

Abstract
Although epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) have been introduced for the treatment of non-small cell lung cancer (NSCLC), the emergence of secondary T790M mutation in EGFR or amplification of the Met proto-oncogene restrain the clinical success of EGFR-TKIs. Since heat shock protein-90 (Hsp90) stabilizes various oncoproteins including EGFR and c-Met, the inhibition of Hsp90 activity appears as a rational strategy to develop anticancer drugs. Despite preclinical efficacy of geldanamycin-anasamycin (GA)-derivatives containing benzoquinone moiety as Hsp90 inhibitors, the hepatotoxicity of these GA-derivatives restricts their therapeutic benefit. We have prepared WK-88 series of GA-derivatives, which lack the benzoquinone moiety. In this study, we have examined the anticancer effects of WK88-1 in Met-amplified- and gefitinib-resistant (HCC827GR) NSCLC cells and its parental HCC827 cells. Treatment with WK88-1 reduced the cell viability in both HCC827 and HCC827GR cells, which was associated with marked decrease in the constitutive expression of Hsp90 client proteins, such as EGFR, ErbB2, ErbB3, Met and Akt. Moreover, WK88-1 attenuated phosphorylation of these Hsp90 client proteins and reduced the anchorage-independent growth of HCC827GR cells. Administration of WK88-1 did not cause hepatotoxicity in animals and significantly reduced the growth of HCC827GR cells xenograft tumors in nude mice. Our study provides evidence that ErbB3 might be a client for Hsp90 in Met-amplified NSCLCs. In conclusion, we demonstrate that inhibition of Hsp90 dampens the activation of EGFR- or c-Met-mediated survival of Met-amplified NSCLCs and that WK88-1 as a Hsp90 inhibitor alleviates gefitinib resistance in HCC827GR cells.
Keywords: Gefitinib, Hsp90, non-benzoquinone GA, non-small cell lung cancer, WK88-1
Lung cancer is one of the most prevalent malignancies and a leading cause of death worldwide. About 85% cases of all lung cancers are non-small cell lung cancer (NSCLC), which is characterized by multiple mutations in the gene encoding epidermal growth factor receptor (EGFR).1 Since the mutated EGFR functions as a constitutively active receptor tyrosine kinase (RTK), several small-molecule tyrosine kinase inhibitors (TKI) have been developed as chemotherapeutic agents for the treatment of NSCLC.2–5 TKIs, such as erlotinib (Tarceva) and gefitinib (Iressa) have been shown to specifically target EGFR and eradicate NSCLCs without causing nonspecific side-effects.6,7 Despite high selectivity of action and proven initial clinical success of TKIs, several studies have demonstrated that acquired resistance to TKIs develops in many patients within a couple of years.8,9 Acquired resistance has been shown to be mainly associated with two genetically conferred mechanisms – a secondary T790M mutation in EGFR and the Met amplification.10–12
Several strategies have been proposed to overcome the acquired chemoresistance of NSCLC. Recently, irreversible EGFR-TKIs have been developed to inhibit the activity of T790M-EGFR,13,14 and combined treatment of NSCLC with EGFR-TKIs and Met-TKIs has been proposed to overcome resistance induced by c-Met amplification.15 Specifically, these approaches have been based on inhibiting the kinase activity of mutant EGFR or Met by selective inhibitors that target mutant receptor conformations, thereby modulating the EGFR or c-Met downstream signaling pathways.14,16 However, these therapeutic approaches often result in renewed drug resistance, because oncogene-addicted tumor cells can readily thwart the anticancer effects of a mono-targeted therapy by activating alternative survival pathways.17,18 Thus a rational strategy to overcome acquired chemotherapy resistance is to develop drugs that can target multiple cancer-related biochemical pathways.
Heat shock protein-90 (Hsp90), a component of a multi-chaperone complex, plays a key role in the stabilization and maturation of various wild type and/or mutant forms of proteins, such as EGFR, c-Met, ErbB2, Raf1, and Akt.19–22 The stabilization and overexpression of these oncoproteins have been implicated in the development and progression of cellular transformation.23–25 The disruption of Hsp90 chaperone activity induces degradation of these client proteins via the ubiquitin-proteasome pathway. Thus, the blockade of Hsp90 activity may inhibit diverse oncogenic signaling pathways. Accumulating evidence indicates that pharmacological inhibition of the Hsp90 activity has emerged as a promising strategy for cancer therapy.26 It is interesting to note that Hsp90 is constitutively expressed in tumor cells at 2–10 fold higher levels than in the normal cells.27 Moreover, Hsp90 exists in tumor cells as an ATP-bound active multi-chaperone complex, while it remains in normal cells in a latent or uncomplexed state.28 This phenomenon coincides with the finding that tumor cell-derived Hsp90 complexes have higher binding affinity to Hsp90 inhibitor, 17-AGG, a 17-allyl amino derivative of geldanamycin anasamycin (GA), as compared with Hsp90 present in normal cells.28 This higher affinity of tumor-derived Hsp90 for its inhibitor compounds suggests the potential selectivity of Hsp90 inhibition for cancer cells over normal cells. Although Hsp90 is an essential chaperone protein for all cells, tumor cells exhibit greater dependence on Hsp90's chaperoning function to restructure numerous unfolded and mutated proteins including EGFR and c-Met, which contribute to the development of chemoresistance in NSCLC. Thus, the targeted inhibition of Hsp90 activity would be a rational approach to overcome acquired resistance and to achieve the therapeutic goal.
