

Abstract
Campomelic dysplasia (CD, OMIM #114290) is a rare autosomal dominant disease characterized with bending and shortness in the long bones of the lower extremities, typical facial features, hypoplastic scapula, costa defect, narrow thorax and pes equinovarus. Campomelic dysplasia occurs with heterozygous mutations in the SOX9 gene in the 17q24 chromosome. The main findings of our four-day old patient included typical facial features, risomelic extremity shortness, angular bending in the long bones of bilateral lower extremities and pes equinovarus. On direct graphies, costa defect and scapula hypoplasia were noted. We showed a missense mutation (c.473C>T [p.A158V]) in the SOX9 gene which had not been reported before in our patient who had the typical clinical findings of CD. The family of the patient was informed about potential future pathologies of this disease and received genetic counseling.
Keywords: Genetic counseling, campomelic dysplasia, SOX9 mutation
Introduction
Campomelic dysplasia (CD, OMIM #114290) which is inherited autosomal dominantly was described by Caffey (1) in 1947 for the first time in a subject who showed angular bending in the extremities, dimples on the skin on these bones and shortness in the long bones. However, its classification as Campomelic nanism was made by Maroteaux et al. (2) in 1971. As the number of the cases reported increased, it was understood that facial signs including pressed and flat face, hypertelorism, short palpebral fissures, cleft palate, small chin and short neck, hypoplastic scapula, costa defect, narrow chest and pes equinovarus were also associated with this disease in addition to the findings described initially (3). In campomelic dysplasia, the fact that XY sex transformations are observed in ¾ of the subjects with XY karyotype is among the improtant findings (4). Campomelic dysplasia occurs with heterozygous mutations localized in the SOX9 gene on the 17q24 chromosome region which encodes the transcription factor which is involved in different steps of development of cartilage tissue and determination of sex (5). In subjects with campomelic dysplasia, the most important reason of mortality rates in the neonatal period is respiratory failure arising from narrow chest, hypoplastic lungs and narrow respiratory tract (3).
This case was presented, because CD is a rare disease and the change found in the SOX9 gene had not been reported before.
Case
The patient information is presented by obtaining informed consent. It was learned that our patient who was born from the fourth pregnancy of a 34-year-old healthy mother who had no consanguinity with her husband as the fourth baby was intubated because of respiratory distress in the early neonatal period. On physical examination, the body weight was found to be 2 500 g (25 p), the height was found to be 41 cm (<3 p), the head circumference was found to be 37 cm (>90 p). Short stature, macrocephaly, rare or bow brows, periorbital fullness, hypertelorism, depressed nasal bridge, low set ears, long philtrum, small chin and short neck were found dismorphologically. In addition, narrow chest, risomelic extremity shortness, dimps on the skin on bilateral tibias, clinodactilia in the fifth fingers in bilateral hands, bilateral sandal gap in the feet, hypoplasic toe nails and pes equinovarus were present (Figure 1a, b).
Figure 1.
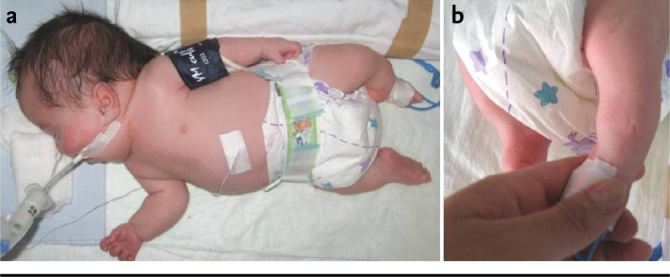
a) Low set ears and hair line, short neck, narrow chest, risomelic extremity shortness and pes equinovarus are noted. b) Dimples on the skin on bilateral tibias are observed
When the laboratory tests of the patient were examined, it was observed that urinalysis, hemogram and biochemical values were within normal limits. Cylindrical short thoracic cage (bell shaped), scapular hypoplasia, 11 pairs of costae which were shorter than normal and inclined downward were observed on plain graphies. Coccyx hypoplasia and bilateral femoral and tibial angulation were observed on hip graphy (Figure 2). On transfontanel ultrasonography, it was observed that the lateral ventricles were larger than normal and congenital anomaly compatible with cavum septum pellisidum et vergae in the middle line was noted.
Figure 2.
