

Abstract
Aim:
This study was planned with the aim of retrospectively reviewing the clinical and laboratory findings and therapies of our patients diagnosed with tuberous sclerosis and redefining the patients according to the diagnostic criteria revised by the 2012 International Tuberous Sclerosis Complex Consensus Group and comparing them with the literature.
Materials and Method:
Twenty patients diagnosed with tuberous sclerosis complex in the Pediatric Neurology Clinic were examined retrospectively in terms of clinical findings and therapies. The diagnoses were compared again according to 1998 and 2012 criteria.
Results:
It was observed that the complaint at presentation was seizure in 17 of 20 patients and hypopigmented spots on the skin in 3 of 20 patients. On the initial physical examination, findings related with the disease were found in the skin in 17 of the patients, in the eye in 5, in the kidneys in 7 and in the brain with imaging in 17. No cardiac involvement was observed in the patients. Infantile spasm was observed in 7 of the patients who presented because of seizure (n=17), partial seizure was observed in 7 and multiple seizure types were observed in 3. It was found that sirolimus treatment was given to 9 of 20 patients because of different reasons, 7 of these 9 patients had epileptic seizures and sirolimus treatment had no effect on epileptic seizures. According to 2012 diagnostic criteria, no marked change occured in the diagnoses of our patients.
Conclusions:
It was observed that the signs and symptoms of our patients were compatible with the literature. Molecular genetic examination was planned for the patients who were being followed up because of probable tuberous sclerosis complex. It was observed that sirolimus treatment had no marked effect on the seizure frequency of our patients.
Keywords: Diagnostic criteria, mammalian target of rapamycin (mTOR), tuberous sclerosis complex
Introduction
Tuberous Sclerosis Complex is the most common single gene disease transmitted by autosomal dominant inheritance and has an incidence of 1/6 000–1/10 000 live births (1). Approximately 2/3 of the cases occur by spontaneous mutation. As a result of mutations in TSC1 (9q34) and TSC2 (16p13.3) genes, respectively, the function of hamartin-tuberin complex is disrupted and clinical findings occur because its inhibitor effect in the mammalian target of rapamycin (mTOR) signaling pathway is disrupted (1, 2). The diagnostic criteria of the disease which was described approximately 150 years ago were redefined in 2012 by the International Tuberous Sclerosis Complex Consensus Group (3). In recent years, mammalian target of rapamycin (mTOR) inhibitors have come into use in treatment of tuberous sclerosis complex (4).
This study was planned to retrospectively review the clinical and laboratory findings and treatment data of our patients diagnosed with tuberous sclerosis complex and redefine the patients according to the diagnostic criteria which were redefined by the International Tuberous sclerosis Complex Consensus Group in 2012 and compare them with the literature.
Material and Methods
Twenty pediatric patients (10 male and 10 female patients) who were followed up with a diagnosis of tuberous sclerosis complex between January 2008 and January 2014 aged between 1 year and 17 years (mean age: 5.9 years) were retrospectively examined in terms of clinical findings and treatment data. The diagnoses were compared again according to 1998 and 2012 diagnostic criteria (Table 1, 2), (5, 6).
Table 1.
Diagnostic criteria for tuberous sclerosis complex (1998)
| Major findings | Minor findings |
|---|---|
|
|
Definite diagnosis: two major or one major+two minor findings
Highly probable diagnosis: one major+one minor finding
Probable diagnosis: one major or two minor findings
Table 2.
Diagnostic criteria for tuberous sclerosis complex (2012)
| A. Genetic diagnostic criteria | |
|
| |
| It is sufficient to show pathogenic TSC mutation from a normal tissue for a definite diagnosis of tuberous sclerosis complex. Pathological mutation is inactivation in the functions of TSC1 and TSC2 proteins (for example; non-frame insertion, deletion or silent mutation which prevent protein synthesis or “missense” mutations which lead to large deletions or functional losses). TSC1 and TSC2 mutations are not sufficent to make a definite diagnosis of TSC (their effects on the functions of proteins are not known). Approximately 10–25% of the individuals with TSC do not have a mutation defined by conventional genetic tests. Lack of demonstration of mutation does not exclude the diagnosis of TSC and has no effect on the use of the clinical diagnostic criteria. | |
|
| |
| B. Clinical diagnostic criteria | |
| Major findings | Minor findings |
|
| |
|
|
|
| |
| Definite diagnosis: 2 major or 1 minor+ ≥2 minor factors | |
| Probable diagnosis: 1 major or ≥2 minor factors | |
Including tubers and cerebral white matter radial migration lines
Presence of lymphangioleiomyomatosis and angiomyolipoma is sufficient for a definite diagnosis
Statistical analysis
The SPSS 17.0 package program was used in the statistical analyses. Descriptive statistics was utilized. The data were summarized as n (%) or mean ± standard deviation.