Practically, Hsp90 inhibitors including GA and its derivatives have been reported to show strong antitumor effect on the growth of NSCLCs with T790M mutation of EGFR.20,29,30 Despite effective preclinical anticancer activities of GA and its derivatives, the clinical success of these molecules are thwarted by their hepatotoxic properties and poor in vivo stability.31,32 Although other classes of Hsp90 inhibitors such as purine scaffold inhibitors or diarylpryrazole compounds have been investigated,33,34 more improved GA-modified Hsp90 inhibitors lacking potential side-effects are being sought. Previous reports suggest that the undesirable toxicity of GA and its derivatives results from the “off target” effects of the benzoquinone moiety.35,36 Therefore, the GA derivative that lacks a benzoquinone moiety may be devoid of toxic effects. In accord with this, we recently reported the development of non-benzoquinone GA (e.g., WK88-1, WK88-2 and WK88-3) derivatives by a mutasynthetic approach and directed biosynthetic method using genetically engineered Streptomyces hygroscopicus (Fig.1).37,38 In the present study, we report the molecular mechanisms of antitumor effects of WK88-1, a new Hsp90 inhibitor, in gefitinib-resistant NSCLC cells with amplified c-Met.
Figure 1.
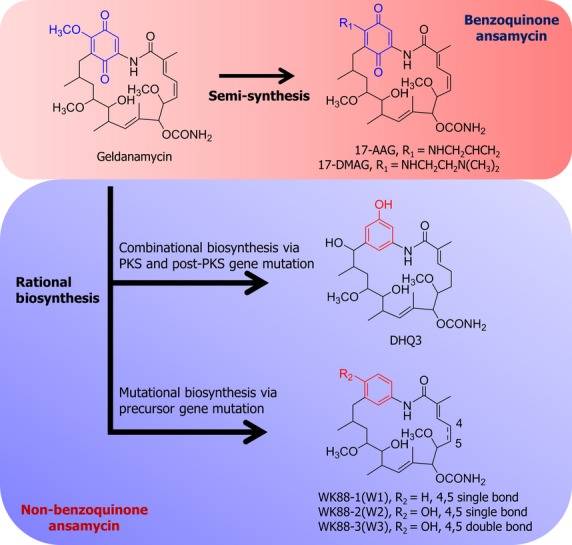
Schematic representation of production of the benzoquinone and non-benzoquinone GA analogs. DHQ3 (15-hydroxyl-17-demethoxyreblastatin) was purified in a combinational mutation with site-directed mutagenesis of the first dehydratase domain of the geldanamycin polyketide synthase (PKS) gene (gelA) and a post-PKS modification gene (gel7) of Streptomyces hygroscopicus JCM4427. WK88-1 (18-dehydroxyl-17-demethoxyreblastatin), WK88-2 (18-hydroxyl-17-demethoxyreblastatin), and WK88-3 (18-hydroxyl-17-demethoxy-4,5-dehydroreblastatin) were purified from a culture of S. hygroscopicus AC2, in which the AHBA synthase gene was disrupted by the kanamycin-resistance gene, supplemented with 3-aminobenzoic acid.
Materials and Methods
Materials
Antibodies specific for phospho-EGFR (Tyr1068; #3777), Met (#4560), phospho-Met (Tyr1234/1235; #3077), ErbB3 (#4754), phospho-ErbB3 (#4791), Akt (#4691), phospho-Akt (Ser473; #4060), Hsp90 (#4874), Hsp70 (#4872), Erk1/2 (#4695), phospho-Erk1/2 (Thr202/Tyr204; #4370), cleaved Caspase-3 (#9661), PARP (#9542) and β-actin (#4970) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody specific for EGFR was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA; sc-03). Gefitinib was purchased from LC Laboratories (Woburn, MA, USA; #G-4408). Geldanamycin was obtained from Enzo Life Sciences (Woburn, MA, USA; #BML-EI280). Fetal bovine serum (FBS), streptomycin, and penicillin were obtained from Thermo Scientific (South Logan, UT, USA). Non-benzoquinone geldanmaycin analogs were produced by following a mutasynthetic approach and a directed biosynthetic method.37,38 DHQ3, a 15-hydroxyl-17-demethoxy non-benzoquinone analog, was prepared from a genetically engineered strain (AC15) of Streptomyces hygroscopicus.38 WK88-1, WK88-2, and WK88-3 were purified from a culture of S. hygroscopicus AC2, in which the AHBA synthase gene was disrupted by the kanamycin-resistance gene, supplemented with 3-aminobenzoic acid.37 All isolated compounds were purified to a minimum purity of 97% as determined by HPLC and confirmed the structure via NMR and LC/MSn analysis.
Cell culture
The human EGFR mutant NSCLC cell lines HCC827 (del E746_A750 in exon 19)39 were maintained in RPMI-1640 with L-glutamine supplemented with 10% FBS and penicillin/streptomycin. Gefitinib-resistant HCC827GR cells were maintained in RPMI-1640 with L-glutamine supplemented with 10% FBS, penicillin/streptomycin, and 1 μM gefitinib according to a previous report.10 All cells were cultured as monolayer at 37°C with 5% CO2 in a humidifier incubator.
Cell proliferation assay
Cell proliferation was determined by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay with CellTiter96 Aqueous One Solution Reagent (Promega, Madison, WI, USA). HCC827 and HCC827GR cells were plated in 96-well flat-bottomed plate at an appropriate cell number according to cell types and allowed to attach for 12 h. The next day, the indicated concentration of compounds or DMSO was added to the wells. Cells were then incubated at 37°C for up to 3 days. After being incubated with compounds, 20 μL of CellTiter96 Aqueous One Solution reagent was added to the wells, and the plate was incubated at 37°C for an additional 1 h. Absorbance at 490 nm was then read on Tecan Infinite F200 Pro plate reader (Promega), and values were expressed as percent of absorbance from cells incubated in DMSO alone.
