

Abstract
Eribulin mesylate is a synthetic macrocyclic ketone analog of the marine sponge natural product halichondrin B and an inhibitor of microtubule dynamics. Some tubulin-binding drugs are known to have antivascular (antiangiogenesis or vascular-disrupting) activities that can target abnormal tumor vessels. Using dynamic contrast-enhanced MRI analyses, here we show that eribulin induces remodeling of tumor vasculature through a novel antivascular activity in MX-1 and MDA-MB-231 human breast cancer xenograft models. Vascular remodeling associated with improved perfusion was shown by Hoechst 33342 staining and by increased microvessel density together with decreased mean vascular areas and fewer branched vessels in tumor tissues, as determined by immunohistochemical staining for endothelial marker CD31. Quantitative RT-PCR analysis of normal host cells in the stroma of xenograft tumors showed that eribulin altered the expression of mouse (host) genes in angiogenesis signaling pathways controlling endothelial cell–pericyte interactions, and in the epithelial–mesenchymal transition pathway in the context of the tumor microenvironment. Eribulin also decreased hypoxia-associated protein expression of mouse (host) vascular endothelial growth factor by ELISA and human CA9 by immunohistochemical analysis. Prior treatment with eribulin enhanced the anti-tumor activity of capecitabine in the MDA-MB-231 xenograft model. These findings suggest that eribulin-induced remodeling of abnormal tumor vasculature leads to a more functional microenvironment that may reduce the aggressiveness of tumors due to elimination of inner tumor hypoxia. Because abnormal tumor microenvironments enhance both drug resistance and metastasis, the apparent ability of eribulin to reverse these aggressive characteristics may contribute to its clinical benefits.
Keywords: Breast cancer, eribulin mesylate, tumor microenvironment, tumor vasculature, vascular remodeling
Tubulin-binding drugs (TBD) are widely used for cancer chemotherapy. Clinical antitumor activities of many TBD, such as taxanes, vinca alkaloids and epothilones, are well established against many types of solid tumors and leukemias.1,2 Targeting tumor vasculature as a therapeutic approach has been strongly supported by preclinical studies and validated by clinical trials.3 Therapeutic vascular targeting encompasses two main strategies: antiangiogenic approaches, which prevent neovascularization thereby inhibiting future tumor growth,4–6 and vascular-disrupting approaches, which cause rapid and selective destruction of established tumor vasculature, leading to secondary tumor cell death.7,8 TBD are generally categorized into microtubule stabilizers (e.g. taxane and epothilones) or destabilizers (e.g. vinca alkaloids and combretastatins),9 and both can interfere with normal mitotic spindle formation in tumor cells, leading to cell cycle arrest and cell death via apoptosis.1 In general, microtubule-stabilizing agents have antiangiogenic actions,5 while microtubule-destabilizing drugs often having vascular disrupting activities associated with collapse of existing vasculature.10,11
Tumor angiogenesis occurs primarily via sprouting of existing capillary vessels. This process involves several steps, beginning with endothelial cell-mediated proteolytic degradation of the basement membrane, migration of endothelial cells from an existing vessel into the surrounding ECM, proliferation of endothelial cells and morphogenesis of cells into tube-like structures.12 TBD show inhibitory activities against many of these steps, including inhibition of proliferation and induction of apoptosis of endothelial cells, both of which contribute to antiangiogenesis effects of TBD.5,6 TBD also inhibit neovessel formation in vitro, probably due to their ability to disrupt one or more of the steps of angiogenesis. TBD can also inhibit the migration of fibroblasts and monocytes.13,14 Identification of the cellular and molecular actions that are responsible for antivascular activities of TBD will be an important objective of future studies.
Eribulin mesylate (eribulin), a nontaxane, synthetic microtubule dynamics inhibitor, is currently in clinical use in many countries for late stage, advanced or metastatic breast cancer.15–17 Eribulin shows preclinical antitumor activity against a wide variety of human cancer types and has a unique mode of action on microtubule dynamics, causing antimitotic, apoptosis-inducing activities.18–22 Known antivascular activities of other TBD prompted us to ask if eribulin might have antivascular (antiangiogenesis or vascular disrupting) activities. In the present study, we used dynamic contrast-enhanced (DCE)-MRI, a technique commonly used to assess antivascular activity in both the preclinical11,23,24 and clinical settings,25–27 and immunohistochemical approaches, to assess morphological changes in tumor vasculature of human breast cancer MX-1 and MDA-MB-231 xenografts. We also performed quantitative RT-PCR (qRT-PCR)-based gene expression profiling, focusing on angiogenesis-related and epithelial–mesenchymal transition (EMT)-related pathways to assess effects of eribulin on normal cells in the tumor microenvironment. Our results show that eribulin has a novel antivascular activity and induces tumor vascular remodeling and provokes phenotypic changes that reduce the abnormality of the tumor microenvironment, thereby reducing drug resistance and metastasis-promoting hypoxia and EMT processes.
Materials and Methods
Compounds, cell lines and animals
Eribulin mesylate (E7389, Halaven; Eisai, Tokyo, Japan) and capecitabine were manufactured at Eisai. The human breast cancer cell line MX-1 was obtained from NCI (Bethesda, MD, USA). The human breast cancer cell line MDA-MB-231 was obtained from ATCC (Rockville, MD, USA). Female nude rats, nude mice and Balb/c mice were obtained from Harlan (Gannat, France) and Charles River Laboratories (Kanagawa, Japan). All studies with laboratory animals were performed according to protocols approved by the presiding Institutional Animal Care and Use Committees, and met all local, governmental and corporate guidelines for the humane care and use of laboratory animals.