Results
It was found that the complaint at presentation was seizure in 85% of the patients and hypopigmented spots on the skin in 15%. On physical examination at presentation, hypopigmented macule was found in 85% of the patients, fascial angiofibroma was found in 40%, “shagreen” patch was found in 10%, fibrous plaque was found in 10% and hyperpigmented macule was found in 10%. No skin lesion was observed in 10% of the patients. Ophtalological examination were found to be normal in 70% of the patients. Retinal hamartoma was found in 25% and papilledema was found in 5% (Table 3). Echocardiography was performed in 90% of the patients and found to be normal. Electrocardiograms of all patients were found to be normal. Renal ultrasonography (USG) was found to be normal in 90% of the patients at presentation. Renal cysts were found in 5% of the patients and millimetric angiomyolipoma was found in 5% (Table 3). On follow-up renal ultrasonography, 25% of the patients had normal findings, 15% of the patients had findings compatible with renal cystic disease, 15% of the patients had findings compatible with renal angiomyolipoma and 5% had findings compatible with renal cyst and angiomyolipoma (Figure 1). Follow-up USG was not performed in 30% of the patients in the last 6 months. At presentation, brain magnetic resonance imaging was found to be normal in 15% of the patients, whereas cortical tubers and supependymal nodules were found in the other patients. Gient cell astrocytoma was found in addition to cortical tubers and supependymal nodules in one patient (Table 3, Figure 2, 3). Thirty five percent of the patients who presented because of seizure presented with infantile spasm, 35% presented with partial seizure and 15% presented with multiple seizure types. On the first electroencephalogram (EEG), hypsarythmia was found in 7 patients (35%), focal epileptic disorder was found in 15% of the patients and multifocal epileptic disorder was found in 15% of the patients. The initial EEG was normal in 7 patients (35%). It was found that vigabatrin was started in all patients with infantile spasm initially and adrenocorticotropic hormone (ACTH) was administered in 2 patients in whom convulsions could not be controlled. It was observed that valproic acid was started in four patients, carbamazepine was started in one patient, levetirecetam was started in four patients and phenobarbital was started in one patient as the initial antiepileptic (Table 4). It was found that 10 patients whose epileptic seizures still continued were being followed up with polytherapy. It was found that sirolimus treatment was given to 9 of 20 patients (45%) because of different reasons and 7 of these patients (77,7%) had epileptic seizures. It was observed that the seizure frequency did not change in 3 of 7 patients who were receiving sirolimus. On the other hand, the seizure frequency increased in 2 patients, decreased in 1 patient and absence of seizures occured in 1 patient (Table 4). It was found that WISC-R Intelligence test was applied to a total of 8 patients who were older than 6 years; one patient was found to be brilliant, two patients were found to have normal intelligence, 2 patients were found to have mild mental retardation, 2 patients were found to have moderate mental retardation and one patient was found to have severe mental retardation.
Table 3.
Demographic properties of the patients and findings at presentation
| Patient number | Age (years)/Gender | Age of onset of complaints (years) | Complaint at presentation | Opthalmological findings at first PEa | Renal USG at first presentation | Brain MRI at first presentation |
|---|---|---|---|---|---|---|
| 1 | 2.4/Male | 0.25 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 2 | 7.1/Female | 0.1 | Hypopigmented spots | Retinal hamartoma | Normal | C.tuber+s.nodule |
| 3 | 8.1/Female | 0.3 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 4 | 7.5/Female | 7 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 5 | 7/Male | 2 | Seizure | Retinal hamartoma | Normal | C.tuber+s.nodule |
| 6 | 2/Male | 0.25 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 7 | 2/Male | 0.8 | Seizure | Normal | Normal | Normal |
| 8 | 5.5/Male | 0.3 | Seizure | Retinal hamartoma | Renal cyst | C.tuber+s.nodule |
| 9 | 2.8/Male | 0.9 | Seizure | Normal | Normal | Normal |
| 10 | 2/Male | 0.25 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 11 | 7.3/Male | 5.1 | Seizure | Papilledema | Normal | SEGA |
| 12 | 17/Female | 1.5 | Hypopigmented spots | Normal | Normal | C.tuber+s.nodule |
| 13 | 8/Female | 1 | Seizure | Retinal hamartoma | Normal | C.tuber+s.nodule |
| 14 | 5.8/Male | 0.25 | Seizure | Normal | Angiomyolipoma | C.tuber+s.nodule |
| 15 | 1/Female | 0.5 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 16 | 2.6/Female | 1 | Seizure | Retinal hamartoma | Normal | C.tuber+s.nodule |
| 17 | 6/Male | 3 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 18 | 5.7/Female | 1 | Seizure | Normal | Normal | Normal |
| 19 | 8.5/Female | 1 | Seizure | Normal | Normal | C.tuber+s.nodule |
| 20 | 11/Female | 6.5 | Hypopigmented spots | Normal | Normal | C.tuber+s.nodule |
PE: physical examination; C.tuber: cortical tuber; S.nodule: solitary nodule
Figure 1.
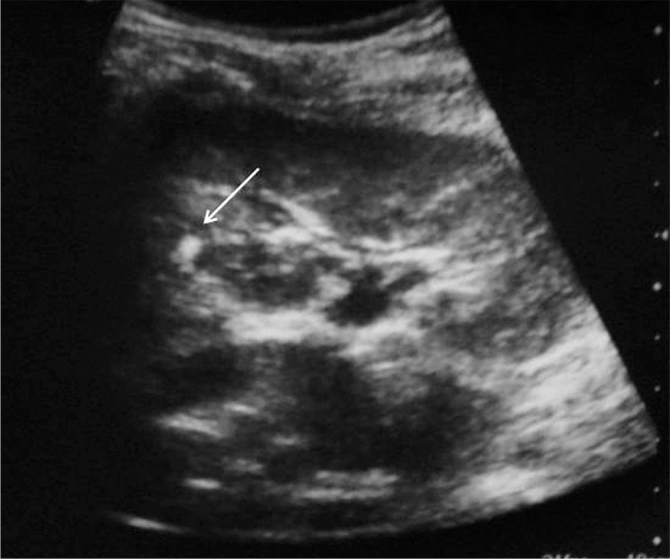
Hyperechogenic appearance compatible with angiomyolipoma on renal USG
Figure 2.