Western blot analysis
After cells were seeded in a 100-mm culture dishes and allowed to attach, compounds were treated for the time indicated. Cells were harvested in ice-cold lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP40) and 20–30 μg of lysate per lane was separated by SDS-PAGE and followed by transferring to a PVDF membrane (Bio-Rad, Hercules, NJ, USA). The membrane was blocked with 5% skim milk in TBS-T, and then incubated with the corresponding antibodies. After binding of an species-specific secondary antibody coupled to horseradish peroxidase, proteins were visualized by SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Waltham, MA, USA) and developed with LAS-3000 (Fuji, Japan) according to the instructions of the manufacturer.
Migration and invasion assay
Cell migration and invasion capacity in NSCLCs were tested using BD cell-culture inserts and/or BD BioCoat Matrigel Invasion Chamber, respectively. The cells were suspended in culture media (1 × 105 cells/mL) and then added 0.5 mL cell suspension (5 × 104 cells/mL) to the upper chambers. After incubation for 24 h (for migration) or 48 h (for invasion), culture media in inserts were carefully removed, and the membrane containing the cells on the lower surface of inserts was fixed using methanol and stained with H&E. The upper surface of the membrane was gently scrubbed with a cotton swab to remove cells. The stained cells were quantified under light microscope.
Soft agar colony formation assay
Cells (8 × 103 cells per well) were suspended in BME (Basal Medium Eagle) (1 mL with 10% FBS, 0.33% agar) and plated over a layer of solidified bottom agar mixture (BME with 10% FBS, 0.5% agar) with indicated concentrations of drugs. The cultures were maintained at 37°C in a 5% CO2 incubator for 6 to 7 days, and the colonies were counted under light microscope.
Flow cytometry
Cell apoptosis was detected with an FITC Annexin V Apoptosis detection kit I (BD Biosciences Pharmingen) in accordance with the supplied protocols. After exposure to indicated compounds for 24 h, cells were collected. Cells were washed twice with cold PBS and then resuspended in 1× binding buffer at a concentration of 1 × 106 cells/mL. 5 μL of FITC Annexin V and PI were added to 100 μL of suspended cells (1 × 105 cells), and the cells were incubated for 15 min at RT in the dark. The analysis was done with a BD FACSVerse flow cytometer with BD FACSuite Software. The fraction of cell population in different quadrants was analyzed using quadrant statistics.
Animals
For in vivo xenograft assay, male BALB/c-nu athymic nude mice (5 weeks old) were obtained from Orient (Seoul, South Korea) and maintained under specific pathogen-free conditions based on the guidelines established by the Seoul National University. Animals were acclimated for 1 week before the study and housed in climate-controlled quarters with a 12-h light/dark cycle. For in vivo hepatotoxicity assay, male C57BL/6 mice (7 weeks old) were obtained from Koatech (Pyungtaek, Gyeonggi, South Korea), housed under specific pathogen-free conditions and acclimated to the local environment for 1 week before use. The animal experiments were conducted using protocols approved by the Institutional Animal Care and Use Committee at Korea Research Institute of Bioscience and Biotechnology (KRIBB).
Xenograft mouse model
Mice were divided into two groups for each cell line: (i) vehicle group (n = 10); (ii) 1 mg/kg of WK88-1 (n = 10). HCC827GR (1 × 106 cells/100 μL) cells were suspended in RPMI-1640 medium and inoculated with 100 μL matrigel subcutaneously into the right flank of each mouse. Vehicle or WK88-1 was injected intraperitoneally three times per week. Tumor volume was calculated according to a standard formula: tumor volume (mm3) = (length × width × height × 0.52). Tumor volume was measured every 3 or 4 days, and tumor weight was recorded after excision on the day of termination of the experiment. Mice were monitored until tumors reached 1 cm3 total volume, at which time mice were euthanized and tumors extracted.
Hepatotoxicity assay
C57BL/6 mice were treated intravenously with vehicle, GA (10 mg/kg), WK88-1 (10 or 30 mg/kg), WK88-2 (10 or 30 mg/kg) or WK88-3 (10 or 30 mg/kg) twice at 24 h intervals (n = 5). Body weight was recorded daily. At the end of experiment, blood was collected from posterior vena cava and plasma was prepared for biochemical analysis. The level of glutamate-oxaloacetate transaminase (GOT) and glutamate-pyruvate transaminase (GPT) in plasma was measured using biochemical analyzer (AU400; Olympus, Tokyo, Japan). For in vivo experiments, GA, WK88-1, WK88-2 and WK88-3 was firstly dissolved in two volumes of absolute ethanol. After the compounds were completely dissolved, two volumes of Cremophor EL were added and final dosing solution was prepared by adding six volumes of 20% hydroxypropyl β-cyclodextrin.
Statistical analysis
Quantitative data are presented as mean value ± SD unless indicated otherwise. The statistical significant of compared measurements was measured using the Student's t-test and P < 0.05 or less calculated from two-tailed test was considered significant.
Results
WK88-1, a non-benzoquinone GA derivative, suppresses the proliferation of gefitinib-resistant NSCLC cells with Met amplification
We have previously designed and synthesized non-benzoquinone GA derivatives by following mutasynthetic and directed biosynthetic approaches.37,38 As shown in Figure1, DHQ3, a 15-hydroxyl-17-demethoxyreblastin, was prepared from a genetically engineered strain (AC15) of S. hygroscopicus38 and WK88-1, WK88-2, and WK88-3 were purified from a culture of S. hygroscopicus AC2, in which the AHBA synthase gene was disrupted by the kanamycin-resistance gene, supplemented with 3-aminobenzoic acid(Fig.1).37 In this study, we examined the possible effects of DHQ3 and the WK88 series of non-benzoquinone GA derivatives in alleviating gefitinib resistance in NSCLC. For this purpose, we used a gefitinib-sensitive HCC827 cells and gefitinib-resistant HCC827GR cell line harboring Met gene amplification. As reported, our data also showed that HCC827 cells were highly sensitive to exposure to gefitinib, whereas HCC827GR cells were relatively resistant to gefitinib treatment (Fig.2a,b). Given that Hsp90 inhibition seems promising to overcome gefitinib resistance, we first assessed the anti-proliferative effects of these compounds in these NSCLC cell lines. Our data revealed that a potent growth-inhibitory effect was observed in NSCLCs, which was treated with WK88 compounds or GA, whereas DHQ3 didn't show any desirable effects (Fig.2a,b). Notably, this anti-proliferative effect of WK88 compounds was evidently observed in HCC827GR cells as well as HCC827 cells, suggesting that WK88 compounds might be a potential alternative of GA to overcome acquired resistance to gefitinib in NSCLCs.