Human breast cancer s.c. xenograft models
MX-1 and MDA-MB-231 cells were cultured with RPMI 1640 medium containing 10% (v/v) FBS. Cultured cells were suspended with 50% (v/v) BD Matrigel (BD Bioscience, San Jose, CA, USA) for s.c. inoculation into nude mice. MDA-MB-231 cells were inoculated into nude rats or mice (1–2 × 107 cells/animal). MX-1 cells were inoculated into nude mice (4 × 106 cells/animal). MX-1 tumors grown in nude mice were surgically excised and smaller tumor fragments were implanted s.c. into nude rats (one 20–30 mg fragment/animal). Inoculations of tumor cells or tumor fragments in nude rats, were performed 24–72 h after whole body irradiation (2 Gy for mice, 5 Gy for rats; γ-source, 60Co; BioMEP SARL, Bretenière, France). Nude mice bearing human breast cancer xenografts were randomized into each treatment group and treatments started on day 1, when mean tumor weights reached approximately 200 mg (9–11 days after inoculation). Tumor volumes were calculated according to the following formula: (width [mm]² × length [mm])/2. Tumor weights were calculated according to the following formula: tumor volume (mm3) × 1 (mg/mm3). Eribulin effects on measured parameters in nude mice were evaluated at the following times: tumor growth, day 8; tumor perfusion, day 6; tumor vascular morphology, day 8; and gene expression levels, day 4, with numbers representing the number of days after the start of treatments on day 1.
Dynamic contrast-enhanced-MRI analysis
A Bruker 4.7T Pharmascan was used for DCE-MRI analysis of nude rats. Anatomic images were acquired under the following conditions: RARE sequence, TE/TR; 38/2500 ms, FOV; 70 × 51 mm, 1.5-mm slices covering the whole tumor. DCE-MRI measurements were conducted under the following conditions: GE FLASH, TE/TR; 3/50 ms, FA 60; 60 × 45 mm, FOV; 108 × 80 matrix; 2-mm slices with a temporal resolution of 4 s/image. Voxel-wise baseline T1 values were estimated according to the method of Fram et al. from measured signal intensities acquired using a FLASH sequence at five different flip angles.28 Along with the Gd relaxivity constant, these T1 estimates were used to estimate the Gd concentration in each voxel at each time point. Tracer uptake curves derived from signal enhancement in selected regions of interest were fitted using a two-compartment kinetic model for determination of volume transfer constants (Ktrans).29 Each rat bearing a xenografted tumor was anesthetized with isofluorane while its respiratory rate was monitored. Image acquisition was started, and then 30 s later, a 0.1 mmol/kg bolus of Magnevist (Bayer Healthcare Pharmaceuticals, Leverkusen, Germany) was injected via the tail vein. We used nude rats for DCE-MRI analysis to evaluate anti-tumor vascular activity against large-sized tumors. When mean tumor volumes on nude rats reached approximately 400 mm3 (16–20 days after inoculation), DCE-MRI analysis was performed 1 day before the start of treatment (day −1) to determine baseline Ktrans values within tumor xenografts. Rats were randomized into vehicle and eribulin groups the next day (day 0) based on baseline Ktrans values and the tumor volumes determined by DCE-MRI on the previous day. Drugs were administered every fourth day starting on day 0, with four repetitions (Q4D×4).
Hoechst 33342 perfusion assay
Tumor vascular perfusion was evaluated in human breast cancer xenograft models by i.v. injection with Hoechst 33342 (5 mg/mL, 0.1 mL/mouse; Life Technologies, Grand Island, NY, USA), a fluorescent stain used to label nucleic acids as a marker for tumor perfusion.11 Tumors were resected 5 min after Hoechst 33342 injection. Cryosections with 10-μm thickness were prepared, and images of Hoechst 33342 staining were captured using a BIOREVO fluorescence microscope (BZ-9000; KEYENCE, Osaka, Japan). Averages of Hoechst-positive areas from each tumor cryosection were calculated using Lumina Vision (version 2.2.2; Mitani Corporation, Tokyo, Japan).
Immunohistochemical analysis
Fragments of resected tumor xenografts from nude mice were fixed in Zinc Formalin Fixative (BD Bioscience) for 1 day and then paraffin-embedded. Cross-sections were prepared and stained with rat anti-mouse CD31 monoclonal antibody (BD Bioscience) using Histofine Simple Stain Mouse MAX PO (Rat) and DAB substrate kits (Nichirei Bioscience, Tokyo, Japan) at Histo Science Laboratory (Tokyo, Japan). Tissue morphology was visualized by H&E staining. Quantification of vascular morphology, microvessel density (MVD) and mean vascular area (MVA) were conducted using the Aperio Microvessel Analysis algorithm (Leica Biosystems, Nussloch, Germany). Vascular morphological profiles in selected regions were measured using Microvessel Analysis v1 software within an Aperio ImageScope (v10.0.36.1805; Leica Biosystems). To calculate the proportion (%) of small and large vessels, a piecewise threshold of vascular area (200 μm2) was selected. For CA9/CD31 double staining, biotinylated anti-mouse CD31 monoclonal antibody (BD Bioscience) with streptavidin-Alexa660 (Life Technologies) and rabbit anti-CA9 antibody (Novus Biologicals, Littleton, CO, USA) with AlexaFluor488 donkey anti-rabbit antibody (Life Technologies) were used. Fluorescent images were captured using a BIOREVO fluorescence microscope.
