

Abstract
The N-oxide derivatives of [2-(3-pyridinyl)-1-hydroxyethylidene-1,1-phosphonocarboxylic acid (or PEHPC) and [2-(3-pyridinyl)-1-ethylidene-1,1-phosphonocarboxylic acid (or PEPC) have been prepared and evaluated for their activity against several enzymes which utilize isoprenoids. The parent pyridines are known inhibitors of GGTase II, but the N-oxide derivatives show no improvement in biological activity in assays with the isolated enzyme. However, the PEHPC N-oxide did induce significant accumulation of intracellular light chain in myeloma cells, consistent with inhibition of Rab geranylgeranylation.
Keywords: pyridyl N-oxides, GGTase II, Rab GGTase, apoptosis, myeloma, bioassay
Graphical Abstract
The Rab family of small GTPases plays key roles in mediating intracellular trafficking events. These proteins are geranylgeranylated by the enzyme geranylgeranyl transferase II (GGTase II) and mutant forms of Rabs that are unable to be geranylgeranylated are mislocalized and therefore nonfunctional.1 We have hypothesized that agents which impair Rab geranylgeranylation, either directly via inhibition of GGTase II,2 or indirectly via depletion of GGPP,3,4 would result in disruption of monoclonal protein secretion in human myeloma cells. We have shown that disruption of Rab geranylgeranylation leads to an accumulation of monoclonal protein in the endoplasmic reticulum, induction of the unfolded protein response pathway, and apoptosis.5,6 Because myeloma cells are so heavily engaged in secretion of antibodies, specific inhibitors of GGTase II may represent a novel therapeutic strategy for treatment of myeloma or other diseases characterized by excessive protein secretion.
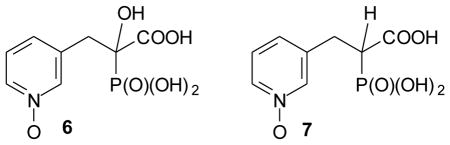
Although several types of GGTase II inhibitors are known,7–10 we were particularly intrigued by the phosphonocarboxylate family.11 Both PEHPC (1, also known as NE-10790) and PEPC (2) can be viewed as analogues of the bisphosphonate risedronate (3).12 However, while risedronate is a potent inhibitor of farnesyl diphosphate synthase (FDPS, IC50 ~ 6 nM)11 and is in clinical use for the treatment of osteoporosis,13 compounds 1 and 2 show little activity against FDPS but do inhibit selectively the downstream enzyme GGTase II. Reported IC50 values for PEHPC against GGTase II range from ~32 to ~600 uM. 14–16 Furthermore, formal deletion of the OH group (i.e. compound 2) does not greatly diminish activity against GGTase II,11 and the enantiomers of 1 differ in activity by only ~4-fold.16 In contrast the phosphono carboxylate 4, also known as IPEHPC and viewed as an analogue of the bisphosphonate minodronate (5),12,17 shows significantly greater potency even as the racemate, and the (+)-enantiomer 4 is ~60-fold more active than the (−)-enantiomer.15 However, the (+)-enantiomer also shows weak inhibition of at least one other enzyme involved in isoprenoid biosynthesis, probably geranylgeranyl diphosphate synthase (GGDPS).16
Studies of IPEHPC have revealed that it inhibits the second geranylgeranylation of Rab proteins,16 which suggests that it might be feasible to inhibit processing of some Rab proteins selectively over others. Because both PEHPC and IPEHPC inhibit this second geranylgeranylation but not the first, they may bind in an enzyme complex that includes both GGTase II and a Rab escort protein (REP) in such a way that the reorganization of the complex necessary for the second transfer is prevented. It has been suggested that complexation occurs in a large carvern in the protein, but the specific structural features that favor this binding are not yet clear. In an effort to advance understanding of structure activity relationships, and ultimately to obtain more potent inhibitors of GGTase II, we have begun to prepare derivatives of PEHPC and PEPC. In this paper we report the preparation and biological activity of the N-oxide derivatives of these compounds. The N-oxide introduces an N-1 substituent while yielding analogues that preserve a lone pair near the placement of the lone pair in the original pyridyl nitrogen, and at the same time are still formally neutral with respect to the pyridyl ring. While other derivatives can be imagined that accomplish both of these goals, they would require significantly more deep-seated changes to the structure. It was our expectation that synthesis and biological evaluation of the N-oxides would encourage, or discourage, preparation of less accessible analogues (or mimics) of these pyridine derivatives. Thus the target compounds became the N-oxides 6 and 7.