Figure 2.
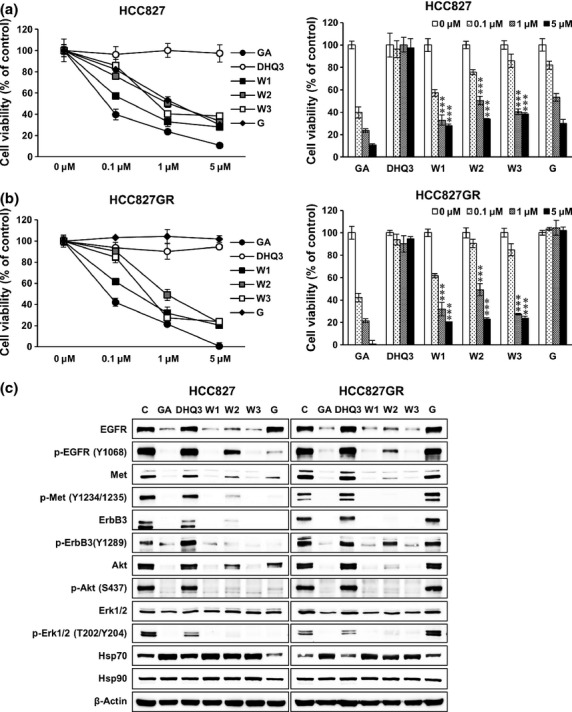
WK88-1 suppresses the proliferation of gefitinib-resistant non-small cell lung cancers (NSCLCs) via the downregulation of oncogenic RTKs. Gefitinib-sensitive HCC827 cells (a) and resistant HCC827GR cells (b) were treated with the indicated concentration of GA derivatives for 3 days, and cell proliferation was estimated using the MTS assay. Cell viability relative to controls was determined after 3 days. Data shown are the representative of 5 independent experiments. Error bars represent the mean ± SD. Statistical significance was determined by the Student's t-test (***P < 0.001). Effect of GA derivatives on the expression or activity of EGFR, Met, ErbB3, and downstream proteins (c). Cells were treated with 1 μM GA derivatives for 24 h and whole cell lysates were assayed by Western blot. β-Actin was used as a loading control. GA, Geldanamycin; G, Gefitinib; W1, WK88-1; W2, WK88-2, W3, WK88-3.
Since hepatotoxicity is a major obstacle in developing GA derivatives as potential chemotherapeutic agents, finding the nontoxic Hsp90 inhibitor might be an important task. Therefore, hematological biochemistry tests were performed to examine hepatotoxicity of WK88 compounds in mice. Our data revealed that intravenous injection with WK88 compounds did not adversely affect body weight in C57BL/6 mice (Table1). Moreover, biochemical evaluation measured by glutamate oxaloacetate transaminase (GOT) and glutamate pyruvate transaminase (GPT) levels clearly showed that WK88-1 and WK88-2 caused no detectable hepatotoxicity, whereas strongly increased GOT/GPT levels were detected in mice treated with WK88-3 (30 mg/kg body weight) (Table1). These increased levels of GOT/GPT in WK88-3-injected mice are comparable to those in GA-injected mice considering lower control levels. Therefore, these results clearly indicate that WK88-1 and 2, but not WK88-3, are relatively safe and nontoxic non-benzoquinone GA derivatives.
Table 1.
Changes in body weight and plasma glutamate-oxaloacetate transaminase (GOT) and glutamate-pyruvate transaminase (GPT) level after treatment with geldanamycin-anasamycin (GA) and non-benzoquinone GA derivatives
| Group (n = 5) | Doses (mg/kg) | Weight change after treatment | GOT (IU/L) | GPT (IU/L) | ||
|---|---|---|---|---|---|---|
| Day 0 | Day 1 | Day 2 | ||||
| Vehicle Control | 0 | 100.0 ± 0.0 | 99.3 ± 2.7 | 99.2 ± 3.3 | 68.0 ± 15.7 | 33.0 ± 5.7 |
| Geldanamycin (GA) | 10 | 100.0 ± 0.0 | 99.8 ± 1.8 | 93.7 ± 6.7 | 23160.0 ± 6523.9 | 18572.0 ± 5735.4 |
| WK88-1 (W1) | 10 | 100.0 ± 0.0 | 99.6 ± 2.4 | 100.6 ± 3.7 | 53.0 ± 12.0 | 31.0 ± 5.5 |
| 30 | 100.0 ± 0.0 | 101.4 ± 1.7 | 98.6 ± 7.9 | 66.0 ± 24.3 | 36.0 ± 6.5 | |
| WK88-2 (W2) | 10 | 100.0 ± 0.0 | 99.8 ± 0.7 | 102.7 ± 4.4 | 56.0 ± 4.2 | 34.0 ± 6.5 |
| 30 | 100.0 ± 0.0 | 101.8 ± 2.9 | 100.6 ± 2.1 | 70.0 ± 18.0 | 68.0 ± 54.4 | |
| WK88-3 (W3) | 10 | 100.0 ± 0.0 | 100.5 ± 2.0 | 99.4 ± 6.6 | 72.5 ± 13.2 | 56.3 ± 18.9 |
| 30 | 100.0 ± 0.0 | 99.5 ± 2.5 | 96.4 ± 3.2 | 2869.0 ± 3521.7 | 3722.0 ± 4419.1 | |
C57BL/6 mice (n = 5) were treated intravenously with vehicle or the indicated concentrations of GA or GA derivatives (WK88-1, WK88-2, and WK88-3). Body weight was recorded daily and plasma level of GOT and GPT was measured by biochemical analysis. Values were expressed as mean ± SD of five determinations.