Quantitative RT-PCR
Total RNA was isolated from resected tumor xenografts in nude mice using an RNeasy Mini Kit (Qiagen, Hilden, Germany) and cDNA was synthesized using a High Capacity cDNA Reverse Transcription Kit (Life Technologies). Then, PCR of the synthesized cDNA was performed with Taqman probe sets for mouse genes related to angiogenesis or EMT (Table S1), a TaqMan Low Density Array card, and a ViiA 7 Real-time PCR System (Life Technologies). Quantitative qRT-PCR data were normalized with ExpressionSuite software v1.0 (Life Technologies). The resultant Ct values were subtracted from the mean Ct values for the normalized genes to provide delta Ct values (ΔCt) for each treatment group, which were then converted to 2−ΔCt values for use in representing and comparing gene expression levels.
ELISA
Resected tumor xenografts from nude mice were immediately frozen in liquid nitrogen, and then crushed, suspended and lysed with sonication in Pierce RIPA Buffer (Thermo Scientific, Yokohama, Japan). Each supernatant was diluted with lysis buffer to adjust the protein concentration to 3 mg/mL, and mouse vascular endothelial growth factor (VEGF) levels were measured using a mouse VEGF Assay Kit (Immuno-Biological Laboratories, Fujioka, Japan).
Statistical analyses
Data were converted to data matrices using Ruby software and, unless otherwise noted, analyzed by the Dunnett multiple comparison test to compare eribulin versus control groups. Ruby and SAS software programs were used for statistical analyses. P-values <0.05 were considered statistically significant.
Results
Dynamic contrast-enhanced-MRI analysis of eribulin effects on tumor vascular function in human breast cancer xenograft models in nude rats
When mean tumor volumes in nude rats bearing human breast cancer MX-1 or MDA-MB-231 xenografts reached approximately 400 mm3 (16–20 days after inoculation), baseline Ktrans values and tumor volumes were measured by DCE-MRI 1 day before randomization (day −1). Rats were randomized into vehicle and eribulin groups the next day (day 0) based on baseline Ktrans values and the tumor volumes. Eribulin (0.1 or 0.3 mg/kg) was administered i.v. to nude rats bearing human breast cancer xenografts at every fourth day from day 0 with four repetitions (Q4D×4). First, antitumor effects of eribulin were confirmed. As shown in Figure1a and b, significant antitumor activity was observed 5–6 days after eribulin treatment and does of 0.3 mg/kg in the MX-1 model and both 0.1 and 0.3 mg/kg in the MDA-MB-231 model compared with the vehicle. In both models, median tumor volumes of the 0.3 mg/kg eribulin groups on day 5 or day 6 were virtually identical to those that had been measured on day −2 or day −1 at prior treatment (MX-1 model, 488 mm3 versus 461 mm3; MDA-MB-231 model, 487 mm3 vs 479 mm3) and then decreased on later days.
Figure 1.
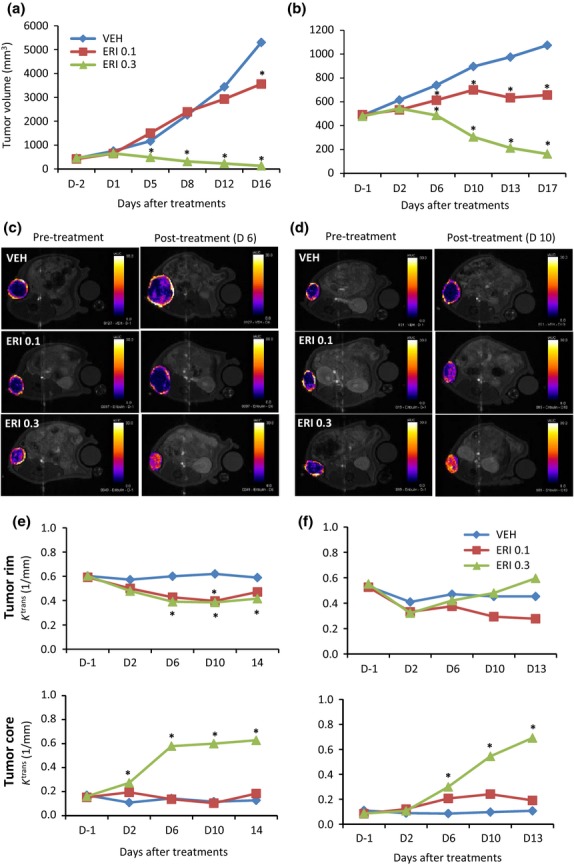
Dynamic contrast-enhanced (DCE)-MRI analysis of eribulin effects on tumor vascular function in human breast cancer xenografts in nude rats. Vehicle (VEH) or eribulin (ERI) at 0.1 or 0.3 mg/kg was administered to nude rats bearing MX-1 or MDA-MB-231 tumor xenografts. Each group consisted of 5 or 6 rats. (a,c,e) MX-1 model; (b,d,f) MDA-MB-231 model. (a,b) Tumor growth curves in nude rats. (c,d) Representative images of initial area under the curve (iAUC) maps (iAUC value in each voxel). (e,f) Average Ktrans values in tumor rim or core regions. DCE-MRI scanning was performed on the indicated days. *P < 0.05 versus VEH.