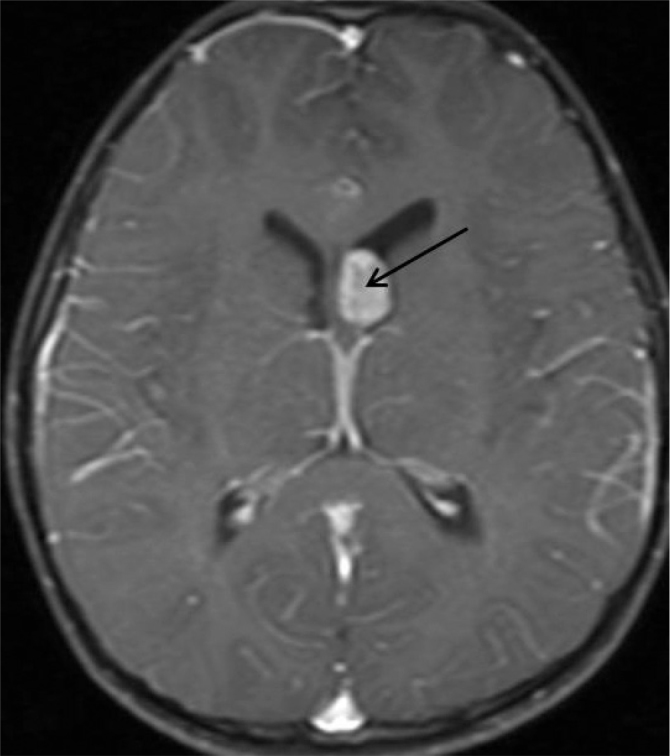
Mass formation compatible with giant cell tumor showing contrast uptake at the level of foramen of Monro on the left side on axial contrast-enhanced T1 weighted brain MRI
Figure 3.
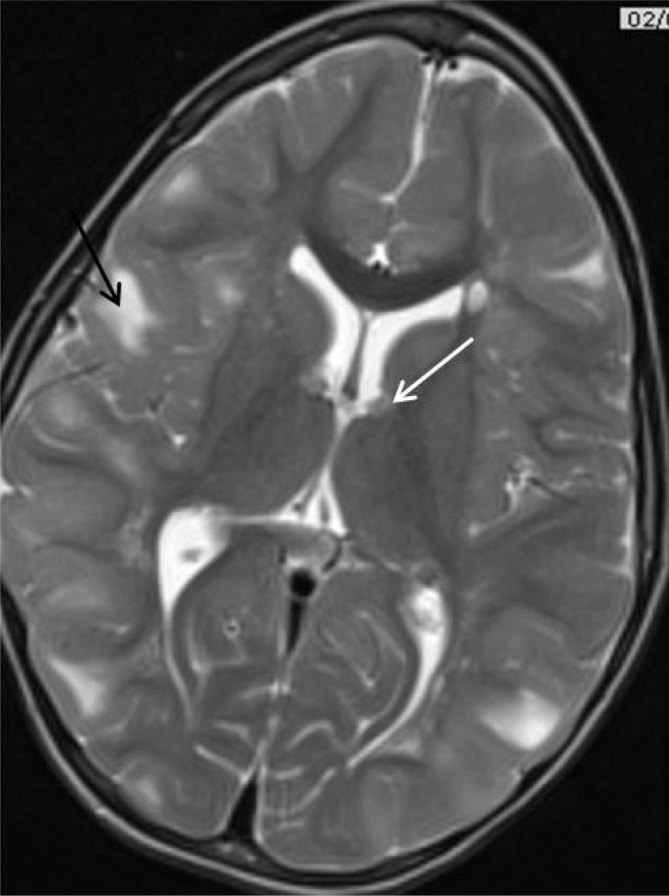
Cortical tubers (black arrow) and subependymal nodules (white arrow) on T2 weighted brain MRI
Table 4.
Seizure type, EEG findings, the antiepileptic drug used initially and relation of use of sirolimus and seizure
| Patient number | Seizure type | Initial EEG | The antiepileptic drug used initially | Sirolimus | Seizure frequency after Sirolimus |
|---|---|---|---|---|---|
| 1 | Infantile spasm | Hpsarrhythymia | Vigabatrin | - | - |
| 2 | No seizure | Normal | - | Received | - |
| 3 | Infantile spasm | Hpsarrhythymia | Vigabatrin | - | - |
| 4 | Partial seizure | Focal epileptic disorder | Carbamazepine | - | - |
| 5 | No seizure | Normal | - | - | - |
| 6 | Infantile spasm | Hpsarrhythymia | Vigabatrin | Received | Increased |
| 7 | Partial seizure | Normal | Sodyum valproat | - | - |
| 8 | Multiple seizure types | Multifocal epileptic disorder | Sodyum valproat | Received | No seizure |
| 9 | Multiple seizure types | Multifocal epileptic disorder | Fenobarbital | - | - |
| 10 | Infantile spasm | Hpsarrhythymia | Vigabatrin | Received | No change |
| 11 | Partial seizure | Normal | Levetirasetam | Received | Increased |
| 12 | Partial seizure | Normal | Sodyum valproat | - | - |
| 13 | Multiple seizure types | Multifocal epileptic disorder | Levetiracetam | - | - |
| 14 | Infantile spasm | Hpsarrhythymia | Vigabatrin | Received | No change |
| 15 | Infantile spasm | Hpsarrhythymia | Vigabatrin | - | - |
| 16 | Infantile spasm | Hpsarrhythymia | Vigabatrin | Received | No change |
| 17 | Partial seizure | Normal | Levetiracetam | - | - |
| 18 | Partial seizure | Focal epileptic disorder | Levetiracetam | - | - |
| 19 | Partial seizure | Normal | Sodium valproate | Received | Decrease by 25% |
| 20 | No seizure | Focal epileptic disorder | - | Received | - |
Seven of 8 patients who were applied 0–6 year Denver II developmental screening test were found to be abnormal (7). Marked retardation was found in language development and individual social development in all subjects who had an abnormal Denver II developmental screening test; mild retardation was found in gross and fine motor areas. Autistic findings were observed in 30% of the patients (8). According to 1998 diagnostic criteria, Seventeen patients were being followed up with a definite diagnosis of tuberous sclerosis complex, one patient was being followed up with a diagnosis of highly probable tuberous sclerosis complex and 2 patients were being followed up with a diagnosis of low probability tuberous sclerosis complex. On the other hand, definite diagnosis was made in 17 patients and a diagnosis of probable tuberous sclerosis was made in three patients according to 2012 diagnostic criteria (Table 5).
