

Abstract
Background
Clostridium difficile and C. sordellii are two anaerobic, spore forming, gram positive pathogens with a broad host range and the ability to cause lethal infections. Despite strong similarities between the two Clostridial strains, differences in their host tissue preference place C. difficile infections in the gastrointestinal tract and C. sordellii infections in soft tissues.
Results
In this study, to improve our understanding of C. sordellii and C. difficile virulence and pathogenesis, we have performed a comparative genomic and phenomic analysis of the two. The global phenomes of C. difficile and C. sordellii were compared using Biolog Phenotype microarrays. When compared to C. difficile, C. sordellii was found to better utilize more complex sources of carbon and nitrogen, including peptides. Phenotype microarray comparison also revealed that C. sordellii was better able to grow in acidic pH conditions. Using next generation sequencing technology, we determined the draft genome of C. sordellii strain 8483 and performed comparative genome analysis with C. difficile and other Clostridial genomes. Comparative genome analysis revealed the presence of several enzymes, including the urease gene cluster, specific to the C. sordellii genome that confer the ability of expanded peptide utilization and survival in acidic pH.
Conclusions
The identified phenotypes of C. sordellii might be important in causing wound and vaginal infections respectively. Proteins involved in the metabolic differences between C. sordellii and C. difficile should be targets for further studies aimed at understanding C. difficile and C. sordellii infection site specificity and pathogenesis.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1663-5) contains supplementary material, which is available to authorized users.
Keywords: Clostridium difficile, Clostridium sordellii, Comparative genomics, Phenotype microarray, Urease
Background
The bacterial class, Clostridia, is typified by gram-positive anaerobes and includes several important human pathogens. The main virulence factors produced by pathogenic Clostridia are secreted toxins. While C. botulinum (botulism) and C. tetani (tetanus) are the best known of these pathogens, other members, in particular C. difficile, have become increasingly notorious due to an accelerating number of documented infections in recent years. In North America and Europe, C. difficile infection (CDI) is now the leading cause of infectious diarrhea [1–3]. CDI can cause a varying range of diseases from mild diarrhea to fulminant colitis and death [4–6]. The primary risk factors of CDI include antibiotic treatment, advanced age, severe underlying illness, prior hospitalization, tube feeding, gastrointestinal surgery, and use of proton-pump inhibitors [7, 8]. C. difficile also has a broad host range and causes infection in agriculturally important animals such as pigs, cattle, horses and chickens [9–13].
Recent studies on the Clostridium genus support a reclassification of C. difficile and the related Cluster XI into a family-level group that is distinct from the current Clostridiaceae family (renaming family genus spp. to Peptostreptococcaceae Peptoclostridium difficile has been suggested) [14]. Along with C. difficile, Cluster XI currently includes several clinically significant members, C. sordellii, Filifactor alocis, and Peptostreptococcus anaerobius [15–17]. C. sordellii infection (CSI), although not as prevalent as CDI, has a very high mortality rate that can often reach 75 % lethality [15]. Both C. sordellii and C. difficile are asymptomatically carried in the gastrointestinal tracts of about 10 % of adult humans [15, 18] and both species also can infect animals [19–22]. In addition, C. sordellii and C. difficile excrete potent toxins with immunological cross-reactivity and similar biological activities [15, 23, 24]. Despite the close similarities in host range and virulence factors, there are two striking differences between C. difficile and C. sordellii. First is that while C. difficile only colonizes the gastrointestinal tract, C. sordellii can colonize both the human gastrointestinal tract and vagina [25]. Secondly, while C. difficile infection affects the host intestine, C. sordellii primarily causes soft tissue infection. The tissue preference of C. sordellii results in CSI being primarily reported among reproductive-age women following natural childbirth, spontaneous, surgical or medical abortions [15, 26]. Wounds from illicit injectable drug use, non-gynecological surgical procedures, penetrating crushing injuries, or traumatic injury in previously healthy men, women, and children can also lead to CSI infection [27–32].
Comparative analyses of closely related bacteria with different infection site specificity and pathogenicity can provide information relevant to understanding adaptation to host environments and mechanisms of infection. Genomic differences can lead to phenotype level changes. In bacteria, phenotypic variations are often related to metabolic changes, which are defined by the ability to utilize various sources of carbon, nitrogen, sulfur, phosphorous, and other essential nutrients. With the development of Phenotype microarrays (PMs), high-throughput determination of a microorganism’s global metabolic phenotype or phenome is now possible [33–37]. In this study, to determine the genomic and phenomic basis for the differences in C. sordellii and C. difficile infections, we have performed a comparative genomic and phenomic analysis of these two species. The global phenome of C. sordellii and C. difficile revealed several differences, most notably in acid resistant growth. We further explored the genomic basis for phenomic differences by determining a draft genome sequence of C. sordellii strain 8483 and comparing it against two C. sordellii genomes (strains ATCC_9714, and VPI_9048) and eight C. difficile genomes (strains 630, BI1, CD196, M68, R20291, 2007855, CF5, and M120). Understanding the differential adaptions to host tissue at the genomic and phenomic level should provide opportunities in the fight against these important infections.
Results and discussion
Nutritional phenomic comparison
In this study, we have performed the comparative analysis of the genome and phenome of the two closely related Clostridial pathogens, C. sordellii and C. difficile. To determine the global phenome of C. sordellii and C. difficile, we used Biolog phenotype microarrays (PMs) which enable whole cellular level determination of bacterial phenotypes [35]. The nutritional PM analysis consisted of 190 assays of carbon source metabolism, 94 assays of phosphorous and sulfur source metabolism, 95 assays of biosynthetic pathways, and 380 assays of nitrogen source metabolism [35]. We have previously analyzed the phenome of six C. difficile strains [38]. We compared those results with phenome of C. sordellii strain 8483 determined in this study. From a total of 759 nutritional phenotype assays, 160 were positive (indicated by a 40 % growth enhancement relative to the control) for C. sordellii strain 8483, while 132 were positive for C. difficile strain 630 (Additional file 1: Table S1 and Additional file 2: Table S2). C. sordellii and C. difficile shared 65 positive phenotypes while 162 positive phenotypes were specific to just one of the two species. A disproportionate number of unshared phenotypes were identified in the carbon and nitrogen source assays (~3.0× unshared vs. shared for carbon and nitrogen combined; ~1.3× unshared vs. shared for other sources combined). The unshared carbon and nitrogen phenotypes are indicative of species specific adaptations to environmentally available nutrient sources.
A basic illustration of the carbon source preference exhibited by each species is depicted in a table listing all 85 positive carbon phenotype conditions (Fig. 1a). A C. sordellii to C. difficile ratio of growth enhancement was used to more directly compare the ability of the two species to utilize specific carbon sources (Fig. 1b). The tested carbon source phenotypes were grouped based on the type of carbon source molecules. C. difficile more effectively utilized typical carbohydrates, while C. sordellii was better able to utilize non-carbohydrates, including amino acids and fatty acids, as carbon sources. Carbon sources grouped as carbohydrates were further divided into molecular sub-types resulting in a similar trend. Most typical saccharides could be utilized more effectively by C. difficile than by C. sordellii, yet saccharides that have been modified for incorporation into nucleic acids or phosphorylated for entering in metabolic pathways can be more easily utilized by C. sordellii. The difference in ability to exploit a broad range of non-carbohydrate molecules as carbon sources could improve the likelihood of C. sordellii to survive within the more complex environment of soft tissue.
Fig. 1.