Analysis of baseline tumor vascular function in both models by DCE-MRI revealed obvious differences in the initial area under the curve (iAUC) map (on day −1) between tumor rim (yellow/red) and core (purple/blue/black) regions (Fig.1c,d), with tumor rims (Ktrans, 0.5–0.6) being substantially better perfused than tumor cores (Ktrans, 0.1–0.2) (Fig.1e,f). Subsequent analyses of eribulin effects using derived parameters (Ktrans and iAUC at 60 s; iAUC60) thus focused on these two regions. In vehicle-treated animals, Ktrans values from both tumor rim and core regions remained stable over time (Fig.1e,f; upper panels). In contrast, treatment with 0.3 mg/kg eribulin led to a dramatic increase of Ktrans in tumor cores observable by day 6 in both models (Fig.1e,f; lower panels). In addition, in the MX-1 model treated with 0.3 mg/kg eribulin, iAUC histogram analysis showed a large rightward shift of the curve representing the tumor core between days 2 and 10 (Fig. S1a), and the curves for tumor rim and core converged to become almost identical between days 6 and 10. In the MDA-MB-231 model, a rightward shift of the tumor core curve occurred between days 6 and 10 at both eribulin doses, with the shift for the 0.3 mg/kg eribulin curve being larger (Fig. S1b). These results confirm that although tumor cores had been poorly vascularized at the time of baseline scanning (day −1), after eribulin treatment, perfusion of tumor cores increased to match those of tumor rims, thus causing a major increase in vascular function throughout the tumor (Fig.1c,f and Fig. S1). These results suggest that eribulin treatment induces tumor vascular remodeling.
Effects of eribulin on tumor perfusion in human breast cancer xenograft models in nude mice
Nude mice bearing MX-1 and MDA-MB-231 xenografts were randomized into vehicle or eribulin groups and treatments started on day 1, when the mean tumor weight reached approximately 200 mg. A single i.v. administration of eribulin on day 1 at doses of 0.3, 1 or 3 mg/kg led to significant antitumor activity observed 7 days after eribulin treatments (on day 8) compared with the vehicle in both xenograft models (Fig.2a,b), confirming the expected antitumor activity of eribulin. To examine the effects of eribulin on tumor perfusion, eribulin was i.v. administered on day 1 and perfusion of Hoechst 33342 was analyzed on day 6 (Fig.2c,d). Fluorescence microscopy revealed greater amounts of Hoechst 33342 staining and increased numbers of tumor vessels in both models after eribulin treatment (Fig.2c) compared with the vehicle. Semi-quantitative analysis of Hoechst 33342 staining indicated that perfused fractions of vascularized areas (Hoechst 33342-positive areas) were significantly higher compared to vehicle at doses of 1.0 or 3.0 mg/kg eribulin in the MX-1 model and at 0.1, 0.3, 1.0 or 3.0 mg/kg in the MDA-MB-231 model (Fig.2d). Because capecitabine is another anticancer agent for metastatic breast cancer,30 this agent was also included as a comparator in this study. Like eribulin, daily capecitabine treatment at 540 mg/kg (maximum tolerated dose) significantly inhibited tumor growth of MDA-MB-231 xenografts in nude mice. However, unlike eribulin, capecitabine failed to increase fractions of Hoechst 33342-positive areas (Fig.2e,f), indicating that the increased tumor perfusion seen with eribulin is not simply dependent on tumor growth inhibition. Consistent with the DCE-MRI results obtained above in tumor xenografts grown in nude rats, these results from Hoechst 33342 perfusion studies in nude mice indicate that a single administration of eribulin is sufficient to trigger substantial increases in tumor vascular perfusion.
Figure 2.
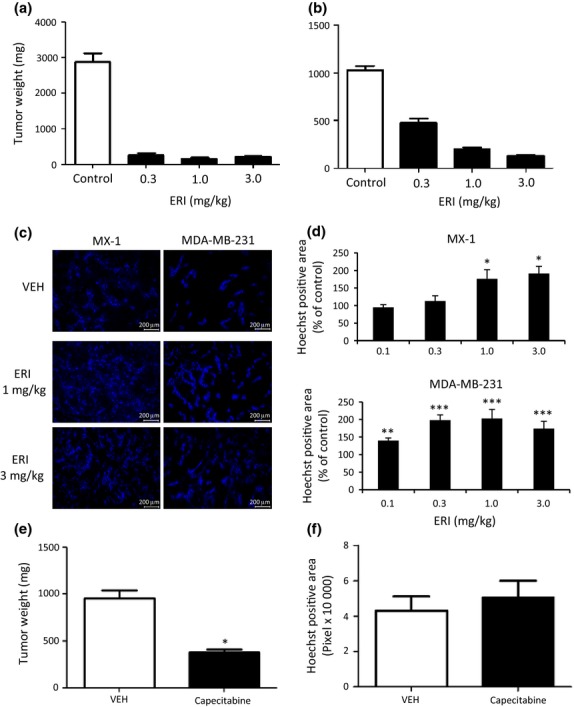
Effects of eribulin on Hoechst 33342 perfusion in human breast cancer xenografts in nude mice. Eribulin i.v. at indicated doses (ERI, a–d), capecitabine p.o. at 540 mg/kg (e,f), or vehicle (VEH) was administered to nude mice bearing MX-1 (b–d) or MDA-MB-231 (a,c–f) xenografts. Each group consisted of 5–6 (a,b,e,f) or 10 (c,d) mice. (a,b,e) Antitumor activity. (c) Representative images of Hoechst 33342 staining (perfusion). (d,f) Quantitative analyses of Hoechst 33342 perfusion. Data represent means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus VEH.