Table 5.
Diagnoses of the subjects according to 1998 and 2012 diagnostic criteria
| Patient number | Diagnosis according to 1998 diagnostic criteria | Diagnosis according to 2012 diagnostic criteria |
|---|---|---|
| 1 | Definite | Definite |
| 2 | Definite | Definite |
| 3 | Definite | Definite |
| 4 | Definite | Definite |
| 5 | Definite | Definite |
| 6 | Definite | Definite |
| 7 | Probable | Probable |
| 8 | Definite | Definite |
| 9 | Probable | Probable |
| 10 | Definite | Definite |
| 11 | Definite | Definite |
| 12 | Definite | Definite |
| 13 | Definite | Definite |
| 14 | Definite | Definite |
| 15 | Definite | Definite |
| 16 | Definite | Definite |
| 17 | Definite | Definite |
| 18 | Highly probable | Probable |
| 19 | Definite | Definite |
| 20 | Definite | Definite |
Discussion
Tuberous sclerosis complex is a disease with highly variable age of onset, disease severity and signs and symptoms which occurs in relation with a specific genotype. In addition, it may have different clinical expressions in the same family (4, 9).
In population-based studies, it has been found that patients diagnosed with tuberous sclerosis complex had one of the characteristic skin lesions with a rate of 81–95% (10). Similarly, at least one of the skin lesions was present in 90% of the patients in our study. In patients with tuberous sclerosis complex, hypomelanotic macules and fibrous plaques occur earlier compared to typical facial angiofibromas or periungual fibromas (11). Hypomelanotic macules were found in 75% of our patients on physical examination at presentation, whereas facial angiofibromas were present in 40%, fibrous plaques were present in 10% and periungual fibromas accompanied hypomelanotic macules in 10%. In tuberous sclerosis complex, each follow-up examination should be performed in detail including nail examination and should be recorded considering that the onset age of the lesions is variable, though the lesions have no risk of malign transformation (10, 11).
Ophtalmological findings in tuberous sclerosis complex include retinal and non-retinal lesions. These lesions usually do not disrupt vision and they do not need to be treated. In a study in which the opthalmological findings of 100 patients with tuberous sclerosis complex aged between 2 and 76 years were examined, retinal hamartoma was found in 44 of the patients (12). Retinal achromic patches were found in 39 of the patients (12). Non-retinal lesions included angiofibroma in the eyelid in 39 patients, non-paralytic strabismus in 5 patients, coloboma in three patients and depigmentation of the iris in 2 patients (12). At presentation, 14 of our patients had normal opthalmological findings, five patients had retinal hamartoma and one patient had papilledema. We think that less opthalmological findings in our patients was related with the fact that the age group was younger and many lesions have the potential of developing in time. Subependymal giant cell astrocytoma (SEGA) and increased intracranial pressure syndrome were found in our patient who was observed to have papilledema.
The characteristic cardiac finding in tuberous sclerosis complex is rhabdomyomas and these tumors are bening tumors which generally occur as multiple lesions (13). Cardiac rhabdomyomas typically develop in the intrauterine period and are generally diagnosed by prenatal USG (6). They are mostly asymptomatic or may become symptomatic in the neonatal period and infancy (8, 12, 13). All rhabdomyomas regress spontaneously including the ones who have become symptomatic (14). Cardiac rhabdomyoma was not found in any of our patients. This was associated with the fact that symptomatic rhabdomyomas were not referred to our center because our center did not include a pediatric cardiology unit and with the possibility that these lesions regressed spontaneously (our patients did not have clinical cardiac symptoms and cardiac examination was performed in the follow-up, but not in the beginning). When rhabdomyomas involve the conduction system, an arrhythmia called Wolf-Parkinson-White syndrome which is permanent for a life time may be observed (10, 13, 14). ECG was normal in all of our patient.