Carbon source utilization microarray. a The list includes all positive carbon source phenotypes (green) for C. sordellii and C. difficile (lanes 1 and 2, respectively). Negative phenotypes are red. b The C. sordellii : C. difficile ratio of growth enhancement for each carbon source phenotype (gray circle) is grouped by carbon source type. The average for each group is indicated (green box). The carbohydrate group was further divided into sub-types
Figures 2a and 2b list all 101 positive nitrogen and peptide nitrogen source phenotype conditions, respectively. The positive phenotype threshold for nitrogen source growth enhancement was achieved for more than 2× as many simple nitrogen source conditions by C. difficile than by C. sordellii; however, C. sordellii was able to reach the positive phenotype threshold for nearly 2× as many peptide nitrogen conditions as C. difficile. For example, C. difficile could better utilize the simple amino alcohol, ethanolamine, which is an abundant nitrogen source in the gut. As C. sordellii was better able to utilize protein building blocks as carbon sources, the fact that this preference extends to nitrogen sources is not surprising. A C. sordellii to C. difficile ratio of growth enhancement was used to compare nitrogen source utilization between the two species for the 246 di-peptide nitrogen source phenotypes. The ratios were grouped by the presence of amino acids in the di-peptide sources and organized into higher level groupings by their expected catabolic pathway leading to nitrogen metabolism as defined by available KEGG pathway enzymes [39] in the C. sordellii genome (Fig. 2c). Because the di-peptide nitrogen source microarray was not comprehensive, the amino acid sample size was variable. For most amino acids, data was obtained from between 22 and 36 di-peptide conditions. Ratios for asparagine (n = 9), glutamine (n = 14), and threonine (n = 15) trended similarly to amino acids within the same grouping. The sample size for cysteine di-peptides (n = 2) was small and only incorporated glycine, but interestingly the growth ratio was strongly dependent on amino acid position. Analysis of only the first amino acid was also included in Fig. 2c since the distributions were generally narrower than when both amino acid positions 1 and 2 were included. Amino acids in group I are likely to follow a pathway that generates pyruvic acid when ammonia is liberated as a nitrogen source (Fig. 2d). The largest bias for C. sordellii growth enhancement was observed for group I amino acids: glycine, serine, and threonine which are linked to the same pathway. The other notable amino acid group (III) contains aspartic acid and asparagine, which are catabolized to oxaloacetate, and are more effectively utilized by C. difficile than by C. sordellii. In comparison to C. difficile, C. sordellii has adapted to the broader use of available peptides (glycine, serine, and threonine are of relatively high abundance in human genes [40]) as nutrient sources, which could help C. sordellii infiltrate soft tissue.
Fig. 2.
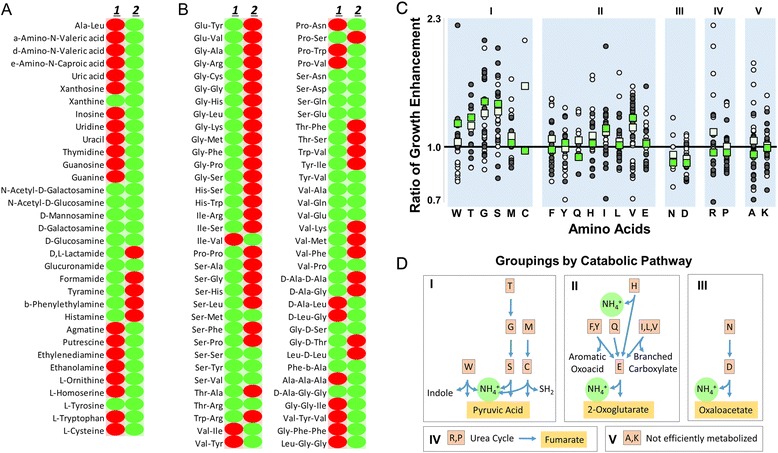
Nitrogen source utilization microarray. The lists include all positive simple (a) and di- and tri-peptide (b) nitrogen source phenotypes (green) for C. sordellii and C. difficile (lanes 1 and 2, respectively). Negative phenotypes are red (c). The C. sordellii : C. difficile ratio of growth enhancement for each dipeptide nitrogen source phenotype is grouped by amino acid (circles; average – white squares). Gray circles indicate the position 1 amino acid (position 1 average – green squares) while white circles indicate the position 2 amino acid. Amino acids are further grouped (I-V) by their expected pathway to nitrogen metabolism (d)
Our analysis finds that, at the phenome level, both of these closely related species share a core group of functions and phenotypes. Our phenotype array results also reveal some key phenotype differences particularly for carbon and nitrogen source utilization that might explain the differences in the primary site of infections caused by these two species. Both glycolysis and amino acid catabolism are differentially regulated in C. difficile suggesting a possible mechanism for how these pathways have evolved to better occupy the respective niche for both C. sordellii and C. difficile [41].
Osmolyte and pH phenomic comparison
Two additional PM arrays were utilized for osmolyte and pH sensitivity (Additional file 1: Table S1). Optimum growth was obtained at pH 6.0 for C. difficile. An analysis of pH-dependent growth normalized to growth at pH 6.0 for each species revealed that C. sordellii also achieved near-optimal growth levels at pH 6.0 (Fig. 3). Further, C. sordellii performed better than C. difficile at off-optimal growth pHs. At pH 4.0, growth of C. sordellii is 2× as robust as C. difficile relative to growth at pH 6.0. Because the normal, healthy pH of the vagina is pH 4.5 or lower, the ability of C. sordellii to grow at acidic pH plays a crucial role in facilitating vaginal infections [42]. Fecal contamination of the vagina during vaginal delivery could provide a source of organisms that may infect vaginal tears, episiotomy sites or ascend to the uterus through the open cervix [15]. Interestingly, C. sordellii was able to maintain similar growth levels from pH 6 through pH 10 while the growth of C. difficile was modestly reduced above pH 7 suggesting that C. sordellii has an improved survivability at both acidic and basic pHs.
Fig. 3.
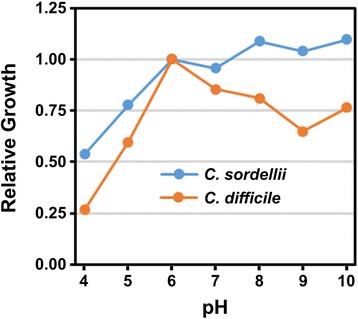
Growth dependence on pH. Phenotype Microarray growth data for the pH range 4–10 was obtained from PM10-A (Additional file 1: Table S1). For each species, the data was normalized to pH 6, which is the optimal growth pH of C. difficile
C. difficile showed better adaptation to growth under 78 % of osmolyte conditions; however, C. sordellii had a growth advantage over C. difficile in 83 % of urea-specific conditions. The combination of low pH and urea led to a > 40 % growth enhancement for C. sordellii over pH 6.0 conditions while the same combination inhibited growth in C. difficile by > 40 % relative to optimal growth at pH 6.0. C. sordellii has previously been shown to exhibit urease activity [43]. Details on the C. sordellii urease gene cluster and its implications for growth improvement under multiple conditions including acidic pH are discussed in the later sections.
Phylogeny
Clostridium species have been classified into different phylogenetic groups with C. sordellii and C. difficile belonging to Clostridial cluster XI [44]. In this study, 134 orthologous genes with greater than 90 % bootstrap support were identified and used to infer phylogenetic relationships of 9 bacterial species from Clostridial clusters I, XI, XII, and XIII. The phylogenetic tree inferred from a concatenation of the 134 genes (Fig. 4) agreed with and further confirmed the ribosomal proteins-based phylogeny reported in a previous study [14], placing Clostridium acidurici (cluster XII) as a sister group of Anaerococcus prevotii and Finegoldia magna (cluster XIII), joined by the cluster XI (C. sordellii, C. difficile, Peptostreptococcus anaerobius, Clostridium sticklandii, and Filifactor alocis), and finally Clostridium perfringens (cluster I) as an outgroup. The concatenation phylogeny demonstrated that the majority (71 %) of genes supported the monophyly of the clade of C. sordellii and C. difficile, suggesting that C. sordellii is a sister group to C. difficile.
Fig. 4.
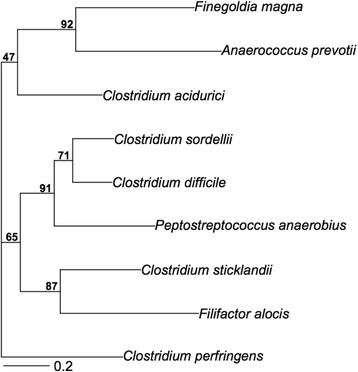
Phylogeny. Maximum likelihood tree obtained from a concatenated nucleotide sequence alignment of the 134 orthologous genes of the 9 bacterial species from Clostridial clusters I, XI, XII, and XIII. The horizontal bar at the base of the figure represents 0.2 substitutions per nucleotide site. The percentages of genes that support the branches of the tree are indicated
Gene repertoire
Species with similar gene contents should have similar functional potential as a whole [45]. Whole gene contents of any genome can be the result of a mixture of different evolutionary events such as vertical inheritance of genes and their duplication, gain and loss events, and their relative contributions may vary among different species [46]. The 22,612 proteins from the 9 bacterial species from Clostridial clusters I, XI, XII, and XIII were classified into 5979 homologous groups or protein families. Of the 5979 protein families, 3578 were present in a single strain, 2401 were present in two or more strains, of which 487 were shared by all the strains.
Cluster analysis of the 9 genomes based on the whole gene content using four agglomeration methods (i.e., single-linkage, complete-linkage, average-linkage, and neighbour-joining clustering) produced four distinct trees (Fig. 5), which were all incongruent with the concatenation phylogeny (Fig. 4). Previous studies also reported that whole gene contents of bacteria do not strictly follow their phylogenetic relationships [47–50] and can lead to a distorted ancestral lineage [51]. Although all four gene-content trees were topologically distinct and none matched the topology of the concatenation phylogeny, C. sordellii and C. difficile ubiquitously formed a single cluster (designated with ** in Fig. 5). Because all four gene-content trees suggest that C. sordellii and C. difficile are more similar to one another than to any other species, a comparison of the genomic differences might provide useful information regarding their important clinical differences.