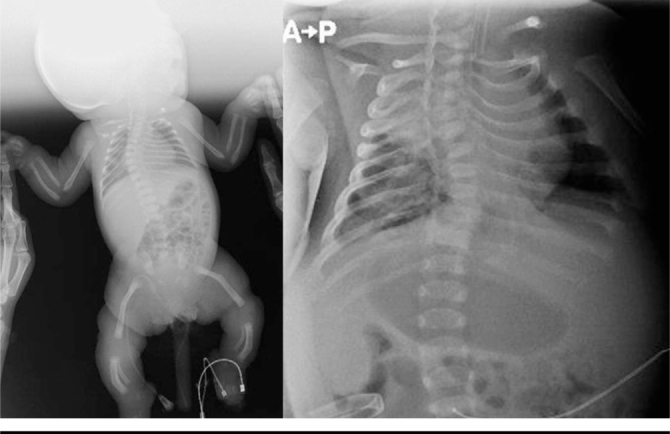
Cylindric short chest cage (bell-shaped), scoliosis, scapular hypoplasia, 11 pairs of costae which are shorter than normal and inclined downward, coccygeal hypoplasia, femoral and tibial angulation are noted on plain graphies
In our patient who was found to have short and angular extremities postnatally, osteogenesis imperfecta was considered primarily in the differential diagnosis. When the patient was evaluated together with the facial findings, spondyloepiphyseal dysplasia congenita and Stickler syndrome were considered. Since the findings of 11 pairs of costae and scapular hypoplasia on plain graphies which were found in our patient were specific for CD, other diseases were excluded. As a result of genetic tests performed for campomelic dysplasia karyotype was found to be 46, XX with high resolution banding (HRB; 650 bands and above) analysis. On molecular analysis directed to the SOX9 gene, c.473C>T (p.A158V) mutation was found heterozygously. The fact that no mutation was found on analyses of the mother and father showed that the mutation was de novo. A prediagnosis of CD which is one of the skeletal dysplasies was made with the present clinical and radiological findings. The diagnosis became definite as a result of molecular analysis.
The patient had respiratory distress which was thought to be related with narrow chest cage. Therefore, the patient was followed up on mechanical ventilator. His enteral feeding was arranged with nasogastric tube and he was followed up in the neonatal intensive care unit.
Discussion
Campomelic dysplasia is one of the fatal skeletal dysplasies which is inherited autosomal dominantly. The incidence of the disease is one in 2 000 000 newborns (6). Facial signs (depressed and flat face, hypertelorism, short palpebral fissures, cleft palate, small chin and short neck), extremity findings (angular binding of the extremities, dimples on the skin on these bones and shortness of the long bones) and radiological findings (hypoplastic scapula, defect of costae, narrow chest) which are important findings of the disease were present in our patient (7).
The incidence of campomelic dysplasia is the same in both sexes. However, female phenotype or ambigious genitalia are observed in the external genitalia in 75% of the subjects with a male genotype. Sex transformation disorder which is observed in the subjects who are gonadally and genetically male has been recently shown to be caused by mutation occuring in the SRY-mediated gene (8). The external genitalia and karyotype of our patient was defined as female and no genital anomaly was found.
The SOX9 gene which is responsible of the disease was defined on the long arm of the 17th chromosome (17q24). The SOX9 gene is involved in differentiation of chondrocytes, in regulation of transcription of anti-Müllerian hormone together with SF1 (steroidogenic factor 1) gene and development of the male genital system by preventing formation of the female reproductive system (9). Changes which occur in the SOX9 gene have been associated with skeletal system disorders, autosomal dominant gender transformation, Pierre Robin sequance and Cooks syndrome (10). In this study, we showed a de novo missense mutation in the High Mobility Group (HMG) region of the SOX9 gene in a patient who had the typical clinical findings of CD. The proband which had a normal 46,XX karyotype and female phenotype findings was found to be heterozygous for de novo c.473C> T (p.A158V) mutation. In the literature search, it was found that A158T change was reported in this region by Preiss et al. (11) in a patient with CD before. In vitro functional expression studies which were conducted in the same study showed that the mutant protein decreased deoxyribonucleic acid binding and transcription activities (11). Therefore, it was predicted that the change we found in the same region acted in the same way and caused to the disease.
Most patients with campomelic dysplasia are born dead or lost due to respiratory distress in a few weeks after delivery. Narrow chest cage, narrow larynx and tracheomalacia are the reasons of respiratory distress which may lead to mortality (12). Cardiac, renal and central nervous system anomalies are among important causes of mortality. However, patients who do not have problems which would lead to respiratory distress and additional serios anomalies can survive. In patients who live up to advanced ages, problems related with the spine occur. Thus, orthopaedic interventions directed to eliminate the bending and shortness in the lower extremities should be performed in the first years of life in children who do not develop serious respiratory problems. Afterwards, the patients should be monitored closely, problems which may develop in the spine should be determined and interventions directed to treatment should be performed (12). In our patient, correction of the bending of the lower extremities is being planned by performing orthopaedic interventions in the early period.