Effects of eribulin on tumor vascular morphology in human breast cancer xenograft models in nude mice
Effects of eribulin on tumor vascular morphology in MX-1 and MDA-MB-231 xenografts in nude mice were evaluated by immunohistochemical staining with antibodies against mouse CD31, an endothelial cell marker. Nude mice bearing xenografted tumors were randomized into treatment groups and treatments started on day 1, when the mean tumor weight reached approximately 200 mg. Eribulin was administered on day 1 and the morphology of tumor vessels was evaluated 7 days after eribulin treatment (day 8) (Fig.3). Eribulin treatment at a dose of 3 mg/kg led to marked changes in CD31-stained tumor vessels in both xenograft types compared with the vehicle: stained areas became smaller, more frequent, without clear lumens, and the number of large branched vascular structures decreased (Fig.3a). Quantitative analysis showed that MVD significantly increased in both tumor xenografts at doses of 1.0 and 3.0 mg/kg, whereas MVA significantly decreased in the MDA-MB-231 model at 0.3, 1.0 or 3.0 mg/kg compared with the vehicle (Fig.3b). The proportion of small vessels (<200 μm2) significantly increased with eribulin at all doses in the MDA-MB-231 model; similar trends were noted in the MX-1 model, although such changes were smaller than those seen in the MDA-MB-231 model. These results demonstrate that eribulin altered the morphology of the tumor vasculature, leading to increased aggregate surface area of the microvasculature, thus explaining the increased tumor perfusion following eribulin treatment observed above.
Figure 3.

Immunohistochemical analysis of tumor vasculature in human breast cancer xenografts in nude mice. Vehicle (VEH) or eribulin (ERI) at indicated doses was i.v. administered to nude mice bearing MX-1 or MDA-MB-231 xenografts. Immunohistochemical analysis was conducted by staining endothelial cells with CD31 with or without H&E staining. Each group consisted of 5–10 mice. (a) Representative images of tumor vasculature (CD31 staining), (b) Quantitative analyses of morphology of tumor vasculature as determined by microvessel density (MVD), mean vascular area (MVA) and proportion of small vessels. Data are means ± SEM. *P < 0.05 versus VEH.
Effects of eribulin on angiogenesis-related and epithelial–mesenchymal transition-related gene expression levels in mouse-derived (host) tumor stroma of human breast cancer xenografts in nude mice
Tumor microenvironments regulate vascular morphology and consist of many non-malignant cells.31 Therefore, we explored the mechanisms underlying the effects of eribulin on non-malignant host (murine) cells in MX-1 and MDA-MB-231 xenograft models by conducting qRT-PCR-based gene expression profiling of mouse-derived tumor stroma. We focused on 84 genes associated with angiogenesis regulatory pathways based on known expression in endothelial cells or pericytes,31–33 as well as 43 EMT-related epithelial or mesenchymal marker genes (Table S1). Four days following eribulin treatment, expression levels of 29 murine genes involved in angiogenesis regulatory pathways were significantly reduced in both MX-1 and MDA-MB-231 xenograft models in a dose-dependent manner (Fig.4a, and Tables S1a, S2). Suppression of host angiogenesis-related genes after eribulin treatment occurred in several different pathways known to regulate angiogenesis, including the VEGF pathway (Vegfa, Pgf, Vegfr1, Vegfr2 and Vegfr3), the Notch pathway (Dll4, Jag1 and Notch4), the Eph pathway (Efnb2, Epha2 and EphB1) and the WNT pathway (Wnt5a, Wnt11, BPM4). These results indicate that eribulin affects key signaling pathways involved in the regulation of angiogenesis within stromal components of the tumor microenvironment. Although no EMT-related epithelial marker genes showed significantly altered expression after eribulin treatment in both models (Tables S1b and S3), 13 EMT-related mesenchymal marker genes, including Snai1, Snai2, Tgfb1, Tgfb2 and Vim, showed decreased expression after eribulin treatment in both models (Fig.4b).
Figure 4.

Effects of eribulin on gene expression and hypoxia of tumor stroma in human breast cancer xenografts in nude mice. Vehicle (VEH) or eribulin (ERI) at indicated doses was administered to nude mice bearing MX-1 or MDA-MB-231 xenografts. Each group consisted of 5 mice. (a,b) Quantitative RT-PCR analyses of mouse genes were performed for (a) angiogenesis-related genes and (b) epithelial–mesenchymal transition (EMT)-related genes that showed significantly altered expression in a dose-dependent manner in both models. Data are the average ratios of ERI treatment to VEH. (c) Levels of mouse vascular endothelial growth factor A protein in tumor tissues, as measured by ELISA. Data are means ± SEM. *P < 0.05 versus VEH. (d) Representative fluorescent images of tumor tissues showing markers of endothelial cells (CD31), perfusion (Hoechst) and hypoxia (CA9).
Although expression levels of VEGF were significantly decreased with eribulin treatment in both xenograft models (Fig.4a), interestingly, they were not decreased in an in vitro angiogenesis model consisting of human umbilical vein endothelial cells co-cultured with human brain vascular pericytes (Fig. S2 and Table S4). Because VEGF is induced by hypoxia,34 we evaluated the relationship between hypoxia and mouse VEGF protein levels in tumor stroma after eribulin treatment. Eribulin treatment at doses of 0.3 and 1.0 mg/kg significantly decreased mouse VEGF protein levels in both tumor xenograft models compared with the vehicle (Fig.4c). Expression of CA9 protein, a marker for hypoxia that is regulated by hypoxia-inducible factor and is found around regions of necrosis,35 also decreased, with 1.0 mg/kg eribulin in the MDA-MB-231 model (Fig.4d). These results indicate that eribulin treatment affected gene expression in angiogenesis-related and EMT-related signaling pathways within the abnormal tumor stroma and reduced the degree of hypoxia in both tumor xenograft models.