Fig. 5.
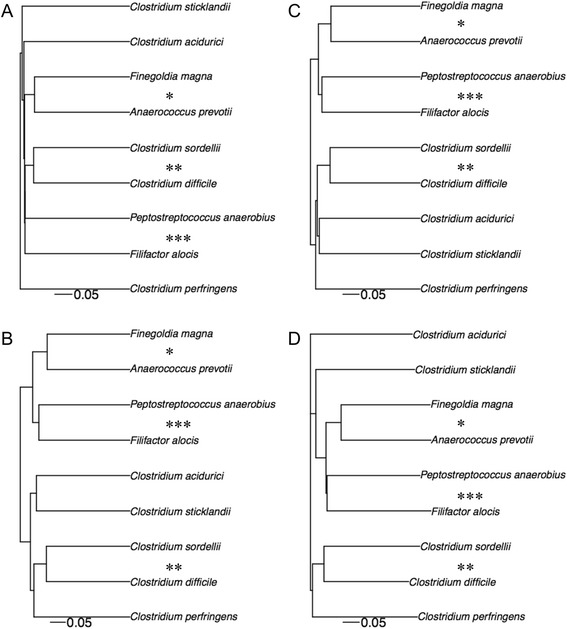
Whole gene content. Trees constructed by cluster analysis of the 9 Clostridial species based on whole gene content using four agglomeration methods: single-linkage a, complete-linkage b, average-linkage c, and neighbour-joining d clustering. Clusters formed by all four methods are identified
A total of 135 protein families were present in C. sordellii and C. difficile but absent in the other 7 species. The protein families included stage III sporulation protein AF, flagellar protein FliZ, and K+-transporting ATPase subunits A (KdpA) and C (KdpC). A total of 296 protein families were present in C. sordellii and C. difficile but absent in the other 3 species (P. anaerobius, C. sticklandii, and F. alocis) in the cluster XI. Within the cluster XI, genome size and the number of protein-coding sequences (CDS) were larger in C. sordellii (3.6 Mbps and 3586 CDS, respectively) and C. difficile (4.3 Mbps and 3758 CDS, respectively) than in the 3 other species (1.9 - 2.7 Mbps and 1871–2573 CDS, respectively), while genome G + C content of C. sordellii (27 %) and C. difficile (29 %) were lower than those of the other 3 species (33 - 36 %) (Fig. 6; Additional file 3: Table S3). Our results suggest that C. sordellii and C. difficile increased their genome size and/or the other cluster XI lineages (P. anaerobius, C. sticklandii, and F. alocis) decreased their genome size after divergence from the common ancestor of cluster XI. The larger genome size of C. difficile and C. sordellii relative to other members of cluster XI may be linked to their unique set of shared genes.
Fig. 6.
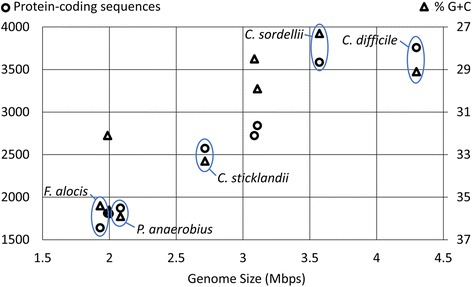
Clostridial genome size. For the 9 Clostridial species analyzed, the number of protein-coding sequences (o) are correlated with genome size (Mbps). The percentage of G + C (∆) is negatively correlated with genome size and is plotted on an inverted axis. The 5 members of Clostridial cluster XI are identified
Gene repertoire comparison between C. sordellii and C. difficile
We compared the gene repertoire between eight C. difficile strains (630, BI1, CD196, M68, R20291, 2007855, CF5, and M120) and three C. sordellii strains (8483, ATCC_9714, and VPI_9048). Proteins from the 11 strains were classified into 4368 homologous groups (protein families); (see Additional file 4: Table S4 for a comprehensive list). Of the 4368 protein families, 928 were present in a single strain, 3440 were present in two or more strains, of which 1395 were shared by all the strains.
We performed gene set enrichment analysis to examine over- and underrepresented functional categories in C. sordellii relative to C. difficile (Additional file 5: Table S5). We calculated the odds ratio (OR) to rank the relative overrepresentation (OR > 1) and underrepresentation (OR < 1) of each functional category, and P-value of Fisher’s exact test. Fig. 7 shows the OR values of SEED subsystems as examples. The SEED subsystem “Urea_decomposition” (OD = 11.9), the Gene Ontology (GO) term “nickel cation binding (GO:0016151)” (OD = Infinite), and the Virulence Factors Database (VFDB) keywords “Acid resistance”, “Colonization”, and “Enzyme” (OD = Infinite) were significantly overrepresented in C. sordellii relative to C. difficile based on Fisher’s exact test after false discovery rate correction for multiple comparisons (FDR < 0.05).
Fig. 7.
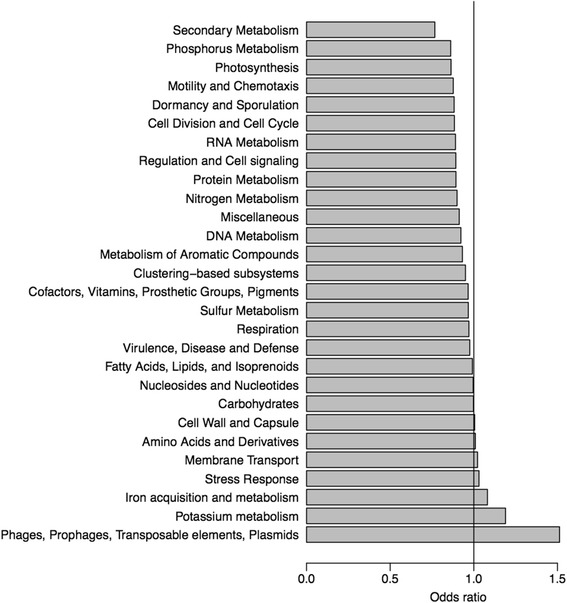
Gene set enrichment. Odds ratio to rank the relative overrepresentation (>1) or underrepresentation (<1) of each SEED category in the C. sordellii genome relative to the C. difficile genome
A total of 738 protein families were present in all the 3 C. sordellii strains but absent in all the 8 C. difficile strains (Additional file 4: Table S4). These C. sordellii-specific genes may have been gained on the branch leading to the C. sordellii strain, and could be linked to its specific environmental adaptation and pathogenesis. They included some genes in the pathogenicity locus; i.e., locus_tag WS9_01807 to WS9_01812 in strain 8483, H476_0268 to H476_0289 in strain VPI_9048, and H477_0262 to H477_0286 for strain ATCC_9714 [52]. They included several amino acid decarboxylases and deaminases (e.g., glutamate decarboxylase [EC:4.1.1.15], histidine decarboxylase [EC:4.1.1.22], and L-serine deaminase) that might enable C. sordellii to grow on peptide nutrient sources in soft tissue. In addition, glutamate decarboxylase and arginine deiminase [EC 3.5.3.6] produce alkaline byproducts that have been suggested to participate in the acid resistance of gram-positive bacteria [53] and improve C. sordellii growth in acidic environments. Two protein families annotated as “KUP system potassium uptake protein (K03549)” and “potassium voltage-gated channel Shab-related subfamily B member 1 (K04885)” were present in C. sordellii strains but absent in C. difficile strains. Proteins involved in potassium transport often play a role in adaptive pH tolerance [54] and may increase broad pH range survival in C. sordellii.
SEED subsystems
The SEED annotation engine defines genes associated with a functional role in a bacterial genome as a subsystem [55]. A SEED subsystem is termed as a generalization of the term “pathway” and is a convenient framework for functional comparisons of bacterial genomes [55]. Of the 4368 protein families, 854 were assigned to the SEED subsystems (Additional file 4: Table S4). Both C. sordellii and C. difficile contained some of the important functions relevant to strain transmission and colonization. For example, both species contained 20 protein families assigned to “Dormancy and Sporulation” including sporulation sigma factor. Genes related to spore coat (cotA, cotB, cotCB, cotD, and cotE) are present in both C. difficile and C. sordellii genomes. This is consistent with the previous reports on C. difficile and C. sordellii spore properties [25, 56, 57]. Of the 14 protein families assigned to “Iron acquisition and metabolism”, hemerythrin-like iron-binding protein was present in C. sordellii but absent in C. difficile. Iron acquisition is essential for growth of pathogenic bacteria during soft tissue infections [58] and is likely to be important for C. sordellii in proliferating in host tissues. It has been shown that Stickland metabolism is important in C. difficile physiology [59]. In C. difficile, several genes located in D-proline reductase operon and glycine reductase operon are involved in Stickland associated metabolism. When C. sordellii genomes were compared to C. difficile genomes, most genes in the prd (prdC, prdR, prdA, prdB, prdD, prdE) and grd (grdA, grdB, grdC, grdD, grdE, and grdX) operons were found to be conserved.