The renal lesions are observed frequently in tuberous sclerosis complex and the frequency increases with age. The most common renal lesions are angiomyolipomas and they have classical and epitheloid histological variants. The classical variant is differentiated by local invasion and the epitheloid variant which is differentiated with local or distant metastases may show malign transformation. Renal cysts, lympangioma and renal cell carcinoma are observed with a lower frequency. Renin-dependent hypertension may be observed in patients with renal lesion. Rarely, chronic renal failure may develop (15, 16). Since the TSC2 gene is close to the autosomal dominant polycystic kidney disease (PCKD), PCKD may also be observed in these patient (17). At presentation, 18 of our patients had normal ultrasonographic findings, simple renal cysts were found in one patient and millimetric angiomyolipoma was found in another patient. On follow-up USG, renal cystic disease was found in 3 patients, renal angiomyolipoma was found in 3 patients and renal cyst and angiomyolipoma were found in one patient. The follow-up period of our patients is short for the present time and we think that renal involvement will increase in the future.
Central nervous system lesions observed in tuberous sclerosis include glioneuronal hamartomas (cortical tubers), white matter heterotypes (dysplastic or demyelinizating white matter lesions), subependymal nodules and SEGA (18, 19). Cortical tubers are considered hamartomatous lesions which are differentiated with abnormal glial, neuronal cells and astrocytosis. They are observed in 80–90% of the patients and it is thought that their sizes and numbers are not related with the clinical picture. They are localized most commonly in the frontal and temporal areas and they are thougth to be responsible of epilepsy and behavioral problems. In contrast to the other tuberous sclerosis complex-related lesions, new tubers do not occur postnatally. Neoplastic transformation is not observed in the tubers and they calcify in time (18, 20, 21, 22). Subependymal nodules are formations smaller than 1 cm localized in the surface of the lateral ventricle and third ventricle which occur in the fetal period. Histologically, they are hamartomatous formations with intensive vascularity including astrocytes surrounded by a thin ependymal cell layer. They are considered not to be related with epilepsy and behavioral problems. They are observed in 80–90% of the patients with tuberous sclerosis complex (18, 20, 21, 22). Subependymal giant cell tumor is observed in 10–15% of the patients and frequently in the first 20 years. Only 6–9% of the patients are symptomatic (1, 19, 23). At presentation, brain MRI findings were found to be normal in three patients, but cortical tubers and subependymal nodules were found in the other patients. In one patient, giant cell astrocytoma was observed in addition to cortical tubers and subependymal nodules.
In tuberous sclerosis complex, seizures are the most common reason for presentation and the most commonly observed medical problems. Epilepsy develops in 75–90% of the patients (10, 24). Sixty percent of the patients who present with seizure present in the first year of life (8, 25). Chu-Shore et al. (25) reported that epilepsy developed in 246 (99%) of 248 patients with tuberous sclerosis complex who had one seizure. At the time of diagnosis, the most common seizure type is infantile spasm which is observed in 36–69% of the patients (26). On the other hand, tuberous sclerosis complex has been shown in approximately 25% of the patients followed up with a diagnosis of infantile spasm (27). Other seizure types including simple partial, complex partial and secondary generalized seizure are also observed (25). In a study performed in our country, it was reported that the age of onset in patients with tuberous sclerosis complex ranged between 3 days and 2,5 years and 76% of the patients were aged one year or younger. In the same study, the most common seizure type was reported to be partial seizure with a rate of 95% and this was followed by infantile spasm and status epilepticus with a rate of 38% (28). Similar to the literature, seizure was observed in 85% of our patients. Infantile spasm was the first seizure type in 41% of our patients. Tuberous sclerosis complex was found in 7 (17,5%) of 40 patients with infantile spasm whom we followed up between the same dates. Partial seizure was observed in 41% of our patients and multiple seizure types were observed in 17.5%. EEG abnormality is observed in approximately 75% of the patients with tuberous sclerosis complex. These include focal or multifocal discharges (48%), hypsarrhythmia (19%) and generalized spike-wave activity (8%) (25, 27). In our study, hypsarrhythmia (41%), focal epileptic disorder (17.5%) and multifocal epileptic disorder (17.5%) were found on the initial EEG. EEG was normal in 41% of the patients. Although epileptic seizures are the most common finding of tuberous sclerosis complex, they are not considered a major diagnostic criterion (7, 8). In treatment of epilepsy, one acts according to the rules of classical epilepsy treatment (25, 26). The classical first-line drug in treatment of infantile spasm is adrenocorticotropic hormone (ACTH), whereas vigabatrin is the first-line drug in tuberous sclerosis complex patients with infantile spasm (29). It was found that vigabatrin was initiated primarily in all of our patients with tuberous sclerosis complex who had infantile spasm and ACTH was given to 2 patients whose spasms could not be controlled. It was observed that valproic acid was started in four patients who had non-infantile spasm seizures, carbamazepine was started in one, levetirecetam was started in four and phenobarbital was started in one as the first antiepileptic drug. It has been found that 10 patients whose epileptic seizures are still continuing are being followed up with polytherapy.
In population-based studies, retardation in cognitive functions have been observed in 44–65% of the patients diagnosed with tuberous sclerosis (8, 24). Cognitive dysfunction is observed more frequently in patients with infantile spasm or resistant seizures (24). In a study conducted with 61 patients with a diagnosis of tuberous sclerosis, it was shown that the ratio of the area of glioneuronal hamartomas to the total brain volume was determinative for cognitive functions rather than the number of glioneuronal hamartomas (30). Intelligence test was performed in a total of 8 patients; 2 patients were found to have mild mental retardation and 3 patients were found to have moderate or severe mental retardation. It was observed that 7 of 8 patients who were applied Denver Developmental test were abnormal. In our study, glioneuronal hamartomas and cognitive functions could not be compared, since the patients did not have MRIs obtained at the same ages.