Synthesis of two N-oxide analogues of PEHPC is shown in Figure 3. Pyridine aldehyde 8 was condensed with ethyl ester 9 to afford the known keto ester 10,18 which was further treated with diethyl phosphite to give the key intermediate 11. Compound 11 was allowed to react with mCPBA to oxidize the pyridine ring and give N-oxide 12 in a reasonable yield. Acid hydrolysis of N-oxide 12 converted it to the corresponding acid 13, but only in low yield. While some carboxylate phosphonate triesters can be converted to the corresponding triacids by prolonged treatment with base,19 that approach was not feasible in this series. Instead, treatment of the triester 12 with base resulted in a smooth rearrangement to the corresponding phosphate 14. Based on analysis of the 31P NMR spectrum, acid catalyzed hydrolysis of phosphate triester 14 gave the corresponding phosphate monoester 15, but this compound was not examined further.
Figure 3. Synthesis of N-oxide analogues of PEHPC.

The GGTase II inhibitor PEPC (2), which has a potency similar to PEHPC,20 was pursued through a parallel synthesis. However, it was challenging to obtain the target molecule 2 following a literature procedure,21 perhaps due to the poor solubility of the commercially available hydrochloride salt 16 in organic solvents. To overcome this issue, the hydrochloride salt was first neutralized by treatment with NaHCO3 to give compound 16 as the free base which was then added to a solution of the anion of triethyl phosphonoacetate (15) in situ.22 The reaction mixture was allowed to stir overnight to form the PEPC precursor 17 in reasonable yield. With the key intermediate 17 in hand, part of the material was converted directly to PEPC (2) by hydrolysis while the remaining material was treated with the mCPBA to generate N-oxide 18. Hydrolysis of triester 18 by treatment with HCl gave the corresponding acid 7. At this point, the known GGTase II inhibitors, PEHPC (1), and PEPC (2) and their new N-oxide analogues 6 and 7 were tested for their relative biological activity.
The N-oxides 6 and 7, as well as the parent compounds 1 and 2, were tested for their ability to inhibit FDPS or GGTase II in in vitro enzyme assays.3 As shown in Table 1, these compounds do not potently inhibit either enzyme. Despite these compounds displaying similar activity against both enzymes, only evidence of GGTase II inhibition was observed in cell culture studies. Immunoblot studies were performed to assess the effects of these compounds on protein geranylgeranylation in RPMI-8226 human myeloma cells. Rap1a is a substrate of GGTase I and an antibody was used which detects only unmodified protein. Rab6 is a representative Rab protein and is therefore a substrate of GGTase II. For Rab6, a Triton X-114 lysis protocol was used to generate a detergent (membrane) fraction.3 With disruption of Rab geranylgeranylation, there is a decrease in membrane-bound protein. Lovastatin, an HMG-CoA reductase inhibitor which globally disrupts protein prenylation, was used as a control. As shown in Figure 5A, none of the bisphosphonates induce an accumulation of unmodified Rap1a. As expected, PEHPC (1) induces a decrease in the amount of membrane-bound Rab6. The N-oxide 6 and PEPC (2) diminished the level of membrane-bound Rab6 to a lesser extent while the N-oxide 7 did not decrease membrane-bound Rab6 levels. As we have demonstrated previously, both lovastatin and PEHPC (1) induce apoptosis (as indicated by PARP and caspase 3 cleavage) as well as ER stress (calnexin cleavage).5,6 Interestingly, although compound 7 does not appear to alter significantly Rab6 levels in the membrane fraction, it does induce cleavage of PARP, caspase 3, and calnexin to a similar extent as the parent compound 2, suggesting there may be off-target effects. Finally, the ability of these compounds to disrupt monoclonal protein trafficking (a functional read-out of impairment of Rab geranylgeranylation3,5) was examined. As shown in Figure 5B, PEPHC and its N-oxide derivative 6 induce an accumulation of intracellular light chain while PEPC and its N-oxide derivative 7 do not significantly alter light chain trafficking, which is consistent with the weaker ability of the latter two compounds to diminish Rab geranylgeranylation.