Virulence Factors Database (VFDB)
We used the Virulence Factors Database (VFDB) [60] to assess the presence of virulence genes in the C. sordellii and C. difficile strains (Additional file 4: Table S4). Toxins A (tcdA) and B (tcdB) were homologous, and the homologous proteins were present in all the 11 genomes of C. difficile and C. sordellii. For strain ATCC_9714, locus_tag H477_0265 (Truncated TcsH) is annotated as toxin A, and H477_0263 (Cytotoxin L) is annotated as “toxin B”. For VPI_9048, locus_tag H476_0269 [cytotoxin L (TcsL)] and H476_0271 [Hemorrhagic toxin (TcsH)] are annotated as toxin B [52]. In the strain C. sordellii 8483 locus tags WS9_01807 to WS9_01812 corresponds to toxin B and locus tag WS9_01787 correspond to toxin A. However, C. sordellii being a draft genomes, the locus tags in C. sordellii strains VPI_9048 and 8483 represent partial sequence of the toxin genes.
Four protein families homologous to collagenase (colA), sialidase (nanH), perfringolysin O (pfoA), and phospholipase C (plc), respectively, from C. perfringens were present in C. sordellii but absent in C. difficile. At the genome level, C. sordellii contains genes encoding enzymes for host tissue lysis and nutrient release during infection (e.g., hyaluronidase and hemolysin) [61]. The presence of these enzymes coupled with the ability to metabolize a larger set of peptides is likely to be a contributing factor in the ability of C. sordellii to cause lethal soft tissue infections. In addition, a cluster of eight genes encoding urease subunits (UreA (λ), UreB (β) and UreC (α)) and urease accessory proteins (UreI, UreE, UreF, UreG and UreH) homologous to known virulence factors of Helicobacter pylori 26695 (Enzyme; Acid resistance; Colonization) were present in C. sordellii but absent in C. difficile.
Clusters of Orthologous Groups (COG)
The Clusters of Orthologous Groups (COG) database [62] defines four major functional categories: “information storage and processing”, “cellular processes and signaling”, “metabolism”, and “poorly characterized”, which are further subdivided into 25 functional categories. Of the 4368 protein families, 1970 were assigned to the COG functional categories (Additional file 4: Table S4). The 211 protein families were assigned to COG functional category E (Amino acid transport and metabolism), of which 36 protein families were present in C. sordellii but absent in C. difficile (Table 1). C. sordellii can better metabolize dipeptides than C. difficile as illustrated in Fig. 2. Several C. sordellii protein families including ABC-type transport systems for dipeptides or amino acid, a variety of peptidases, and amino acid degradation enzymes could be responsible. A C. sordellii L-serine deaminase might facilitate not only improved utilization of serine as a nitrogen source but also improved glycine and threonine metabolism within the di-peptide analysis. A branched chain amino acid aminotransferase from C. sordellii might explain the improved metabolism of isoleucine and valine. The abundance in amino acid associated functions are consistent with our finding in the phenotype level that C. sordellii has more capacity to use a wider range of amino acids and peptides as nutrient sources than C. difficile.
Table 1.
Protein families assigned to COG functional category E (Amino acid transport and metabolism) that are present in C. sordellii strain 8483 but absent in C. difficile strain 630
| COG functional annotation |
| COG0076E|Glutamate decarboxylase and related PLP-dependent proteins |
| COG0115EH|Branched-chain amino acid aminotransferase/4-amino-4-deoxychorismate lyase |
| COG0346E|Lactoylglutathione lyase and related lyases |
| COG0477GEPR|Permeases of the major facilitator superfamily |
| COG0493ER|NADPH-dependent glutamate synthase beta chain and related oxidoreductases |
| COG0549E|Carbamate kinase |
| COG0697GER|Permeases of the drug/metabolite transporter (DMT) superfamily |
| COG0703E|Shikimate kinase |
| COG0747E|ABC-type dipeptide transport system, periplasmic component |
| COG0757E|3-dehydroquinate dehydratase II |
| COG0804E|Urea amidohydrolase (urease) alpha subunit |
| COG0831E|Urea amidohydrolase (urease) gamma subunit |
| COG0832E|Urea amidohydrolase (urease) beta subunit |
| COG0834ET|ABC-type amino acid transport/signal transduction systems, periplasmic component/domain |
| COG1104E|Cysteine sulfinate desulfinase/cysteine desulfurase and related enzymes |
| COG1410E|Methionine synthase I, cobalamin-binding domain |
| COG1703E|Putative periplasmic protein kinase ArgK and related GTPases of G3E family |
| COG1760E|L-serine deaminase |
| COG2235E|Arginine deiminase |
| COG2755E|Lysophospholipase L1 and related esterases |
| COG2856E|Predicted Zn peptidase |
| COG2866E|Predicted carboxypeptidase |
| COG2986E|Histidine ammonia-lyase |
| COG2987E|Urocanate hydratase |
| COG3033E|Tryptophanase |
| COG3191EQ|L-aminopeptidase/D-esterase |
| COG3227E|Zinc metalloprotease (elastase) |
| COG3643E|Glutamate formiminotransferase |
| COG4401E|Chorismate mutase |
| COG4448E|L-asparaginase II |
| COG4608E|ABC-type oligopeptide transport system, ATPase component |
| COG0141E|Histidinol dehydrogenase |
Urease gene cluster in environmental adaptation
Historically, a high level of urease activity has been used in the positive taxonomic identification of C. sordellii, although a number of urease-negative C. sordellii isolates have been identified [43]. A cluster of eight genes in C. sordellii strain 8483 was found to encode urease subunits: UreA (λ), UreB (β), UreC (α), UreI, UreE, UreF, UreG, and UreH (Fig. 8a). The organization of the urease gene cluster is similar to that of Helicobacter sp. [63, 64], yet the entire gene cluster was absent in C. difficile. Homologues to the eight urease genes are also present in C. perfringens; however, several of these urease genes are positioned on a large plasmid [65]. The set of urease proteins play an important function in acid resistance while also contributing a significant nitrogen source [53, 63]. UreA (λ), UreB (β), and UreC (α) form the core urease complex involved in conversion of urea to ammonia and carbonic acid [66]. Cytoplasmic urease activity substantially increases the intracellular pH and is essential for survival of many acidophilic pathogens including Helicobacter pylori. The proteins in the C. sordellii urease complex are highly homologous to C. perfringens and H. pylori (Fig. 8a) and share the highest degree of conservation near the enzyme active site (Fig. 8b). UreI forms a urea channel that allows efficient entrance of urea into the cytoplasm for urease degradation [67]. UreI is also homologous to C. perfringens and H. pylori with a high degree of conservation for residues lining the pore (Fig. 8c). The relatedness of proteins from the C. sordellii urease gene cluster to other pathogen urease proteins (particularly near functional residues) suggests a similar role in low pH tolerance (Fig. 9). In addition, urea is a byproduct of protein degradation creating a potential nitrogen source during infection. The two major phenotypes that provide C. sordellii the opportunity to colonize the vagina and soft tissue are the ability to survive below pH 4.5 and to utilize peptides as nitrogen sources. These two functions likely make the urease gene cluster important in establishing and maintaining C. sordellii infections. Future studies directed at disrupting the C. sordellii urease gene cluster (i.e., gene knockouts) should better define the gene cluster’s role in the low pH tolerance exhibited by C. sordellii.
Fig. 8.