Autistic behavioral disorders are frequently observed problems in patients diagnosed with tuberous sclerosis complex. In different series, behavioral problems have been reported with a rate of 40–90%. These problems are more prominent in patients who have mental retardation or resistant seizures (31). In some studies, it was reported that glioneuronal hamartomas localized in the temporal area were related with autistic findings (32). However, it was reported that the number and localization of glioneuronal hamartomas were not related with autistic findings in some other studies (33). In our study, autistic findings were observed in 30% of the patients.
In tuberous sclerosis complex, the function of hamartin-tuberin complex is disrupted and clinical findings occur because its inhibitor effect in the mammalian target of rapamycin (mTOR) signaling pathway is disrupted (1, 2). mTOR is a kinase which has a key function in catabolic and anabolic metabolisms (34). Classically, the safety and reliability of mTOR inhibitors have been proven in patients with cancer (33). In selected patients with tuberous sclerosis complex, mTOR inhibitors have been started to be used in SEGAs and cardiac rhabdomyomas or angiomyolipomas (36). Vigabatrin is a dose-dependent enzyme inhibitor of gamma aminobutiric acid transaminase (GABA-T). It shows its action by increasing the GABA level. It is especially efficient in tuberous sclerosis patients with infantile spasm (29, 37). It has also been shown to be efficient in partial seizures in patients with tuberous sclerosis (38). It has not been elucidated why it is very efficient in infantile spasm in patients with tuberous sclerosis. However, vigabatrin has been shown to be a partial mTOR inhibitor in tuberous sclerosis mouse models (39). This property may explain why vigabatrin is more efficient in epilepsy related with tuberous sclerosis complex. Again, the efficieny of evarilimus which is a mTOR inhibitor was examined in patients with tuberous sclerosis complex who had resistant epilepsy and the frequency of seizure was found to be decreased in 4 of 6 patients (40). In another study in which 7 patients with tuberous sclerosis complex who received everolimus or sirolimus treatment were examined, it was reported that mTOR inhibitors were efficient in resistant epilepsy (41). In a study performed in our country, it was reported that rapamycine was given to 7 of 86 patients with a diagnosis of tuberous sclerosis complex with different reasons, 5 of these patients had resistant seizures and all patients were free of seizure after a 6-month treatment period (42). Sirolimus treatment was given to 45% of the patients becasue of SEGA or angiomyolipomas. Seven of these 9 patients were found to have epileptic seizures at the same time. No marked change was observed in the patients who received sirolimus.
The difference of the diagnostic criteria redefined by the International Tuberous Sclerosis Complex Group in 2012 compared to 1998 diagnostic criteria is that the genetic results were included in the criteria and the number of diagnosis groups decreased from three (definite, high probability and low probability) to two (definite, probable) (Table 1, 2) (7, 8). In our study, the diagnosis of the patients who had a definite diagnosis did not change, whereas the patients who were being followed up with probable and high probability diagnosis all shifted to the probable diagnosis group according to 2012 criteria. Molecular genetic examination was planned in the patients who were diagnosed with probable tuberous sclerosis complex according to 2012 diagnostic criteria.
Conclusively, it was observed that the signs and symptoms of our patients were compatible with the literature. No marked change occured in the diagnoses of our patients according to 2012 diagnostic criteria. Molecular genetic testing was planned in the patients who were being followed up with a diagnosis of probable tuberous sclerosis complex. It was observed that sirolimus treatment had no marked effect on the frequency of seizures in our patients. The efficiency of mTOR inhibitors in epileptic children with a diagnosis of tuberous sclerosis complex will be determined with large-scale, multi-center, prospective studies.
Acknowledgments
Authors would like to thank Prof. Faik Sarıalioğlu for his valuable contributions to this study.
Footnotes
Ethics Committee Approval: Ethics committee approval was not received due to the retrospective nature of this study.
Informed Consent: Written informed consent was obtained from the parents of the patients who participated in this study.
Conflict of Interest: No conflict of interest was declared by the authors.
