392.x. [DOI] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Castedo M, Ferri K, Roumier T, Metivier D, Zamzami N, Kroemer G. Quantitation of mitochondrial alterations associated with apoptosis. J Immunol Methods. 2002;265:39–47. doi: 10.1016/S0022-1759(02)00069-8. [DOI] [PubMed] [Google Scholar]
- Weinberg JM, Saikumar P. Mitochondrial function. Methods Mol Med. 2003;86:351–371. doi: 10.1385/1-59259-392-5:351. [DOI] [PubMed] [Google Scholar]
- Ballinger SW, Van Houten B, Jin GF, Conklin CA, Godley BF. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp Eye Res. 1999;68:765–772. doi: 10.1006/exer.1998.0661. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403. doi: 10.1016/S0014-4835(03)00023-X. [DOI] [PubMed] [Google Scholar]
- Weigel AL, Handa JT, Hjelmeland LM. Microarray analysis of H2O2-, HNE-, or tBH-treated ARPE-19 cells. Free Radic Biol Med. 2002;33:1419–1432. doi: 10.1016/S0891-5849(02)01082-1. [DOI] [PubMed] [Google Scholar]
- Baker MA, He SQ. Elaboration of cellular DNA breaks by hydroperoxides. Free Radic Biol Med. 1991;11:563–572. doi: 10.1016/0891-5849(91)90137-R. [DOI] [PubMed] [Google Scholar]
- Scott JA, Fischman AJ, Homcy CJ, Fallon JT, Khaw BA, Peto CA, Rabito CA. Morphologic and functional correlates of plasma membrane injury during oxidant exposure. Free Radic Biol Med. 1989;6:361–367. doi: 10.1016/0891-5849(89)90080-4. [DOI] [PubMed] [Google Scholar]
- Kim JM, Bae HR, Park BS, Lee JM, Ahn HB, Rho JH, Yoo KW, Park WC, Rho SH, Yoon HS, Yoo YH. Early mitochondrial hyperpolarization and intracellular alkalinization in lactacystin-induced apoptosis of retinal pigment epithelial cells. J Pharmacol Exp Ther. 2003;305:474–481. doi: 10.1124/jpet.102.047811. [DOI] [PubMed] [Google Scholar]
- Li L, Tao J, Davaille J, Feral C, Mallat A, Rieusset J, Vidal H, Lotersztajn S. 15-deoxy-Delta 12,14-prostaglandin J2 induces apoptosis of human hepatic myofibroblasts. A pathway involving oxidative stress independently of peroxisome-proliferator-activated receptors. J Biol Chem. 2001;276:38152–38158. doi: 10.1074/jbc.M101980200. [DOI] [PubMed] [Google Scholar]
- Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J Biol Chem. 2001;276:12076–12083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- Wang YL, Frauwirth KA, Rangwala SM, Lazar MA, Thompson CB. Thiazolidinedione activation of peroxisome proliferator-activated receptor gamma can enhance mitochondrial potential and promote cell survival. J Biol Chem. 2002;277:31781–31788. doi: 10.1074/jbc.M204279200. [DOI] [PubMed] [Google Scholar]
- Chang JY, Liu L. Peroxisome proliferator-activated receptor agoinsts prevent 25-OH-cholesterol induced c-jun activation and cell death. BMC Pharmacol. 2001;1:10. doi: 10.1186/1471-2210-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–126. doi: 10.1016/S0076-6879(84)05016-3. [DOI] [PubMed] [Google Scholar]
- Nourooz-Zadeh J. Ferrous ion oxidation in presence of xylenol orange for detection of lipid hydroperoxides in plasma. Methods Enzymol. 1999;300:58–62. doi: 10.1016/s0076-6879(99)00113-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.