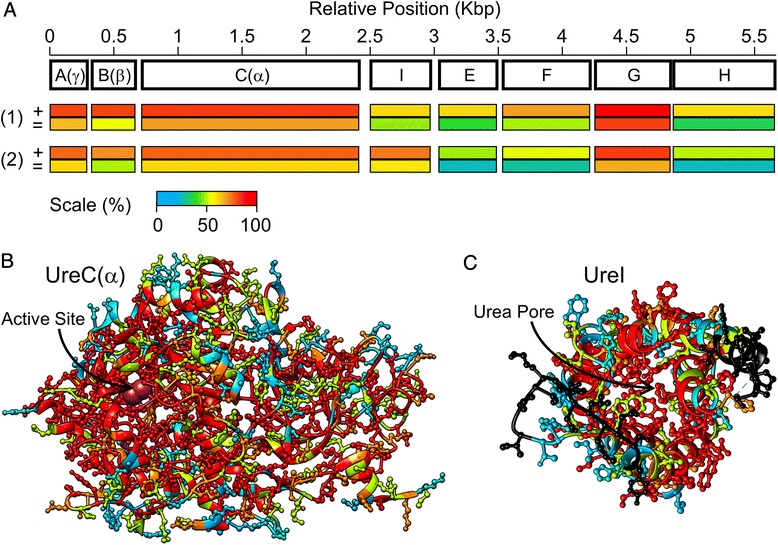
Urease gene cluster. a The size and organization of C. sordellii urease genes is illustrated. Overall homology of individual proteins to (1) H. pylori and (2) C. perfringens is depicted by colored bars for (+) % similarity and (=) % identity located below each gene. The scale is defined with red > 90 % and cyan < 20 %. b The urease enzyme structure from H. pylori (PDBID: 1E9Z) [66] and the urea channel structure from H. pylori (PDBID: 3UX4) [67] are both shown to illustrate the degree of conservation with corresponding C. sordellii urease proteins near the functional sites. Using the same scale for a, residues colored in red are conserved across H. pylori, C. perfringens and C. sordellii, while residues colored in cyan are not conserved
Fig. 9.
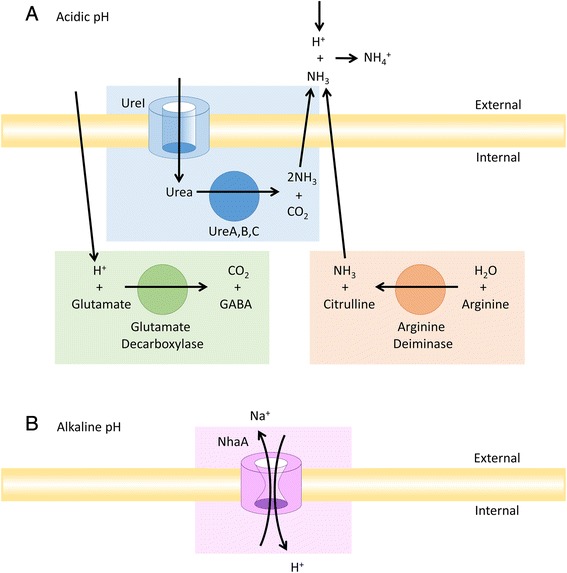
Possible mechanisms of acid adaptation. Several C. sordellii genes could be utilized to sequester protons and improve the internal and surrounding pH. a The expected contributions of urease, glutamate decarboxylase, and arginine deiminase to increasing the cytoplasmic pH under acidic conditions are depicted. b The NhaA Na+/H+ antiporter is expected to improve alkaline adaptation
Other genes involved in environmental stress tolerance
Several other C. sordellii genes that are absent from C. difficile could play a role in adaptation to acidic conditions (Table 1; Fig. 9a). Glutamate decarboxylase converts glutamate to γ-aminobutyric acid (GABA) which absorbs a proton during the reaction. Arginine deiminase generates ammonia which can also absorb a proton and create a more alkaline internal, periplasmic, or local pH in an acidic environment. Some bacteria can accumulate high cytoplasmic potassium levels under acidic stress. Potassium transport is expected to play a role in adaptation to acidic environments by maintaining the membrane potential for optimum bioenergetics homeostasis [68, 69], yet the exact mechanism of how the various transporters and channels work together to support the internal pH is not fully understood. Potassium homeostasis is also pivotal in the osmotic stress response.
Similar growth of C. sordellii strain 8483 from pH 6 to a more alkaline pH range was observed (Fig. 3). A homologue of the Na+/H+ antiporter, NhaA, was identified in the genome of C. sordellii strains 8483, ATCC 9714 and VPI 9048 but was identified only in C. difficile strain F501. In Escherichia coli and Salmonella enterica, NhaA has been implicated as a mechanism for maintaining internal pH homeostasis under alkaline conditions by catalyzing H+ uptake for a preferred pHout range of 6.5 to 8.5 [70, 71]. The presence of NhaA in all C. sordellii strains but absence from most C. difficile strains could explain the ability of C. sordellii to maintain growth levels at high pH (Fig. 9b). Finally, some of the observed phenotypic differences could be multifactorial and related to differences in the gene expression levels of many genes. As is the case with the activation of toxin genes in C. difficile, gene expression levels are likely to be correlated to several components in the bacterium's nutritional environment, such as the presence of sugars, amino acids, and fatty acids [72–74].
Conclusions
The related pathogens, C. difficile and C. sordellii, were compared through the analysis of phenomic and genomic datasets. While C. difficile infections have been well studied, significantly less information regarding C. sordellii infections is available. In particular, the current study focused on uncovering the basis for C. sordellii‘s preference for infecting both soft tissues and the vagina, while not infecting the gastrointestinal tract (a major clinical difference between C. difficile and C. sordellii). A comparison of the phenome between C. difficile and C. sordellii revealed that C. sordellii had adapted to survive under conditions that require the procurement of resources from host tissue. In addition, C. sordellii can withstand more acidic pH than C. difficile thereby allowing it to survive in the low pH environment of the vagina. The complementary genomic analysis revealed a large number of proteins present in C. sordellii but not in C. difficile that are likely to play an adaptive role in metabolism and pH tolerance. In this context, the urease gene cluster is described in detail. The phenomic and genomic comparison between C. difficile and C. sordellii should provide guidance for the development of targeted treatments for Clostridial infections.
Methods
Bacterial culturing and phenotype microarray experiments
The global nutritional phenome of C. sordellii strain 8483 was measured using Biolog Phenotype microarrays (PMs). C. sordellii strain 8483 is a human blood isolate obtained from the United Sates Centers for Disease Control and Prevention. PM Technology consists of different PM panels, of which PMs 1–8 are linked to nutrient utilization (metabolism) and PMs 9–10 are related to chemical sensitivity. We have used PMs 1–8 to analyze the global nutritional phenome of C. sordellii and C. difficile and PMs 9–10 to test osmolyte and pH sensitivities. All experiments were conducted in a Bactron IV anaerobic chamber (Shell Lab, OR). Prior to PM experiments, C. sordellii 8483 was grown in anaerobic Brain Heart Infusion (BHI) broth. PM experiments were performed following standard Biolog Inc. protocol [35]. Briefly, 300 μl of the bacteria grown in BHI broth was plated on Biolog Universal blood agar plates and was incubated overnight at 37 °C. A 40 % transmittance cell suspension in Biolog solution IF-0a was then prepared by re-suspending bacteria grown on Biolog Universal blood agar plates. This suspension was then diluted with Biolog mix B at a ratio of 1:16 and then transferred to each of the 96-well PM microplates (a set of 95 substrates and one blank well). The inoculation volume was 100 μl/well. The plates were incubated at 37 °C for 48 h. The optical density (OD) values were then measured at 750 nm using an ELISA reader. Each experiment was performed as biological triplicates.
For statistical analysis of the PM data, means of the replicates were taken. For normalizing the data between strains, each PM well’s mean was divided with the mean of the respective plates negative control. The value of each well was then compared using ANOVA to the negative control value of the respective plate. A PM well was considered positive if its value was 40 % higher than the negative control at 5.0 % significance level. The Model SEED database [75] was then used to predict the genome scale metabolic phenotype of C. sordellii and C. difficile. For phenotype comparisons of C. difficile with C. sordellii, sum of positive phenotypes of C. difficile strains [38] was taken. We compared the predicted metabolic phenotypes with the positive PM results. This comparison showed that at 40 % growth increase cut off from negative control, false positives are completely avoided.
Genomic DNA isolation, genome sequencing and data collation
For isolating genomic DNA, C. sordellii strain 8483 was streaked on BHI agar plate and incubated anaerobically at 37 °C overnight. A single colony from this plate was then used to inoculate BHI broth and was incubated at anaerobic conditions for 12 h. From 1.0 ml of this culture, following manufacturer’s protocol, genomic DNA was isolated using MasterPure™ Gram Positive DNA Purification Kit (Epicenter Biotechnologies, Madison, WI). Roche/454 pyrosequencing, involving paired-end reads from the FLX sequencer, was used to determine the genome sequence of C. sordellii strain 8483 with sequencing coverage of 35x. The sequences were assembled De novo using Newbler Software Release: 2.5.3. Genome annotation for the strain was done by the National Center for Biotechnology Information (NCBI) Prokaryotic Genomes Automatic Annotation Pipeline. The C. sordellii whole genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AJXR00000000. The version described in this paper is the first version, AJXR01000000.