Author Contributions: Concept - İ.E.; Design - İ.E.; Supervision - İ.E.; Funding - İ.E., N.Y., A.E., O.A.; Materials - İ.E., T.S., S.S., A.E., N.Y.; Data Collection and/or Processing - İ.E., S.Ş., Ş.D., T.S.; Analiz ve/veya yorum / Analysis and/or Interpretation - İ.E.; Literature Review - İ.E., T.Ş., S.Ş.; Writer - İ.E.; Critical Review - İ.E., N.Y.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Hallett L, Foster T, Liu Z, et al. Burden of disease and unmet needs in tuberous sclerosis complex with neurological manifestations: systematic review. Curr Med Res. doi: 10.1185/03007995.2011.586687. http://dx.doi.org/10.1185/03007995.2011.586687 [DOI] [PubMed] [Google Scholar]
- 2.Schwartz RA, Fernández G, Kotulska K, Jóźwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189–202. doi: 10.1016/j.jaad.2007.05.004. http://dx.doi.org/10.1016/j.jaad.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49:243–54. doi: 10.1016/j.pediatrneurol.2013.08.001. http://dx.doi.org/10.1016/j.pediatrneurol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Northrup H, Wheless JW, Bertin TK, Lewis RA. Variability of expression in tuberous sclerosis. J Med Genet. 1993;30:41–3. doi: 10.1136/jmg.30.1.41. http://dx.doi.org/10.1136/jmg.30.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roach E, Gomez M, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624–8. doi: 10.1177/088307389801301206. http://dx.doi.org/10.1177/088307389801301206. [DOI] [PubMed] [Google Scholar]
- 6.Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–54. doi: 10.1016/j.pediatrneurol.2013.08.001. http://dx.doi.org/10.1016/j.pediatrneurol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anlar B, Bayoğlu BU, Yalaz K. DENVER II gelişimsel tarama testi “Türk Çocuklarına Uyarlanması ve Standardizasyonu”. Gelişimsel Çocuk Nörolojisi Derneği, Ankara. 2009:5–6. [Google Scholar]
- 8.Volkmar FR, McPartland JC. From Kanner to DSM-5: Autism as an evolving diagnostic concept. Annu Rev Clin Psychol. 2014;10:193–212. doi: 10.1146/annurev-clinpsy-032813-153710. http://dx.doi.org/10.1146/annurev-clinpsy-032813-153710. [DOI] [PubMed] [Google Scholar]
- 9.Smalley SL, Burger F, Smith M. Phenotypic variation of tuberous sclerosis in a single extended kindred. J Med Genet. 1994;31:761–5. doi: 10.1136/jmg.31.10.761. http://dx.doi.org/10.1136/jmg.31.10.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yates JR, Maclean C, Higgins JN, et al. The Tuberous Sclerosis 2000 Study: presentation, initial assessments and implications for diagnosis and management. Arch Dis Child. 2011;96:1020–5. doi: 10.1136/adc.2011.211995. http://dx.doi.org/10.1136/adc.2011.211995. [DOI] [PubMed] [Google Scholar]
- 11.Webb DW, Clarke A, Fryer A, Osborne JP. The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol. 1996;135:1–5. http://dx.doi.org/10.1111/j.1365-2133.1996.tb03597.x. [PubMed] [Google Scholar]
- 12.Rowley SA, O’Callaghan FJ, Osborne JP. Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol. 2001;85:420–3. doi: 10.1136/bjo.85.4.420. http://dx.doi.org/10.1136/bjo.85.4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jóźwiak S, Domańska-Pakieła D, Kwiatkowski DJ, Kotulska K. Multiple cardiac rhabdomyomas as a sole symptom of tuberous sclerosis complex: case report with molecular confirmation. J Child Neurol. 2005;20:988–9. doi: 10.1177/08830738050200121101. http://dx.doi.org/10.1177/08830738050200121101. [DOI] [PubMed] [Google Scholar]
- 14.Webb DW, Thomas RD, Osborne JP. Cardiac rhabdomyomas and their association with tuberous sclerosis. Arch Dis Child. 1993;68:367–70. doi: 10.1136/adc.68.3.367. http://dx.doi.org/10.1136/adc.68.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006;70:1777–82. doi: 10.1038/sj.ki.5001853. http://dx.doi.org/10.1038/sj.ki.5001853. [DOI] [PubMed] [Google Scholar]
- 16.Nelson CP, Sanda MG. Contemporary diagnosis and management of renal angiomyolipoma. J Urol. 2002;168:1315–25. doi: 10.1016/S0022-5347(05)64440-0. http://dx.doi.org/10.1016/S0022-5347(05)64440-0. [DOI] [PubMed] [Google Scholar]
- 17.Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80. doi: 10.1086/316951. http://dx.doi.org/10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizuguchi M, Takashima S. Neuropathology of tuberous sclerosis. Brain Dev. 2001;23:508–15. doi: 10.1016/s0387-7604(01)00304-7. http://dx.doi.org/10.1016/S0387-7604(01)00304-7. [DOI] [PubMed] [Google Scholar]
- 19.Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology. 2004;63:1457–61. doi: 10.1212/01.wnl.0000142039.14522.1a. http://dx.doi.org/10.1212/01.WNL.0000142039.14522.1A. [DOI] [PubMed] [Google Scholar]
- 20.Altman NR, Purser RK, Post MJ. Tuberous sclerosis: characteristics at CT and MR imaging. Radiology. 1988;167:527–32. doi: 10.1148/radiology.167.2.3357966. http://dx.doi.org/10.1148/radiology.167.2.3357966. [DOI] [PubMed] [Google Scholar]
- 21.Kingsley DP, Kendall BE, Fitz CR. Tuberous sclerosis: a clinicoradiological evaluation of 110 cases with particular reference to atypical presentation. Neuroradiology. 1986;28:38–46. doi: 10.1007/BF00341764. http://dx.doi.org/10.1007/BF00341764. [DOI] [PubMed] [Google Scholar]
- 22.Goodman M, Lamm SH, Engel A, Shepherd CW, Houser OW, Gomez MR. Cortical tuber count: a biomarker indicating neurologic severity of tuberous sclerosis complex. J Child Neurol. 1997;12:85–90. doi: 10.1177/088307389701200203. http://dx.doi.org/10.1177/088307389701200203. [DOI] [PubMed] [Google Scholar]