WK88-1 suppresses the expression of RTKs and downstream signaling in NSCLCs
To explore the underlying mechanism by which WK88 compounds inhibit cell growth in NSCLCs, we examined their effects by analyzing the expression of Hsp90's client proteins in HCC827 and HCC827GR cell lines. As expected, treatment with 1 μM gefitinib inhibited phosphorylation of EGFR and subsequent activation of Akt and Erk1/2 in gefitinib-sensitive HCC827 cells, but not in gefitinib-resistant HCC827GR cells (Fig.2c). On the other hand, WK88 compounds showed a robust downregulation of total levels of EGFR, Met, ErbB3, and Akt proteins in all tested cells together with upregulation of co-chaperone HSP70, suggesting that WK88 compounds effectively target the Hsp90. Also, downregulation of client proteins was paralleled by dephosphorylation events, and subsequent inhibition of downstream Akt and Erk1/2 (Fig.2c). Among tested compounds, WK88-1 and WK88-3 was shown to have prominent Hsp90 inhibitory effect similar to GA in modulating the expression of Hsp90's client proteins, whereas WK88-2 was shown to have modest effects compared with WK88-1 or WK88-3. Considering the potential hepatotoxicity of WK88-3 in tested animals, WK88-1 might be a desirable Hsp90 inhibitor that could circumvent gefitinib-resistance in NSCLCs without causing toxicity.
WK88-1 promotes the degradation of oncogenic RTKs including ErbB3 in NSCLCs
Engelman et al.10 have revealed that Met amplification leads to persistent activation of PI3K/Akt by maintaining ErbB3 phosphorylation in an EGFR- and ErbB2-independent manner. This supports a crucial role for ErbB3 in the survival of drug-resistant NSCLCs with Met amplification. In this context, we reasoned that ErbB3 might be a crucial target in Met-amplified HCC827GR cells. Based on the notion that both EGFR and ErbB2 are well-known targets for Hsp90, we investigated whether ErbB3 also could be regulated by proteasomal degradation. To investigate whether WK88-1 downregulates ErbB3 through this mechanism, protein stability of ErbB3 was examined by protein turnover assay using cycloheximide in HCC827GR cells. Our data revealed that a half-life of ErbB3 was markedly decreased by co-treatment with cyclohexidmide and WK88-1, which is similar to the case of other Hsp90 clients, EGFR, ErbB2, and Met (Fig.3a). We also performed a recovery experiment using proteasomal inhibitor, MG132. WK88-1 markedly decreased the protein levels of ErbB3 together with other Hsp90 clients, whereas pretreatment with MG132 markedly recovered protein abundance of these receptors (Fig.3b). This result suggests that ErbB3 is regulated by Hsp90-mediated proteasomal degradation pathway similar with EGFR and other client proteins. Therefore, we conclude that the overall degradation of ErbB3 as well as EGFR, ErbB2, and Met by a novel Hsp90 inhibitor, WK88-1, might provide a clue for overcoming gefitinib resistance in Met-amplified NSCLCs.
Figure 3.

WK88-1 augments the instability of oncogenic RTKs in non-small cell lung cancers (NSCLCs). Cells were treated with cycloheximide (CHX) in combination with WK88-1 for the indicated time and then subjected to Western blot (a). Proteasome inhibitor MG132 restored oncogenic RTKs after WK88-1 treatment. Cells were treated with MG132 for 2 h prior to WK88-1 treatment and then subjected to Western blot (b).
Effects of WK88-1 on the migration, invasion, and colony formation in NSCLCs
To check whether the WK88-1 has anti-metastatic activity, we next performed migration and invasion assay in HCC827 and HCC827GR cells. As shown in Figure4(a), treatment with gefitinib markedly reduced the migration and invasion in HCC827 cells, but not in gefitinib-resistant HCC827GR cells (Fig.4a,b). However, treatment with WK88-1 strongly abrogated the migratory and invasive capacities of both gefitinib-sensitive and resistant cell lines (Fig.4a,b). Notably, the representative microphotograph clearly shows that a significant inhibition of migration and invasion was observed in Met-amplified HCC827GR cells following treatment with WK88-1. The soft-agar colony formation assay also revealed that treatment with WK88-1 inhibited the anchorage-independent growth of HCC827GR cells, indicating that the malignant potential of HCC827GR cells might be blunted by WK88-1 (Fig.4c).
Figure 4.