We used Bioperl version 1.6.1 [76] and G-language Genome Analysis Environment version 1.8.13 (http://www.g-language.org) [77–79] for sequence data analysis, and R version 3.1.0 for statistical computing (http://www.R-project.org) [80]. For comparative analysis, bacterial genome sequences in GenBank format [81] were retrieved from the NCBI FTP site (ftp://ftp.ncbi.nih.gov/) and from the PATRIC [82] FTP site (ftp://ftp.patricbrc.org/patric2/genomes/). Protein-coding sequences were retrieved from the bacterial genomes. Homologous proteins were identified by the BLAST (Basic Local Alignment Search Tool) program [83] with an E value cutoff of 1e-5 and a minimum aligned sequence length coverage of 50 % of a query sequence.
Phylogenetic analysis
A group of orthologous proteins was built by all-against-all protein sequence comparison using BLASTP followed by FastOrtho with default parameters (http://enews.patricbrc.org/fastortho/), which is a reimplementation of the OrthoMCL program [84]. We used the 351 ortholog groups shared by all the strains and contained only a single copy from each strain. These orthologs were aligned as follows: i.e., nucleotide sequences are translated into amino acid sequences, aligned with MUSCLE [85, 86], back translated into nucleotide sequences, and ambiguous regions (containing gaps and poorly aligned) were eliminated with Gblocks [87, 88]. The orthologs with more than 50 % of their regions removed are disregarded from the phylogenetic analysis. This retained 346 reliably aligned orthologs from a set of the 351 orthologs. A phylogenetic tree for each of the 346 orthologous genes (gene tree) was reconstructed using RAxML [89] with the GTRGAMMA model. A majority-rule consensus (extended) of the gene trees was constructed using consense program of PHYLIP 3.69 [90]. Because the selection of genes with stronger phylogenetic signal reduced incongruence [91], we analyzed the data set of comprising genes whose bootstrap consensus trees showed average bootstrap support across all internodes that was greater than or equal to 90 % (134 genes). The alignments from the set of the 134 orthologous genes were concatenated, and a tree search was performed using RAxML with the same settings as for the individual gene trees. Phylogenetic trees were drawn using DendroPy [92] and the R package APE (Analysis of Phylogenetics and Evolution) [93].
Gene repertoire analysis
A group of homologous proteins (protein family) was built by all-against-all protein sequence comparison using BLASTP followed by Markov clustering (MCL) [94] with an inflation factor of 1.2 using MCLBLASTLINE (http://micans.org/mcl/). To detect missed protein-coding sequences due to differences in gene finding algorithms [95], we performed TBLASTN homology searches of each strain’s proteins against the other strain’s whole nucleotide sequence. The resulting gene content (binary data, 1 or 0, representing presence or absence of each protein family) is shown in Additional file 4: Table S4.
We used Jaccard distance (one minus Jaccard coefficient) to measure a distance between two genomes based on binary data, 1 or 0, representing the presence or absence of each protein family for each genome (gene content). The resulting distance matrix was subject to a neighbour-joining clustering and hierarchical clustering with three agglomeration methods (i.e., single-, complete-, and average-linkage clustering), and dendrograms were drawn to visualize the clustering results.
Gene functional annotation
We assigned functional annotations to each protein family by merging all the functional annotations of proteins belonging to the same family. To gain different aspects and maximize coverage, protein families were annotated by multiple databases. We performed BLASTP searches of protein sequences against NCBI nr (non-redundant) database, COG [62], KEGG [39], UniProtKB/Uniref90 [96], Virulence Factors Database (VFDB) [60], and assigned the functional annotations of the most similar protein sequences in each database. We converted protein_ID to subsystems (Category, Subcategory, Subsystem, and Role) in SEED database [97]. We also searched protein sequences against the Pfam library of hidden Markov models (HMMs) [98] using HMMER, and mapped Gene Ontology (GO) terms to Pfam entries using the ‘pfam2go’ mapping provided by the GO consortium [99].
Availability of supporting Data
The data sets supporting the results of this article are included within the article and its additional files. The genome sequence data for the C. sordellii whole genome shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession AJXR00000000. The version described in this paper is the first version, AJXR01000000. The phylogenetic trees described in this manuscript have been deposited to TreeBase. Access to the data is available upon publication at http://purl.org/phylo/treebase/phylows/study/TB2:S17636.
Acknowledgement
This work was partially supported with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract, N01-AI-30054, Project No. ZC005-06 and ZC008-09 and a grant (NYCV-478820) from the USDA Animal Health and Disease Research Program.
Abbreviations
- BHI
Brain Heart Infusion
- colA
collagenase
- CDI
C. difficile infectio
- CSI
C. sordellii infection
- GO
Gene Ontology
- GABA
γ-aminobutyric acid
- HMMs
hidden Markov models
- MCL
Markov clustering
- pfoA
perfringolysin O
- PMs
Phenotype microarrays
- plc
phospholipase C
- CDS
protein-coding sequences
- nanH
sialidase
- VFDB
Virulence Factors Database
Additional files
Comparison of nutritional, osmolyte, and pH phenotype values tested using Biolog Phenotype microarrays (PMs) for C. sordellii strain 8483 and C. difficile strain 630. Phenotypes are colored green (positive) and red (negative).
Positive counts for the 854 nutritional phenotypes tested using Biolog Phenotype microarrays (PMs) for C. sordellii strain 8483 and C. difficile strain 630.
Genomic features for bacterial species analyzed.
Gene content table for eight C. difficile strains (630, BI1, CD196, M68, R20291, 2007855, CF5, and M120) and three C. sordellii strains (8483, ATCC_9714, and VPI_9048). The first 11 columns contain the protein family identification number (Family No.), locus_tag, amino acid length (Laa), functional annotations from different databases (GenBank, COG, KEGG, SEED, VFDB, Pfam, GO, and UniProt). The remaining columns show binary data (1 or 0) for presence or absence of each protein family for each strain.
Database categories that are over- or underrepresented in C. sordellii 8483 relative to C. difficile 630. a = the number of C. sordellii protein families in this category, b = the number of C. sordellii protein families not in this category, c = the number of C. difficile protein families in this category, d = the number of C. difficile protein families not in this category, odds ratio = ad/bc, P-value obtained by Fisher’s exact test, and false discovery rate (FDR) adjusted p-value.
Footnotes
Joy Scaria, Haruo Suzuki and Christopher P. Ptak contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JS, HS, and Y-FC conceived the study and participated in its design. JS, HS, and J-WC carried out the genomic and phenomic experiments. JS, HS, and CPP analyzed and interpreted the data. JS, HS, CPP, YZ, X-KG, and Y-FC have been involved in drafting the manuscript. All authors read and approved the final manuscript.
Contributor Information
Joy Scaria, Email: joy.scaria@sdstate.edu.
Haruo Suzuki, Email: haruo@yamaguchi-u.ac.jp.
Christopher P. Ptak, Email: cpp6@cornell.edu
Jenn-Wei Chen, Email: kurohagi2012@gmail.com.
Yongzhang Zhu, Email: yz335@cornell.edu.
Xiao-Kui Guo, Email: xkguo@shsmu.edu.cn.
Yung-Fu Chang, Phone: +1 607-253-2675, Email: yc42@cornell.edu.