Table 1.
| FDPS IC50 (mM) |
GGTase II IC50 (mM) |
|
|---|---|---|
| 1 (PEHPC) | 0.2 | 0.7 |
| 6 | 2 | 1.8 |
| 2 (PEPC) | 1 | 1.1 |
| 7 | >2 | 0.8 |
Figure 5. Effects of PEHPC derivatives in myeloma cells.

RPMI-8226 cells were incubated for 48 hours in the presence or absence of lovastatin (20 μM, Lov), PEHPC (5 mM), PEPC (5 mM), or the N-oxides 6 and 7 (5 mM). A) Cells were lysed using RIPA buffer to generate whole cell lysate or with Triton X-114 to generate a detergent (membrane) fraction and immunoblot analysis was performed. The Rap1a antibody detects only unmodified protein. β-Tubulin was used as a loading control for whole cell lysate and calnexin was used as the loading control for the detergent fraction. * Denotes the PARP cleavage product while ** denotes the calnexin cleavage product. The gels are representative of two independent studies. B) Intracellular lambda light chain concentrations were determined via ELISA. Data are expressed as percentage of control (mean + SD, n=3). The * denotes p<0.05 per unpaired two-tailed t-test and compares treated cells to untreated control cells.
While it is somewhat disappointing that the new N-oxides are not more potent inhibitors of GGTase II in assays with the isolated enzyme, at the same time it is significant that the PEHPC N-oxide 6 does have cellular activity consistent with inhibition of Rab geranylgeranylation. This suggests that larger substituents at the pyridyl nitrogen might be tolerated or even afford greater potency. Studies along these lines are underway and will be reported in due course.
Supplementary Material
Figure 1. Pyridyl bisphosphonates and the corresponding carboxy phosphonates.
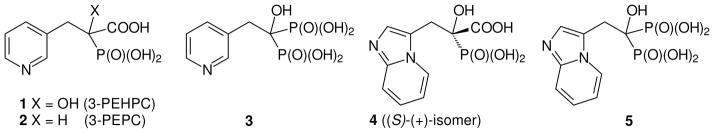
Figure 2. N-Oxide derivatives of PEHPC (6) and PEPC (7).
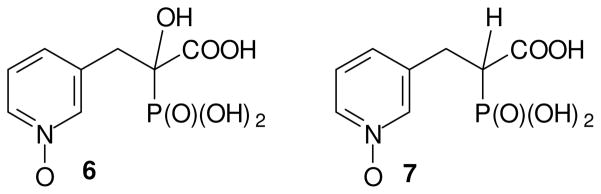
Figure 4. Synthesis of PEPC and its N-oxide analogue.

Acknowledgments
Financial support from the NIH (R01CA-172070), the American Society of Hematology (a Scholar Award to S.A.H), and the Roy J. Carver Charitable Trust is gratefully acknowledged.
Footnotes
Supplementary data (representative experimental procedures, NMR spectra, and bioassay protocols) associated with this article can be found in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Gomes AQ, Ali BR, Ramalho JS, Godfrey RF, Barral DC, Hume AN, Seabra MC. Mol Biol Cell. 2003;14:1882. doi: 10.1091/mbc.E02-10-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou X, Hartman SV, Born EJ, Smits JP, Holstein SA, Wiemer DF. Bioorg Med Chem Lett. 2013;23:764. doi: 10.1016/j.bmcl.2012.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou X, Ferree SD, Wills VS, Born EJ, Tong H, Wiemer DF, Holstein SA. Bioorg Med Chem. 2014;22:2791. doi: 10.1016/j.bmc.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shull LW, Wiemer AJ, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2006;14:4130. doi: 10.1016/j.bmc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Holstein SA, Hohl RJ. Leukemia Res. 2011;35:551. doi: 10.1016/j.leukres.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Born EJ, Hartman SV, Holstein SA. Blood Cancer Journal. 2013;3 doi: 10.1038/bcj.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stigter EA, Guo Z, Bon RS, Wu YW, Choidas A, Wolf A, Menninger S, Waldmann H, Blankenfeldt W, Goody RS. J Med Chem. 2012;55:8330. doi: 10.1021/jm300624s. [DOI] [PubMed] [Google Scholar]