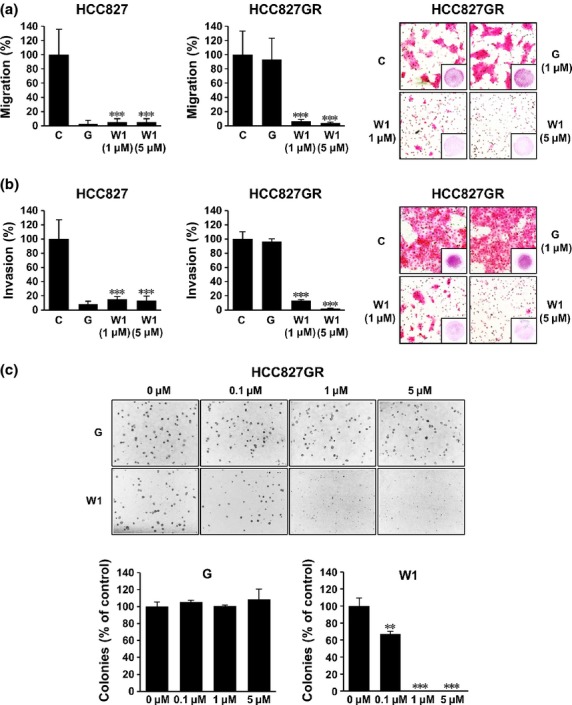
WK88-1 suppresses migration, invasion and anchorage-independent growth of non-small cell lung cancers (NSCLCs). WK88-1 suppresses migration and invasion of NSCLCs in the Boyden chamber migration (a) or Matrigel invasion assay (b). After incubation for 24 h (migration) or 48 h (invasion) with the indicated concentration of drugs, live cells that migrate or invade to the lower surface were fixed, stained, and counted using light microscopy. Random areas were scanned (five areas per membrane of the well) in cells on the lower surface of membrane in both the Boyden and Matrigel chamber. Error bars represent the mean ± SD of five areas. The representative photographs for migration (upper right) and invasion (bottom right) of HCC827GR cells were shown. WK88-1 suppresses anchorage-independent growth of HCC827GR cells (c). Colony formation of HCC827GR cells after exposure to the increasing concentration of WK88-1 for 7 days. Random areas were scanned (five areas per well, three wells per set) in colonies grown in soft agar. Error bars represent the mean ± SD of 15 areas. Statistical significance was determined by the Student's t-test (**P < 0.01, ***P < 0.001).
WK88-1 induces apoptosis and inhibits xenograft tumor growth of gefitinib-resistant NSCLC cells in nude mice
Incubation with WK88-1, but not gefitinib, induced apoptosis in gefitinib-resistant HCC827GR cells in a concentration-dependent manner as revealed by Annexin-V staining and flow-cytometry analyses (Fig.5a). Treatment with WK88-1 also increased the expression of cleaved caspase-3 and cleaved PARP (Fig.5b), indicating the potential of WK88-1 in inducing death signaling in gefitinib-resistant NSCLCs with Met amplification. We next examined whether WK88-1 overcomes resistance to gefitinib in vivo. To investigate the in vivo antitumor activity of WK88-1, gefitinib-resistant HCC827GR cells were subcutaneously transplanted into nude mice. Treatment with WK88-1 (1 mg/kg body weight) caused a significant decrease in tumor volume (Fig.5c) and weight (Fig.5d), suggesting that treatment with WK88-1 could circumvent gefitinib resistance in NSCLCs.
Figure 5.
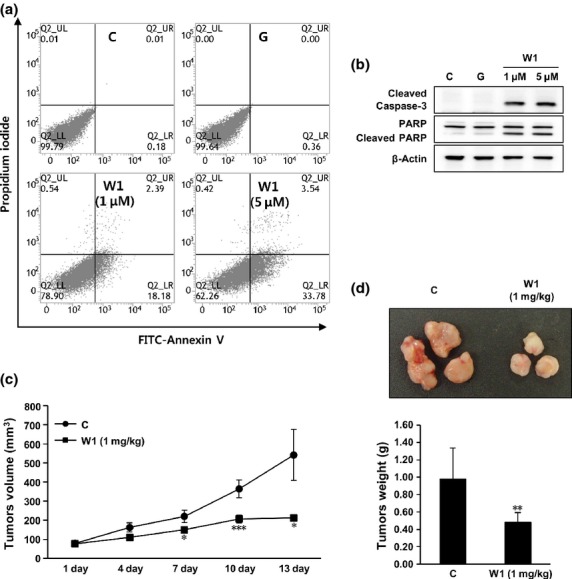
Anti-tumor activity of WK88-1 in a mouse xenograft model. Flow-cytometry analysis with annexin V staining was performed in WK88-1-treated HCC827GR cells (a). Cleaved caspase-3 and PARP was examined using Western blots after exposure to the indicated concentration of WK88-1 in HCC827GR cell lines (b). The average volume of HCC827GR-derived tumors from control and WK88-1-treated mice was plotted 15 days after inoculation (c). Statistical significance was determined by the Student's t-test (*P < 0.05; **P < 0.01, ***P < 0.001). Representative photographs and weights of HCC827GR -derived tumors formed in nude mice (d). Ten mice were used for each group and mice were sacrificed 4 weeks after cell injection. Tumors were weighted after sacrificing mice on the final day of the study. The asterisk indicate a significant decrease in tumor weight between the control group and the group treated with WK88-1 (**P < 0.01).
Discussion
Intracellular signaling networks comprising multiple RTKs and their downstream molecules may confer the molecular complexity and compensatory pathway against TKIs-mediated inhibition of tumor cell growth.40 In particular, members of the EGFR family, including EGFR, ErbB2, ErbB3, ErbB4 and Met, have been reported to crosstalk with each other, thereby developing resistance to EGFR-TKIs.41 For example, NSCLCs with activating mutations in EGFR develop acquired resistance to EGFR-TKIs by recruiting Met and subsequent activation of ErbB3-PI3K-Akt axis,10 indicating a central role for ErbB3 in acquiring chemoresistance by tumor cells. Also, a recent report showed that inhibiting EGFR with gefitinib also inhibits phosphorylation of ErbB2, ErbB3, and Met in gefitinib-sensitive cells.40 Moreover, EGFR has been shown to regulate Met expression at multiple levels.42,43 Consequently, it is crucial to define the exact mechanism by which drug resistance develops and find an integrative strategy for simultaneous blocking of both oncogene activation and potential redundant cell signaling pathways. Because of these signaling complexities and an increasing trend of drug-resistance, a molecular target that plays a central role in diverse oncogenic signaling pathways have long been sought. One of the promising anticancer drug targets is Hsp90, a molecular chaperone that promotes the proliferation and growth of cancer cells, at least in part, through the stabilization and maturation of various oncoproteins. In NSCLCs, Hsp90 has been shown to stabilize key oncogenic proteins including EGFR, ErbB2, Raf-1, Met, and Akt.19–22 Thus, attempts have been made to develop anticancer therapies by targeting Hsp90 because blockade of the Hsp90 activity would cause simultaneous inhibition of cell survival pathways mediated by aforementioned oncoproteins.