References
- 1.Cohen SH, Gerding DN, Johnson S, Kelly CP, Loo VG, McDonald LC, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA) Infect Control Hosp Epidemiol. 2010;31(5):431–455. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 2.Gerding DN. Global epidemiology of Clostridium difficile infection in 2010. Infect Control Hosp Epidemiol. 2010;31(Suppl 1):S32–S34. doi: 10.1086/655998. [DOI] [PubMed] [Google Scholar]
- 3.Chitnis AS, Holzbauer SM, Belflower RM, Winston LG, Bamberg WM, Lyons C, et al. Epidemiology of community-associated Clostridium difficile infection, 2009 through 2011. JAMA internal medicine. 2013;173(14):1359–1367. doi: 10.1001/jamainternmed.2013.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly CP, LaMont JT. Clostridium difficile–more difficult than ever. N Engl J Med. 2008;359(18):1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 5.Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330(4):257–262. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- 6.Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7(7):526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 7.O’Connor JR, Johnson S, Gerding DN. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology. 2009;136(6):1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 8.O’Keefe SJ. Tube feeding, the microbiota, and Clostridium difficile infection. World J Gastroenterol. 2010;16(2):139–142. doi: 10.3748/wjg.v16.i2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Songer JG, Anderson MA. Clostridium difficile: an important pathogen of food animals. Anaerobe. 2006;12(1):1–4. doi: 10.1016/j.anaerobe.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Songer JG. The emergence of Clostridium difficile as a pathogen of food animals. Anim Health Res Rev. 2007;5(02):321–326. doi: 10.1079/AHR200492. [DOI] [PubMed] [Google Scholar]
- 11.Hammitt MC, Bueschel DM, Keel MK, Glock RD, Cuneo P, DeYoung DW, et al. A possible role for Clostridium difficile in the etiology of calf enteritis. Vet Microbiol. 2008;127(3–4):343–352. doi: 10.1016/j.vetmic.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jhung MA, Thompson AD, Killgore GE, Zukowski WE, Songer G, Warny M, et al. Toxinotype V Clostridium difficile in humans and food animals. Emerg Infect Dis. 2008;14(7):1039–1045. doi: 10.3201/eid1407.071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Songer JG, Trinh HT, Killgore GE, Thompson AD, McDonald LC, Limbago BM. Clostridium difficile in retail meat products, USA, 2007. Emerg Infect Dis. 2009;15(5):819–821. doi: 10.3201/eid1505.081071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yutin N, Galperin MY. A genomic update on clostridial phylogeny: Gram-negative spore formers and other misplaced clostridia. Environ Microbiol. 2013;15(10):2631–2641. doi: 10.1111/1462-2920.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aldape MJ, Bryant AE, Stevens DL. Clostridium sordellii infection: epidemiology, clinical findings, and current perspectives on diagnosis and treatment. Clin Infect Dis. 2006;43(11):1436–1446. doi: 10.1086/508866. [DOI] [PubMed] [Google Scholar]
- 16.Schlafer S, Riep B, Griffen AL, Petrich A, Hubner J, Berning M, et al. Filifactor alocis - involvement in periodontal biofilms. BMC Microbiol. 2010;10:66. doi: 10.1186/1471-2180-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, He JZ, He ZL, Zhou Y, Yuan MT, Xu X, et al. Phylogenetic and functional gene structure shifts of the oral microbiomes in periodontitis patients. ISME J. 2014;8(9):1879–1891. doi: 10.1038/ismej.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riggs MM, Sethi AK, Zabarsky TF, Eckstein EC, Jump RL, Donskey CJ. Asymptomatic carriers are a potential source for transmission of epidemic and nonepidemic Clostridium difficile strains among long-term care facility residents. Clin Infect Dis: Off Pub Infect Dis Soc Am. 2007;45(8):992–998. doi: 10.1086/521854. [DOI] [PubMed] [Google Scholar]
- 19.Clark S. Sudden death in periparturient sheep associated with Clostridium sordellii. Vet Rec. 2003;153(11):340. [PubMed] [Google Scholar]
- 20.Lewis CJ, Naylor RD. Sudden death in sheep associated with Clostridium sordellii. Vet Rec. 1998;142(16):417–421. doi: 10.1136/vr.142.16.417. [DOI] [PubMed] [Google Scholar]
- 21.De Groot B, Dewey CE, Griffin DD, Perino LJ, Moxley RA, Hahn GL. Effect of booster vaccination with a multivalent clostridial bacterin-toxoid on sudden death syndrome mortality rate among feedlot cattle. J Am Vet Med Assoc. 1997;211(6):749–753. [PubMed] [Google Scholar]
- 22.Al-Mashat RR, Taylor DJ. Clostridium sordellii in enteritis in an adult sheep. Vet Rec. 1983;112(1):19. doi: 10.1136/vr.112.1.19. [DOI] [PubMed] [Google Scholar]
- 23.Popoff MR. Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect Immun. 1987;55(1):35–43. doi: 10.1128/iai.55.1.35-43.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez RD, Wilkins TD. Comparison of Clostridium sordellii toxins HT and LT with toxins A and B of C. difficile. J Med Microbiol. 1992;36(1):30–6. doi: 10.1099/00222615-36-1-30. [DOI] [PubMed] [Google Scholar]
- 25.Liggins M, Ramirez N, Magnuson N, Abel-Santos E. Progesterone analogs influence germination of Clostridium sordellii and Clostridium difficile spores in vitro. J Bacteriol. 2011;193(11):2776–2783. doi: 10.1128/JB.00058-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meites E, Zane S, Gould C, Investigators Cs Fatal Clostridium sordellii infections after medical abortions. N Engl J Med. 2010;363(14):1382–1383. doi: 10.1056/NEJMc1001014. [DOI] [PubMed] [Google Scholar]
- 27.Bangsberg DR, Rosen JI, Aragon T, Campbell A, Weir L, Perdreau-Remington F. Clostridial myonecrosis cluster among injection drug users: a molecular epidemiology investigation. Arch Intern Med. 2002;162(5):517–522. doi: 10.1001/archinte.162.5.517. [DOI] [PubMed] [Google Scholar]
- 28.Kimura AC, Higa JI, Levin RM, Simpson G, Vargas Y, Vugia DJ. Outbreak of necrotizing fasciitis due to Clostridium sordellii among black-tar heroin users. Clin Infect Dis. 2004;38(9):e87–e91. doi: 10.1086/383471. [DOI] [PubMed] [Google Scholar]
- 29.Brett MM, Hood J, Brazier JS, Duerden BI, Hahne SJ. Soft tissue infections caused by spore-forming bacteria in injecting drug users in the United Kingdom. Epidemiol Infect. 2005;133(4):575–582. doi: 10.1017/S0950268805003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Browdie DA, Davis JH, Koplewitz MJ, Corday L, Leadbetter AW. Clostridium sordellii infection. J Trauma. 1975;15(6):515–518. doi: 10.1097/00005373-197506000-00011. [DOI] [PubMed] [Google Scholar]
- 31.Mory F, Lozniewski A, Guirlet MN, Guidat D, Bresler L, Weber M, et al. Severe sepsis caused by Clostridium sordellii following liver biopsy in a liver transplant recipient. Clin Infect Dis. 1995;21(6):1522–1523. doi: 10.1093/clinids/21.6.1522. [DOI] [PubMed] [Google Scholar]
- 32.Adamkiewicz TV, Goodman D, Burke B, Lyerly DM, Goswitz J, Ferrieri P. Neonatal Clostridium sordellii toxic Omphalitis. Pediatr Infect Dis J. 1993;12(3):253–257. doi: 10.1097/00006454-199303000-00020. [DOI] [PubMed] [Google Scholar]
- 33.Bochner BR, Gadzinski P, Panomitros E. Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 2001;11(7):1246–1255. doi: 10.1101/gr.186501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keymer DP, Miller MC, Schoolnik GK, Boehm AB. Genomic and phenotypic diversity of coastal Vibrio cholerae strains is linked to environmental factors. Appl Environ Microbiol. 2007;73(11):3705–3714. doi: 10.1128/AEM.02736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bochner BR. Global phenotypic characterization of bacteria. FEMS Microbiol Rev. 2009;33(1):191–205. doi: 10.1111/j.1574-6976.2008.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JW, Scaria J, Chang YF. Phenotypic and transcriptomic response of auxotrophic Mycobacterium avium subsp. paratuberculosis leuD mutant under environmental stress. PLoS One. 2012;7(6):e37884. doi: 10.1371/journal.pone.0037884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lei XH, Bochner BR. Using phenotype microarrays to determine culture conditions that induce or repress toxin production by Clostridium difficile and other microorganisms. PLoS One. 2013;8(2):e56545. doi: 10.1371/journal.pone.0056545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scaria J, Chen JW, Useh N, He H, McDonough SP, Mao C, et al. Comparative nutritional and chemical phenome of Clostridium difficile isolates determined using phenotype microarrays. Int J Infect Dis. 2014;27:20–25. doi: 10.1016/j.ijid.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Echols N, Harrison P, Balasubramanian S, Luscombe NM, Bertone P, Zhang ZL, et al. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002;30(11):2515–2523. doi: 10.1093/nar/30.11.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scaria J, Janvilisri T, Fubini S, Gleed RD, McDonough SP, Chang YF. Clostridium difficile Transcriptome analysis using pig ligated loop model reveals modulation of pathways not modulated in vitro. J Infect Dis. 2011;203(11):1613–1620. doi: 10.1093/infdis/jir112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caillouette JC, Sharp CF, Zimmerman GJ, Roy S. Vaginal pH as a marker for bacterial pathogens and menopausal status. Am J Obstet Gynecol. 1997;176(6):1270–1275. doi: 10.1016/S0002-9378(97)70345-4. [DOI] [PubMed] [Google Scholar]