- 23.Nishio S, Morioka T, Suzuki S, Kira R, Mihara F, Fukui M. Subependymal giant cell astrocytoma: clinical and neuroimaging features of four cases. J Clin Neurosci. 2001;8:31–4. doi: 10.1054/jocn.2000.0767. http://dx.doi.org/10.1054/jocn.2000.0767. [DOI] [PubMed] [Google Scholar]
- 24.Joinson C, O’Callaghan FJ, Osborne JP, et al. Learning disability and epilepsy in an epidemiological sample of individuals with tuberous sclerosis complex. Psychol Med. 2003;33:335–44. doi: 10.1017/s0033291702007092. http://dx.doi.org/10.1017/S0033291702007092. [DOI] [PubMed] [Google Scholar]
- 25.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–41. doi: 10.1111/j.1528-1167.2009.02474.x. http://dx.doi.org/10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riikonen R, Simell O. Tuberous sclerosis and infantile spasms. Dev Med Child Neurol. 1990;32:203–9. doi: 10.1111/j.1469-8749.1990.tb16926.x. http://dx.doi.org/10.1111/j.1469-8749.1990.tb16926.x. [DOI] [PubMed] [Google Scholar]
- 27.Westmoreland BF. The electroencephalogram in tuberous sclerosis. In: Gomez MR, Sampson JR, Whittemore VH, editors. Tuberous sclerosis complex: developmental perspectives in psychiatry. 3rd ed. New York: Oxford University Press; 1999. p. 63. [Google Scholar]
- 28.Saltık S, Karatoprak YE, Taşel B. Tüberoskleroz kompleksi tanılı hastalarda epilepsinin özellikleri ve klinik seyri. Turk Arch Ped. 2013:123–30. [Google Scholar]
- 29.Pellock JM, Hrachovy R, Shinnar S, et al. Infantile spasms: a US consensus report. Epilepsia. 2010;51:2175–89. doi: 10.1111/j.1528-1167.2010.02657.x. http://dx.doi.org/10.1111/j.1528-1167.2010.02657.x. [DOI] [PubMed] [Google Scholar]
- 30.Jansen FE, Vincken KL, Algra A, et al. Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology. 2008;70:916–23. doi: 10.1212/01.wnl.0000280579.04974.c0. http://dx.doi.org/10.1212/01.wnl.0000280579.04974.c0. [DOI] [PubMed] [Google Scholar]
- 31.Kopp CM, Muzykewicz DA, Staley BA, et al. Behavior problems in children with tuberous sclerosis complex and parental stress. Epilepsy Behav. 2008;13:505–10. doi: 10.1016/j.yebeh.2008.05.010. http://dx.doi.org/10.1016/j.yebeh.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Bolton PF, Park RJ, Higgins JN, et al. Neuro-epileptic determinants of autism spectrum disorders in tuberous sclerosis complex. Brain. 2002;125:1247–55. doi: 10.1093/brain/awf124. http://dx.doi.org/10.1093/brain/awf124. [DOI] [PubMed] [Google Scholar]
- 33.Walz NC, Byars AW, Egelhoff JC, Franz DN. Supratentorial tuber location and autism in tuberous sclerosis complex. J Child Neurol. 2002;17:830–2. doi: 10.1177/08830738020170111401. http://dx.doi.org/10.1177/08830738020170111401. [DOI] [PubMed] [Google Scholar]
- 34.Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev. 2006;5:671–8. doi: 10.1038/nrd2062. http://dx.doi.org/10.1038/nrd2062. [DOI] [PubMed] [Google Scholar]
- 35.Atkins MB, Hidalgo M, Stadler WM, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of sirolimus kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22:909–18. doi: 10.1200/JCO.2004.08.185. http://dx.doi.org/10.1200/JCO.2004.08.185. [DOI] [PubMed] [Google Scholar]
- 36.Kohrman MH. Emergingtreatments in the management of tuberous sclerosis complex. Pediatr Neurol. 2012;46:267–75. doi: 10.1016/j.pediatrneurol.2012.02.015. http://dx.doi.org/10.1016/j.pediatrneurol.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Hsieh DT, Jennesson MM, Thiele EA. Epileptic spasms in tuberous sclerosis complex. Epilepsy Res. 2013;106:200–10. doi: 10.1016/j.eplepsyres.2013.05.003. http://dx.doi.org/10.1016/j.eplepsyres.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 38.Friedman D, Bogner M, Parker-Menzer K, Devinsky O. Vigabatrin for partial-onset seizure treatment in patients with tuberous sclerosis complex. Epilepsy Behav. 2013;27:118–20. doi: 10.1016/j.yebeh.2012.12.033. http://dx.doi.org/10.1016/j.yebeh.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 39.Zhang B, McDaniel SS, Rensing NR, Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS ONE. 2013;8:e57445. doi: 10.1371/journal.pone.0057445. http://dx.doi.org/10.1371/journal.pone.0057445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiegand G, May TW, Ostertag P, Boor R, Stephani U, Franz DN. Everolimus in tuberous sclerosis patients with intractable epilepsy: a treatment option? Eur J Paediatr Neurol. 2013;17:631–8. doi: 10.1016/j.ejpn.2013.06.002. http://dx.doi.org/10.1016/j.ejpn.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Cardamone M, Flanagan D, Mowat D, Kennedy SE, Chopra M, Lawson JA. Mammalian target of rapamycin inhibitors for intractable epilepsy and subependymal giant cell astrocytomas in tuberous sclerosis complex. J Pediatr. 2014;164:1195–200. doi: 10.1016/j.jpeds.2013.12.053. http://dx.doi.org/10.1016/j.jpeds.2013.12.053. [DOI] [PubMed] [Google Scholar]
- 42.Canpolat M, Per H, Gumus H, et al. Rapamycin has a beneficial effect on controlling epilepsy in children with tuberous sclerosis complex: results of 7 children from a cohort of 86. Childs Nerv Syst. 2014;30:227–40. doi: 10.1007/s00381-013-2185-6. http://dx.doi.org/10.1007/s00381-013-2185-6. [DOI] [PubMed] [Google Scholar]