Although a number of GA-derivatives, such as 17-AAG and 17-DMAG have been developed as Hsp90 inhibitors with remarkable preclinical success, the major limitations of these molecules are their hepatotoxic activity, which is likely to be caused by the benzoquinone moiety present in their structure. We, therefore, have prepared several GA-derivatives lacking the benzoquinone group in their structure and evaluated their anticancer activities in both gefitinib-sensititive and-resistant NSCLCs. Among the non-benzoquinone GA-derivatives, WK88-1, was found to have no hepatotoxic effects, while the WK88-3 appears to show hepatotoxicity. Though WK88-2 did not exhibit hepatotoxicity but showed only a marginal effect in suppressing tumor NSCLC cells growth. We, therefore, further examined the molecular details underlying the antitumor effects of non-benzoquinone GA-derivative, WK88-1.
Our data clearly shows that WK88-1 induces simultaneous degradation of RTKs and inhibition of downstream signaling pathways, thereby suppressing the proliferation of HCC827GR cells. This is further supported by the inhibitory effect of the compound on the anchorage-independent growth of the gefitinib-resistant NSCLC cells, and the reduced growth of HCC827GR cells xenograft tumors in nude mice. Furthermore, the inhibition of the migration and invasion of HCC827GR cells by treatment with WK88-1 suggests that this novel Hsp90 inhibitor is effective in alleviating the EGFR-TKI resistance. Whereas WK88-1 showed antiproliferative effects in HCC827GR cells, its structural analog DHQ3 failed to affect cell viability. Considering the prominent inhibitory effect of DHQ3 against Hsp90 in vitro,37 it would be worthwhile to examine whether the membrane permeability of WK88-1 and DHQ3 differs in relation to their chemical properties.
One of the molecular targets for overcoming EGFR-TKI resistance is to inactivate signaling mediated via Met, which has been implicated in a wide range of human malignancies inducing NSCLC.44 In addition, acquired resistance to EGFR-TKIs in NSCLC has been reported to emerge by recruiting Met and subsequent activation of ErbB3.10 Based on the crucial roles of ErbB3-PI3K-Akt axis in Met-amplified HCC827GR cells, we sought to determine whether treatment with WK88-1 could diminish the expression levels of these oncoproteins. Our findings show that treatment with WK88-1 downregulates the protein levels of ErbB3 together with other well-known Hsp90 client proteins including EGFR, ErbB2, Met, and Akt, suggesting that ErbB3 is a potential substrate for Hsp90 as reported.45,46 Therefore, these findings provide insight as to how the treatment with WK88-1 may block alternative cell survival pathways in Met-amplified HCC827GR cell lines (Fig.6).
Figure 6.
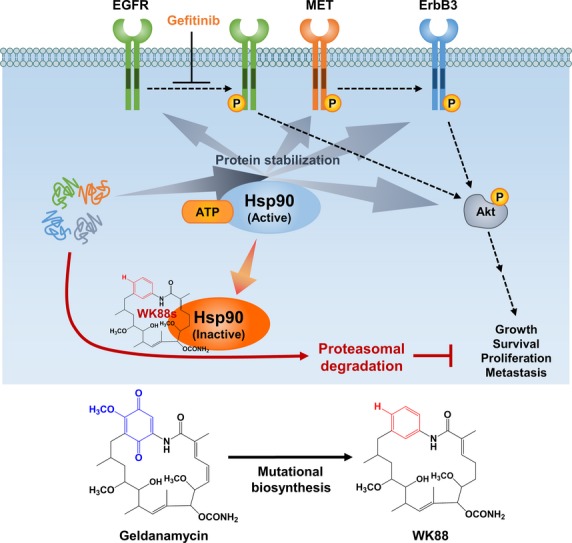
Simplified diagram of mechanism for anti-tumor activity of WK88-1 in non-small cell lung cancers (NSCLCs). WK88-1 as a potential alternative of GA for overcoming acquired resistance to gefitinib in NSCLCs.
In conclusion, our findings clearly demonstrate that treatment with WK88-1 lead to a significant reduction in vivo tumor growth in a mouse xenograft model as well as migration, invasion, and colony formation in gefitinib-resistant NSCLCs. Our data also reveal that intraperitoneal administration of WK88-1 in mice does not elicit any undesirable toxicity, suggesting that WK88-1 might be a less toxic inhibitor for Hsp90. Therefore, our data suggest that WK88-1 is a potential alternative of GA for overcoming acquired resistance to gefitinib in NSCLCs.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A1A1010735) and by College of Pharmacy-specialized Research Fund (from institute for new drug development) of Keimyung University in 2012.
Disclosure Statement
The authors have no conflict of interest.