- 43.Brooks JB, Dowell VR, Farshy DC, Armfield AY. Further studies on differentiation of Clostridium sordellii from clostridium-Bifermentans by gas chromatography. Can J Microbiol. 1970;16(11):1071–8. doi: 10.1139/m70-181. [DOI] [PubMed] [Google Scholar]
- 44.Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994;44(4):812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- 45.Dutilh BE, Backus L, Edwards RA, Wels M, Bayjanov JR, van Hijum SA. Explaining microbial phenotypes on a genomic scale: GWAS for microbes, Brief Funct Genomics. 2013;12(4):366–80. [DOI] [PMC free article] [PubMed]
- 46.Galperin MY, Koonin EV. Who’s your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18(6):609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- 47.Ogura Y, Ooka T, Iguchi A, Toh H, Asadulghani M, Oshima K, et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc Natl Acad Sci U S A. 2009;106(42):17939–17944. doi: 10.1073/pnas.0903585106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki H, Lefebure T, Hubisz MJ, Pavinski Bitar P, Lang P, Siepel A, et al. Comparative genomic analysis of the Streptococcus dysgalactiae species group: gene content, molecular adaptation, and promoter evolution. Genome Biol Evol. 2011;3:168–185. doi: 10.1093/gbe/evr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki H, Lefebure T, Pavinski Bitar P, Stanhope MJ. Comparative genomic analysis of the genus Staphylococcus including Staphylococcus aureus and its newly described sister species Staphylococcus simiae. BMC Genomics. 2012;13(1):38. doi: 10.1186/1471-2164-13-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kunin V, Ahren D, Goldovsky L, Janssen P, Ouzounis CA. Measuring genome conservation across taxa: divided strains and united kingdoms. Nucleic Acids Res. 2005;33(2):616–621. doi: 10.1093/nar/gki181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;21(1):108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- 52.Reddy ARS, Girinathan BP, Zapotocny R, Govind R. Identification and characterization of Clostridium sordellii toxin gene regulator. J Bacteriol. 2013;195(18):4246–4254. doi: 10.1128/JB.00711-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cotter PD, Hill C. Surviving the acid test: responses of gram-positive bacteria to low pH. Microbiol Mol Biol Rev. 2003;67(3):429–453. doi: 10.1128/MMBR.67.3.429-453.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trchounian A, Kobayashi H. Kup is the major K+ uptake system in Escherchia coli upon hyper-osmotic stress at a low pH. FEBS Letters. 1999;447(2–3):144–148. doi: 10.1016/S0014-5793(99)00288-4. [DOI] [PubMed] [Google Scholar]
- 55.Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33(17):5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paredes-Sabja D, Shen A, Sorg JA. Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol. 2014;22(7):406–416. doi: 10.1016/j.tim.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Permpoonpattana P, Phetcharaburanin J, Mikelsone A, Dembek M, Tan S, Brisson MC, et al. Functional characterization of Clostridium difficile spore coat proteins. J Bacteriol. 2013;195(7):1492–503. doi: 10.1128/JB.02104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hammer ND, Skaar EP. Molecular Mechanisms of Staphylococcus aureus Iron Acquisition. In: Gottesman S, Harwood CS, editors. Annual review of microbiology, Vol 65. Palo Alto: Annual Reviews; 2011. pp. 129–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bouillaut L, Self WT, Sonenshein AL. Proline-dependent regulation of Clostridium difficile Stickland metabolism. J Bacteriol. 2013;195(4):844–854. doi: 10.1128/JB.01492-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40(Database issue):D641–D645. doi: 10.1093/nar/gkr989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aronoff DM. Clostridium novyi, sordellii and tetani: mechanisms of disease. Anaerobe. 2013;24:98–101. doi: 10.1016/j.anaerobe.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 62.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mobley HLT, Island MD, Hausinger RP. Molecular-biology of microbial Ureases. Microbiol Rev. 1995;59(3):451–480. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gueneau P, Loiseaux-De Goer S. Helicobacter: molecular phylogeny and the origin of gastric colonization in the genus. Infect Genet Evol. 2002;1(3):215–223. doi: 10.1016/S1567-1348(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 65.Dupuy B, Daube G, Popoff MR, Cole ST. Clostridium perfringens urease genes are plasmid borne. Infect Immun. 1997;65(6):2313–2320. doi: 10.1128/iai.65.6.2313-2320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ha NC, Oh ST, Sung JY, Cha KA, Lee MH, Oh BH. Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Biol. 2001;8(6):505–509. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 67.Strugatsky D, McNulty R, Munson K, Chen CK, Soltis SM, Sachs G, et al. Structure of the proton-gated urea channel from the gastric pathogen Helicobacter pylori. Nature. 2013;493(7431):255–8. doi: 10.1038/nature11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stingl K, Brandt S, Uhlemann EM, Schmid R, Altendorf K, Zeilinger C, et al. Channel-mediated potassium uptake in Helicobacter pylori is essential for gastric colonization. EMBO J. 2007;26(1):232–241. doi: 10.1038/sj.emboj.7601471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ochrombel I, Ott L, Kramer R, Burkovski A, Marin K. Impact of improved potassium accumulation on pH homeostasis, membrane potential adjustment and survival of Corynebacterium glutamicum. Biochimica Et Biophysica Acta-Bioenergetics. 2011;1807(4):444–450. doi: 10.1016/j.bbabio.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 70.Krulwich TA, Sachs G, Padan E. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol. 2011;9(5):330–343. doi: 10.1038/nrmicro2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lentes CJ, Mir SH, Boehm M, Ganea C, Fendler K, Hunte C: Molecular Characterization of the Na+/H+/- Antiporter NhaA from Salmonella Typhimurium. PLoS One. 2014;9(7):e101575. [DOI] [PMC free article] [PubMed]
- 72.Antunes A, Camiade E, Monot M, Courtois E, Barbut F, Sernova NV, Rodionov DA, Martin-Verstraete I, Dupuy B: Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Res. 2012;40(21):10701–18. [DOI] [PMC free article] [PubMed]
- 73.Soutourina OA, Monot M, Boudry P, Saujet L, Pichon C, Sismeiro O, et al. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet. 2013;9(5):e1003493. doi: 10.1371/journal.pgen.1003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bouillaut L, Dubois T, Sonenshein AL, Dupuy B. Integration of metabolism and virulence in Clostridium difficile. Res Microbiol. 2015;166(4):375–383. doi: 10.1016/j.resmic.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28(9):977–982. doi: 10.1038/nbt.1672. [DOI] [PubMed] [Google Scholar]
- 76.Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12(10):1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arakawa K, Mori K, Ikeda K, Matsuzaki T, Kobayashi Y, Tomita M. G-language Genome Analysis Environment: a workbench for nucleotide sequence data mining. Bioinformatics. 2003;19(2):305–306. doi: 10.1093/bioinformatics/19.2.305. [DOI] [PubMed] [Google Scholar]
- 78.Arakawa K, Suzuki H, Tomita M. Computational genome analysis using the G-language system. Genes, Genomes Genomics. 2008;2(1):1–13. [Google Scholar]
- 79.Arakawa K, Tomita M. G-language System as a platform for large-scale analysis of high-throughput omics data. J Pesticide Sci. 2006;31(3):282–288. doi: 10.1584/jpestics.31.282. [DOI] [Google Scholar]
- 80.R_Core_Team: R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2014 (http://www.R-project.org).
- 81.Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. GenBank. Nucleic Acids Res. 2013;41(Database issue):D36–42. [DOI] [PMC free article] [PubMed]
- 82.Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, Dalay O, et al. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 2011;79(11):4286–4298. doi: 10.1128/IAI.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 88.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 89.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 90.Felsenstein J. PHYLIP - phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 91.Salichos L, Rokas A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature. 2013;497(7449):327–331. doi: 10.1038/nature12130. [DOI] [PubMed] [Google Scholar]
- 92.Sukumaran J, Holder MT. DendroPy: a Python library for phylogenetic computing. Bioinformatics. 2010;26(12):1569–1571. doi: 10.1093/bioinformatics/btq228. [DOI] [PubMed] [Google Scholar]
- 93.Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 94.van Dongen S: Graph Clustering by Flow Simulation. PhD thesis. University of Utrecht; 2000.
- 95.Poptsova MS, Gogarten JP. Using comparative genome analysis to identify problems in annotated microbial genomes. Microbiology. 2010;156(Pt 7):1909–1917. doi: 10.1099/mic.0.033811-0. [DOI] [PubMed] [Google Scholar]
- 96.UniProt_Consortium Update on activities at the universal protein resource (UniProt) in 2013. Nucleic Acids Res. 2013;41(Database issue):D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2014;42(Database issue):D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gene_Ontology_Consortium Gene Ontology annotations and resources. Nucleic Acids Res. 2013;41(Database issue):D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]