Effects of prior treatment with eribulin on antitumor activity of capecitabine in the MDA-MB-231 human breast cancer xenograft model in nude mice
The results above led us to investigate whether eribulin-induced increases in tumor core perfusion could lead to better inner tumor delivery of subsequently administered drugs, with a potential outcome of enhanced antitumor efficacy of such administrations. To test this hypothesis, we evaluated the antitumor activity of 540 mg/kg capecitabine in the MDA-MB-231 model with or without prior treatment with 1.0 mg/kg eribulin. Results indicated that prior treatment with eribulin (Fig. S3) significantly enhanced the antitumor activity of capecitabine compared with that achieved without prior treatment (Fig.5), consistent with the idea that increased perfusion of tumor cores leads to enhanced delivery of subsequently administered drugs, resulting in better antitumor efficacy.
Figure 5.
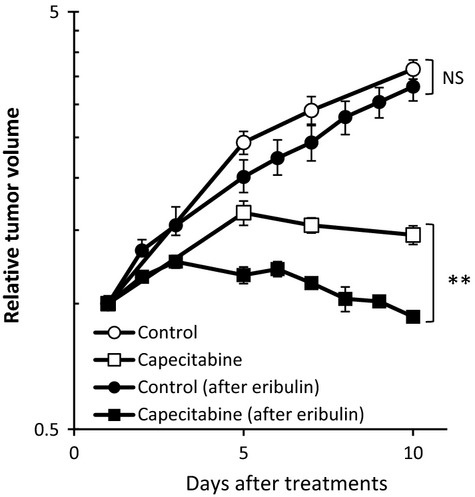
Effects of prior treatment with eribulin on antitumor activity of capecitabine in the MDA-MB-231 human breast cancer xenograft model in nude mice. When the mean MDA-MB-231 xenograft tumor volumes reached approximately 200 mg (day 1), nude mice were randomly divided into control (non-treatment), capecitabine (540 mg/kg, QD8, p.o.) and eribulin (1.0 mg/kg,QD1, i.v.) groups. Following treatment in the eribulin group, when tumor volumes had recovered to the initial target level of tumor size (i.e. approximately 200 mg; on day 12, see Fig. S3), eribulin-treated mice were then randomly divided into new control (eribulin-pretreated) and capecitabine (eribulin-pretreated) groups as if on a new day 1. Each group consisted of 6 mice. Data are means ± SEM. *P < 0.05 versus after eribulin treatment by Student's t-test. NS, not significant.
Discussion
The present studies were undertaken to examine the potential effects of eribulin on tumor vascular function in two different models of human breast cancer xenografts, MX-1 and MDA-MB-231. First, we employed DCE-MRI to determine antivascular activities of eribulin in the two models in nude rats. Unexpectedly, eribulin robustly increased Ktrans in the tumor core, which had previously been poorly vascularized at the time of baseline scanning (day −1). Moreover, iAUC histogram analysis after eribulin treatment showed almost completely overlapping curves for tumor rim and core regions, indicating that eribulin-induced remodeling of tumor vasculature had led to uniform tumor perfusion across all tumor regions. Such remodeling was readily observed 5 days after treatments, a time at which eribulin-treated tumor volumes were similar to those seen before eribulin treatment. Two classes of TBD have been defined based on their predominant antivascular activities: antiangiogenic and vascular-disrupting.9 Our results, showing that eribulin increases inner tumor core perfusion, fall outside of previously-described phenomena, suggesting that eribulin's effects may represent a new, previously-undescribed category of vascular modulating activities of TBD.
Consistent with DCE-MRI data obtained in nude rats, analysis of Hoechst 33342 staining, a functional marker for perfusion, confirmed that eribulin caused higher perfusion in both MX-1 and MDA-MB-231 xenograft models in nude mice. Immunohistochemical analysis with anti-mouse CD31 antibody indicated that eribulin decreased the proportion of large branched tumor vessels and increased the proportion of small vessels, with the overall result of increased MVD and decreased MVA. Such conditions would be expected to lead to increased perfusion per unit of vascular area, and are consistent with the DCE-MRI and Hoechst 33342 staining results above.
Vascular networks in tumors frequently show abnormal, complex branched patterns with a lack of typical arterial and venous network hierarchy, resulting in a high geometrical resistance to blood flow.7,36,37 After treatment with antiangiogenesis agents such as bevacizumab, tumor vessels are often described as having been temporarily “normalized,” effects that are accompanied by decreased MVD.38 In contrast, our studies reveal that eribulin treatment leads to increased, not decreased, MVD; in this regard, eribulin shares some characteristics of tumor vasculature “normalization,” such as reduced vessel diameters. Our data suggest that the vascular remodeling activity of eribulin is part of a longer-term phenotypic change of the tumor microenvironment rather than the temporary “normalization” effects that have been described for other antiangiogenesis agents such as bevacizumab. Importantly, the increased MVD induced by eribulin did not stimulate growth of MX-1 or MDA-MB-231 xenografts, indicating that eribulin's vascular remodeling activity in the context of tumor microenvironment is separable from eribulin's direct inhibitory effects on tumor growth.
Eribulin decreased the expression of mouse genes associated with the VEGF, Notch and Eph signaling pathways within stromal compartments of tumor microenvironment in both xenograft models examined. These pathways are known to be involved in sequential processes of vascular branching, starting with tip cell formation and ending with maturation of newly formed vessels.39 In in vitro vascular networks established by co-culturing HUVEC with HBVP, eribulin decreased the expression of Dll4, Notch4 and Efnb2 (Fig. S2), demonstrating that eribulin directly regulates angiogenesis signaling pathways in vascular cells. These in vitro cell-based results support the hypothesis that eribulin-induced tumor vascular remodeling and increased tumor perfusion is caused by altered expression levels of tumor vasculature regulatory genes in host tumor stromal components.