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] J Clin Oncol. 2003;21:2237–46. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Lynch TJ, Adjei AA, Bunn PA, Jr, et al. Summary statement: novel agents in the treatment of lung cancer: advances in epidermal growth factor receptor-targeted agents. Clin Cancer Res. 2006;12:4365s–71s. doi: 10.1158/1078-0432.CCR-06-1005. [DOI] [PubMed] [Google Scholar]
- Perez-Soler R, Chachoua A, Hammond LA, et al. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J Clin Oncol. 2004;22:3238–47. doi: 10.1200/JCO.2004.11.057. [DOI] [PubMed] [Google Scholar]
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2011;28:357–60. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–32. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–4. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Du R, Jiang S, et al. Dual MET-EGFR combinatorial inhibition against T790M-EGFR-mediated erlotinib-resistant lung cancer. Br J Cancer. 2008;99:911–22. doi: 10.1038/sj.bjc.6604559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Shimamura T, Ji H, et al. Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell. 2007;12:81–93. doi: 10.1016/j.ccr.2007.06.005. [DOI] [PubMed] [Google Scholar]
- McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70:1625–34. doi: 10.1158/0008-5472.CAN-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha Dias S, Friedlos F, Light Y, Springer C, Workman P, Marais R. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 2005;65:10686–91. doi: 10.1158/0008-5472.CAN-05-2632. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65:6401–8. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- Webb CP, Hose CD, Koochekpour S, et al. The geldanamycins are potent inhibitors of the hepatocyte growth factor/scatter factor-met-urokinase plasminogen activator-plasmin proteolytic network. Cancer Res. 2000;60:342–9. [PubMed] [Google Scholar]
- Yang S, Qu S, Perez-Tores M, et al. Association with HSP90 inhibits Cbl-mediated down-regulation of mutant epidermal growth factor receptors. Cancer Res. 2006;66:6990–7. doi: 10.1158/0008-5472.CAN-06-1042. [DOI] [PubMed] [Google Scholar]
- Chiosis G. Targeting chaperones in transformed systems–a focus on Hsp90 and cancer. Expert Opin Ther Targets. 2006;10:37–50. doi: 10.1517/14728222.10.1.37. [DOI] [PubMed] [Google Scholar]
- Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10:283–90. doi: 10.1016/j.molmed.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, Neckers K. Heat-shock protein 90 inhibitors as novel cancer chemotherapeutics – an update. Expert Opin Emerg Drugs. 2005;10:137–49. doi: 10.1517/14728214.10.1.137. [DOI] [PubMed] [Google Scholar]
- Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–7. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- Gooljarsingh LT, Fernandes C, Yan K, et al. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc Natl Acad Sci U S A. 2006;103:7625–30. doi: 10.1073/pnas.0602650103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Pratilas CA, Hanrahan AJ, Halilovic E, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68:9375–83. doi: 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Li D, Ji H, et al. Hsp90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008;68:5827–38. doi: 10.1158/0008-5472.CAN-07-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausville EA, Tomaszewski JE, Ivy P. Clinical development of 17-allylamino, 17-demethoxygeldanamycin. Curr Cancer Drug Targets. 2003;3:377–83. doi: 10.2174/1568009033481831. [DOI] [PubMed] [Google Scholar]
- Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–15. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- Brough PA, Aherne W, Barril X, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem. 2008;51:196–218. doi: 10.1021/jm701018h. [DOI] [PubMed] [Google Scholar]
- Chiosis G, Lucas B, Shtil A, Huezo H, Rosen N. Development of a purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and induce the degradation of Her2 tyrosine kinase. Bioorg Med Chem. 2002;10:3555–64. doi: 10.1016/s0968-0896(02)00253-5. [DOI] [PubMed] [Google Scholar]
- Banerji U, Sain N, Sharp SY, et al. An in vitro and in vivo study of the combination of the heat shock protein inhibitor 17-allylamino-17-demethoxygeldanamycin and carboplatin in human ovarian cancer models. Cancer Chemother Pharmacol. 2008;62:769–78. doi: 10.1007/s00280-007-0662-x. [DOI] [PubMed] [Google Scholar]
- Cysyk RL, Parker RJ, Barchi JJ, Jr, Steeg PS, Hartman NR, Strong JM. Reaction of geldanamycin and C17-substituted analogues with glutathione: product identifications and pharmacological implications. Chem Res Toxicol. 2006;19:376–81. doi: 10.1021/tx050237e. [DOI] [PubMed] [Google Scholar]
- Kim W, Lee D, Hong SS, et al. Rational biosynthetic engineering for optimization of geldanamycin analogues. ChemBioChem. 2009;10:1243–51. doi: 10.1002/cbic.200800763. [DOI] [PubMed] [Google Scholar]
- Kim W, Lee JS, Lee D, et al. Mutasynthesis of geldanamycin by the disruption of a gene producing starter unit: generation of structural diversity at the benzoquinone ring. ChemBioChem. 2007;8:1491–4. doi: 10.1002/cbic.200700196. [DOI] [PubMed] [Google Scholar]
- Amann J, Kalyankrishna S, Massion PP, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005;65:226–35. [PubMed] [Google Scholar]
- Guo A, Villen J, Kornhauser J, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–7. doi: 10.1073/pnas.0707270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizaki J, Okamoto I, Sakai K, Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer. 2011;105:807–13. doi: 10.1038/bjc.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breindel JL, Haskins JW, Cowell EP, Zhao M, Nguyen DX, Stern DF. EGF Receptor Activates MET through MAPK to Enhance Non-Small Cell Lung Carcinoma Invasion and Brain Metastasis. Cancer Res. 2013;73:5053–65. doi: 10.1158/0008-5472.CAN-12-3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Nilsson MB, Saintigny P, et al. Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene. 2010;29:2616–27. doi: 10.1038/onc.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Camphausen K, Tofilon PJ. Inhibition of Hsp90: a multitarget approach to radiosensitization. Clin Cancer Res. 2007;13:4326–30. doi: 10.1158/1078-0432.CCR-07-0632. [DOI] [PubMed] [Google Scholar]
- Gerbin CS, Landgraf R. Geldanamycin selectively targets the nascent form of ERBB3 for degradation. Cell Stress Chaperones. 2010;15:529–44. doi: 10.1007/s12192-009-0166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]