- 8.Deraeve C, Guo Z, Bon RS, Blankenfeldt W, DiLucrezia R, Wolf A, Menninger S, Stigter EA, Wetzel S, Choidas A, Alexandrov K, Waldmann H, Goody RS, Wu YW. J Am Chem Soc. 2012;134:7384. doi: 10.1021/ja211305j. [DOI] [PubMed] [Google Scholar]
- 9.Bon RS, Guo Z, Stigter EA, Wetzel S, Menninger S, Wolf A, Choidas A, Alexandrov K, Blankenfeldt W, Goody RS, Waldmann H. Angew Chem, Int Ed. 2011;50:4957. doi: 10.1002/anie.201101210. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe M, Fiji HD, Guo L, Chan L, Kinderman SS, Slamon DJ, Kwon O, Tamanoi F. J Biol Chem. 2008;283:9571. doi: 10.1074/jbc.M706229200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coxon FP, Ebetino FH, Mules EH, Seabra MC, McKenna CE, Rogers MJ. Bone. 2005;37:349. doi: 10.1016/j.bone.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 12.Ebetino FH, Hogan AML, Sun S, Tsoumpra MK, Duan X, Triffitt JT, Kwaasi AA, Dunford JE, Barnett BL, Oppermann U, Lundy MW, Boyde A, Kashemirov BA, McKenna CE, Russell RGG. Bone. 2011;49:20. doi: 10.1016/j.bone.2011.03.774. [DOI] [PubMed] [Google Scholar]
- 13.Nuti R. Clinical Cases in Mineral and Bone Metabolism. 2014;11:208. [PMC free article] [PubMed] [Google Scholar]
- 14.Coxon FP, Helfrich MH, Larijani B, Muzylak M, Dunford JE, Marshall D, McKinnon AD, Nesbitt SA, Horton MA, Seabra MC, Ebetino FH, Rogers MJ. J Biol Chem. 2001;276:48213. doi: 10.1074/jbc.M106473200. [DOI] [PubMed] [Google Scholar]
- 15.Baron RA, Tavare R, Figueiredo AC, Blazewska KM, Kashemirov BA, McKenna CE, Ebetino FH, Taylor A, Rogers MJ, Coxon FP, Seabra MC. J Biol Chem. 2009;284:6861. doi: 10.1074/jbc.M806952200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna CE, Kashemirov BA, Blazewska KM, Mallard-Favier I, Stewart CA, Rojas J, Lundy MW, Ebetino FH, Baron RA, Dunford JE, Kirsten ML, Seabra MC, Bala JL, Marma MS, Rogers MJ, Coxon FP. J Med Chem. 2010;53:3454. doi: 10.1021/jm900232u. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi M, Sakamoto S, Kawamuki K, Kurihara H, Nakahara H, Isomura Y. Chem Pharm Bull (Tokyo) 1998;46:1703. doi: 10.1248/cpb.46.1703. [DOI] [PubMed] [Google Scholar]
- 18.Horner L, Renth EO. Justus Liebigs Ann Chem. 1967;703:37. [Google Scholar]
- 19.Holstein SA, Cermak DM, Wiemer DF, Lewis K, Hohl RJ. Bioorg Med Chem. 1998;6:687. doi: 10.1016/s0968-0896(98)00034-0. [DOI] [PubMed] [Google Scholar]
- 20.Marma MS, Xia Z, Stewart C, Coxon F, Dunford JE, Baron R, Kashemirov BA, Ebetino FH, Triffitt JT, Russell RGG, McKenna CE. J Med Chem. 2007;50:5967. doi: 10.1021/jm0702884. [DOI] [PubMed] [Google Scholar]
- 21.Marma MS, Xia Z, Stewart C, Coxon F, Dunford JE, Baron R, Kashemirov BA, Ebetino FH, Triffitt JT, Russell RGG, McKenna CE. Journal of Medicinal Chemistry. 2007;50:5967. doi: 10.1021/jm0702884. [DOI] [PubMed] [Google Scholar]
- 22.Barney RJ, Wasko BM, Dudakovic A, Hohl RJ, Wiemer DF. Bioorg Med Chem. 2010;18:7212. doi: 10.1016/j.bmc.2010.08.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.