Tumor microenvironment is markedly abnormal compared to that of nonmalignant tissues, and such abnormalities can enhance tumor metastasis and resistance to anticancer drugs.32,33 Eribulin did not inhibit tumor growth of murine breast cancer 4T1 cells at the mammary fat pad in the syngeneic mouse model (Fig. S4), because 4T1 cells expressed p-glycoprotein, called MDR, and showed low sensitivity to eribulin (IC50 is approximately 100 nM; data not shown). However, effects of eribulin on abnormal tumor microenvironment might contribute to a decrease in the development of lung metastasis of murine breast cancer 4T1 grown at the mammary fat pad (Fig. S4).
In summary, eribulin treatment results in improved tumor perfusion in human breast cancer tumor models, an effect driven by vascular remodeling associated with increased numbers of smaller functional microvessels. The overall result of eribulin-induced increased tumor perfusion is removal of the hypoxic stress associated with abnormal tumor microenvironments, and increased ability of subsequently administered drugs to reach tumor areas that had previously been poorly perfused. Combined, such effects might contribute to increased antitumor activities of subsequent treatments with an anticancer drug (Fig.5), enhancement of eribulin activities when combined with other agents, decreased development of drug resistance, and decreased tendency for tumors to metastasize. Further translational studies are currently underway to determine whether and how the eribulin-induced effects on the tumor vasculature and microenvironment revealed in the current studies might contribute to the overall clinical benefits of eribulin therapy.
Acknowledgments
We thank Tyler Teceno, Denise Welsh, Galina Kuznetsov, Namita Kumar and Shanqin Xu for their experimental contributions, Kentaro Matsuura for statistical analyses, and Ken Ito, Mamoru Yanagimachi, Mitsuhiro Ino, Masao Iwata and Jesse Chow for helpful discussions. We also thank Bruce Littlefield for critical reading of the manuscript.
Disclosure Statement
YF, PJM, KN, and YO are employees of Eisai Inc. and XT is a employee of Oncodesign S.A. Other authors are employee of Eisai Co, Ltd.
Supporting Information
Fig. S1. dynamic contrast-enhanced (DCE)-MRI analysis of eribulin with initial area under the curve (iAUC) histograms.
Fig. S2. Effects of eribulin on gene expression in pericyte-covered vascular networks in vitro.
Fig. S3. Antitumor activity of prior treatment with eribulin in the MDA-MB-231 xenograft model.
Fig. S4. Effect of eribulin on 4T1 lung metastases.
Table S1. Alteration of mouse gene expression in tumor stroma of xenografted human tumors treated with eribulin.
Table S2. Mouse gene expression in angiogenesis-related signaling pathways in tumor stroma.
Table S3. Mouse gene expression in epithelial–mesenchymal transition (EMT)-related signaling pathways in tumor stroma.
Table S4. Gene expression in angiogenesis-related signaling pathways in pericyte-covered vascular networks in vitro.
References
- Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–39. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- Belotti D, Vergani V, Drudis T, et al. The microtubule-affecting drug paclitaxel has antiangiogenic activity. Clin Cancer Res. 1996;2:1843–9. [PubMed] [Google Scholar]
- Grant DS, Williams TL, Zahaczewsky M, Dicker AP. Comparison of antiangiogenic activities using paclitaxel (taxol) and docetaxel (taxotere) Int J Cancer. 2003;104:121–9. doi: 10.1002/ijc.10907. [DOI] [PubMed] [Google Scholar]
- Pasquier E, Honore S, Pourroy B, et al. Antiangiogenic concentrations of paclitaxel induce an increase in microtubule dynamics in endothelial cells but not in cancer cells. Cancer Res. 2005;65:2433–40. doi: 10.1158/0008-5472.CAN-04-2624. [DOI] [PubMed] [Google Scholar]
- Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–35. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- Hinnen P, Eskens FA. Vascular disrupting agents in clinical development. Br J Cancer. 2007;96:159–65. doi: 10.1038/sj.bjc.6603694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EL. Antivascular actions of microtubule-binding drugs. Clin Cancer Res. 2009;15:2594–601. doi: 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozer GM, Prise VE, Wilson J, et al. Mechanisms associated with tumor vascular shut-down induced by combretastatin A-4 phosphate: intravital microscopy and measurement of vascular permeability. Cancer Res. 2001;61:6413–22. [PubMed] [Google Scholar]
- Salmon HW, Siemann DW. Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin Cancer Res. 2006;12:4090–4. doi: 10.1158/1078-0432.CCR-06-0163. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Zakhireh B, Malech HL. The effect of colchicine and vinblastine on the chemotactic response of human monocytes. J Immunol. 1980;125:2143–53. [PubMed] [Google Scholar]
- Liao G, Nagasaki T, Gundersen GG. Low concentrations of nocodazole interfere with fibroblast locomotion without significantly affecting microtubule level: implications for the role of dynamic microtubules in cell locomotion. J Cell Sci. 1995;108:3473–83. doi: 10.1242/jcs.108.11.3473. [DOI] [PubMed] [Google Scholar]
- Cortes J, O'Shaughnessy J, Loesch D, et al. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomized study. Lancet. 2011;377:914–23. doi: 10.1016/S0140-6736(11)60070-6. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Lemery SJ, Yuan W, et al. Eribulin mesylate for the treatment of patients with refractory metastatic breast cancer: use of a “physician's choice” control arm in a randomized approval trial. Clin Cancer Res. 2012;18:1496–505. doi: 10.1158/1078-0432.CCR-11-2149. [DOI] [PubMed] [Google Scholar]