Conclusively, the family was told that no mutation was found on the SOX9 gene analysis performed on the peripheral blood samples of the mother and father and this change which occured de novo had a possibility of reccurence of 5% in the other pregnancies (in relation with potential germ mutations in the mother or father) when published and unpublished cases were considered.
Footnotes
Informed Consent: Written informed consent was obtained from the parents of the patient who participated in this case.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - K.K.; Design - K.K., Z.Y.; Supervision - K.K.; Funding - K.K., Z.Y.; Materials - K.K., E.Y.; Data Collection and/or Processing - K.K., E.Y.; Analysis and/or Interpretation - K.K., G.S.; Literature Review - K.K., Z.Y.; Writer - K.K., Z.Y.; Critical Review - K.K., Z.Y., E.Y.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Caffey JP. Prenatal bowing and thickening of tubular bones, with multiple cutaneous dimples in arms and legs: a congenital syndrome of mechanical origin. Am J Dis Child. 1947;74:543–62. doi: 10.1001/archpedi.1947.02030010557001. http://dx.doi.org/10.1001/archpedi.1947.02030010557001. [DOI] [PubMed] [Google Scholar]
- 2.Maroteaux P, Spranger J, Opitz W, et al. Le syndrome campomelique. Presse Med. 1971;22:1157–62. [PubMed] [Google Scholar]
- 3.Houston CS, Opitz JM, Spranger JW, et al. The campomelic syndrome: review, report of 17 cases, and follow-up on the currently 17-year-old boy first reported by Maroteaux et al. in 1971. Am J Med Genet. 1983;15:3–28. doi: 10.1002/ajmg.1320150103. http://dx.doi.org/10.1002/ajmg.1320150103. [DOI] [PubMed] [Google Scholar]
- 4.Mansour S, Hall CM, Pembrey ME, Young ID. A clinical and genetic study of campomelic dysplasia. J Med Genet. 1995;32:415–20. doi: 10.1136/jmg.32.6.415. http://dx.doi.org/10.1136/jmg.32.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster JW, Dominguez-Steglich MA, Guioli S, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372:525–30. doi: 10.1038/372525a0. http://dx.doi.org/10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- 6.Orioli M, Castilla EE, Barbosa-Neto JG. The birth prevalance rates for skeletal dysplasias. J Med Genet. 1986;23:328–32. doi: 10.1136/jmg.23.4.328. http://dx.doi.org/10.1136/jmg.23.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gugliantini P, Maragliano G, Piscione M, Licata G. Constitutional osteochondrodysplasias identifiable at birth. A short review on the state of the art in radiodiagnosis in the late 20th century. Radiol Med. 1999;97:116–20. [PubMed] [Google Scholar]
- 8.Meyer J, Sudbeck P, Held M, et al. Mutational analysis of the SOX9 gene in campomelic dysplasia and autosomal sex reversal: lack of genotype/phenotype correlations. Hum Molec Genet. 1997;6:91–8. doi: 10.1093/hmg/6.1.91. http://dx.doi.org/10.1093/hmg/6.1.91. [DOI] [PubMed] [Google Scholar]
- 9.De Santa Barbara P, Bonneaud N, Boizet B, et al. Direct interaction of SRY-related protein SOX9 and steroidogenic factor 1 regulates transcription of the human anti-Müllerian hormone gene. Mol Cell Biol. 1998;18:6653–65. doi: 10.1128/mcb.18.11.6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benko S, Fantes JA, Amiel J, et al. Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nature Genet. 2009;41:359–64. doi: 10.1038/ng.329. http://dx.doi.org/10.1038/ng.329. [DOI] [PubMed] [Google Scholar]
- 11.Preiss S, Argentaro A, Clayton A, et al. Compound effects of point mutations causing campomelic dysplasia/autosomal sex reversal upon SOX9 structure, nuclear transport, DNA binding, and transcriptional activation. J Biol Chem. 2001;276:27864–72. doi: 10.1074/jbc.M101278200. http://dx.doi.org/10.1074/jbc.M101278200. [DOI] [PubMed] [Google Scholar]
- 12.Thomas S, Winter RB, Lonstein JE. The treatment of progressive kyphoscoliosis in campomelic dysplasia. Spine. 1997;22:1330–7. doi: 10.1097/00007632-199706150-00010. http://dx.doi.org/10.1097/00007632-199706150-00010. [DOI] [PubMed] [Google Scholar]