- Pean E, Klaar S, Berglund EG, et al. The European medicines agency review of eribulin for the treatment of patients with locally advanced or metastatic breast cancer: summary of the scientific assessment of the committee for medicinal products for human use. Clin Cancer Res. 2012;18:4491–7. doi: 10.1158/1078-0432.CCR-11-3075. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Kamath K, Manna T, et al. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol Cancer Ther. 2005;4:1086–95. doi: 10.1158/1535-7163.MCT-04-0345. [DOI] [PubMed] [Google Scholar]
- Kuznetsov G, Towle MJ, Cheng H, et al. Induction of morphological and biochemical apoptosis following prolonged mitotic blockage by halichondrin B macrocyclic ketone analog E7389. Cancer Res. 2004;64:5760–6. doi: 10.1158/0008-5472.CAN-04-1169. [DOI] [PubMed] [Google Scholar]
- Towle MJ, Salvato KA, Wels BF, et al. Eribulin induces irreversible mitotic blockade: implications of cell-based pharmacodynamics for in vivo efficacy under intermittent dosing conditions. Cancer Res. 2011;71:496–505. doi: 10.1158/0008-5472.CAN-10-1874. [DOI] [PubMed] [Google Scholar]
- Towle MJ, Salvato KA, Budrow J, et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001;61:1013–21. [PubMed] [Google Scholar]
- Towle MJ, Nomoto K, Asano M, Kishi Y, Yu MJ, Littlefield BA. Broad spectrum preclinical antitumor activity of eribulin (Halaven(R)): optimal effectiveness under intermittent dosing conditions. Anticancer Res. 2012;32:1611–9. [PubMed] [Google Scholar]
- Nielsen T, Bentzen L, Pedersen M, et al. Combretastatin A-4 phosphate affects tumor vessel volume and size distribution as assessed using MRI-based vessel size imaging. Clin Cancer Res. 2012;18:6469–77. doi: 10.1158/1078-0432.CCR-12-2014. [DOI] [PubMed] [Google Scholar]
- Galbraith SM, Maxwell RJ, Lodge MA, et al. Combretastatin A4 phosphate has tumor antivascular activity in rat and man as demonstrated by dynamic magnetic resonance imaging. J Clin Oncol. 2003;21:2831–42. doi: 10.1200/JCO.2003.05.187. [DOI] [PubMed] [Google Scholar]
- O'Connor JP, Jackson A, Parker GJ, Jayson GC. DCE-MRI biomarkers in the clinical evaluation of antiangiogenic and vascular disrupting agents. Br J Cancer. 2007;96:189–95. doi: 10.1038/sj.bjc.6603515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor JP, Jackson A, Parker GJ, Roberts C, Jayson GC. Dynamic contrast-enhanced MRI in clinical trials of antivascular therapies. Nat Rev Clin Oncol. 2012;9:167–77. doi: 10.1038/nrclinonc.2012.2. [DOI] [PubMed] [Google Scholar]
- Patterson DM, Zweifel M, Middleton MR, et al. Phase I clinical and pharmacokinetic evaluation of the vascular-disrupting agent OXi4503 in patients with advanced solid tumors. Clin Cancer Res. 2012;18:1415–25. doi: 10.1158/1078-0432.CCR-11-2414. [DOI] [PubMed] [Google Scholar]
- Fram EK, Herfkens RJ, Johnson GA, et al. Rapid calculation of T1 using variable flip angle gradient refocused imaging. Magn Reson Imaging. 1987;5:201–8. doi: 10.1016/0730-725x(87)90021-x. [DOI] [PubMed] [Google Scholar]
- Tofts PS, Brix G, Buckley DL, et al. Estimating kinetic parameters from dynamic contrast-enhanced T(1)-weighted MRI of a diffusible tracer: standardized quantities and symbols. J Magn Reson Imaging. 1999;10:223–32. doi: 10.1002/(sici)1522-2586(199909)10:3<223::aid-jmri2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Fornier MN. Approved agents for metastatic breast cancer. Semin Oncol. 2011;38(Suppl 2):S3–10. doi: 10.1053/j.seminoncol.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M. Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene. 2013;33:1743–54. doi: 10.1038/onc.2013.121. [DOI] [PubMed] [Google Scholar]
- Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–18. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke VG, LeBleu VS, Keskin D, et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell. 2012;21:66–81. doi: 10.1016/j.ccr.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Wykoff CC, Beasley NJ, Watson PH, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83. [PubMed] [Google Scholar]
- Konerding MA, Malkusch W, Klapthor B, et al. Evidence for characteristic vascular patterns in solid tumours: quantitative studies using corrosion casts. Br J Cancer. 1999;80:724–32. doi: 10.1038/sj.bjc.6690416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevick EM, Jain RK. Geometric resistance to blood flow in solid tumors perfused ex vivo: effects of tumor size and perfusion pressure. Cancer Res. 1989;49:3506–12. [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. dynamic contrast-enhanced (DCE)-MRI analysis of eribulin with initial area under the curve (iAUC) histograms.
Fig. S2. Effects of eribulin on gene expression in pericyte-covered vascular networks in vitro.
Fig. S3. Antitumor activity of prior treatment with eribulin in the MDA-MB-231 xenograft model.
Fig. S4. Effect of eribulin on 4T1 lung metastases.
Table S1. Alteration of mouse gene expression in tumor stroma of xenografted human tumors treated with eribulin.
Table S2. Mouse gene expression in angiogenesis-related signaling pathways in tumor stroma.
Table S3. Mouse gene expression in epithelial–mesenchymal transition (EMT)-related signaling pathways in tumor stroma.
Table S4. Gene expression in angiogenesis-related signaling pathways in pericyte-covered vascular networks in vitro.